

Abstract
Frontotemporal lobar degeneration is a highly familial disease and the most common known genetic cause is the repeat expansion mutation in the gene C9orf72. We have identified 2 brothers with an expansion mutation in C9orf72 using Southern blotting that is undetectable using repeat-primed polymerase chain reaction. Sequencing using high concentrations of DNA denaturants of a bacterial artificial chromosome clone obtained from one of the brothers identified a 10-base pair deletion adjacent to the expansion that presumably confers strong secondary structure that interferes with the genotyping. Using an alternative method, we have identified missed expansion carriers in our cohort, and this number has increased by approximately 25%. This observation has important implications for patients undergoing genetic testing for C9orf72.
Keywords: Frontotemporal lobar degeneration, FTLD, C9orf72, Repeat expansion, ALS
1. Introduction
Frontotemporal lobar degeneration (FTLD) is a common neurodegenerative disease that usually affects people in midlife (Snowden et al., 2007). It is clinically heterogenous with 3 main subtypes: behavioral variant frontotemporal dementia (bvFTD), progressive nonfluent aphasia, and semantic dementia. It is also pathologically heterogenous with about 50% of cases showing neuronal cytoplasmic inclusions consisting of hyperphosphorylated tau protein. Approximately 45% of cases have neuronal cytoplasmic inclusions that lack tau, but instead contain the DNA- and RNA-binding protein TDP-43. The remaining 5% cases have inclusions that consist of another DNA- and/or RNA-binding protein, fused in sarcoma (Mackenzie et al., 2011).
Up to 40% of patients with FTLD present with a family history of this disease and in some instances also with amyotrophic lateral sclerosis (ALS). Much progress in the understanding of the genetic basis of FTLD has been made in the past 15 years, with 3 main genes causing autosomal dominant forms of disease having been identified. These are tau gene (Hutton et al., 1998), progranulin (Baker et al., 2006), and most recently a hexanucleotide repeat expansion in C9orf72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011), the latter being the most common genetic cause of FTLD and ALS identified to date. Despite being extremely large, sometimes over 20 kb, the expansion is relatively easy to genotype using repeat-primed polymerase chain reaction (PCR). This method has been widely used to establish the frequency of the expansion in populations worldwide (Majounie et al., 2012). In addition, this methodology is presently used in a clinical genetics setting to aid diagnosis of FTLD and ALS. The expansion has been shown to be the most common in Northern European populations, probably because of a founder effect (Mok et al., 2012). Currently, 3 potential mechanisms have been proposed by which the expansion leads to disease, these being haploinsufficiency, toxic RNA in the form of nuclear foci, and dipeptide-repeat proteins (DPR) which are repeat associated non-ATG translated from the expansion (Ash et al., 2013; Mori et al., 2013).
When establishing the presence and frequency of the C9orf72 repeat expansion in our own clinical and autopsy patient cohorts with various diseases, we detected 2 patients (brothers, one with bvFTD and the other ALS) that were positive for DPR pathology in both cerebral cortex and cerebellum (Supplementary Fig. 1) but had no expansion when tested with repeat-primed PCR. Despite repeated re-extraction of DNA from both blood and brain tissues in both patients, we could not detect more than 6 repeats using previously published repeat-primed PCR (Renton et al., 2011). Consequently, a Southern blot was performed with DNA extracted from brain tissue. This showed both patients had an expansion of approximately 1500–3000 repeats. As the repeat-primed PCR methodology is currently being used in a clinical genetics setting to diagnose disease presence in patients and to test family members for disease risk, it is vital that such methodology is robust in providing a correct genotype. Here, we describe the characterization of the C9orf72 expansion in these 2 patients to understand why we were unable to detect the expansion when using previously validated repeat-primed PCR (Renton et al., 2011).
2. Methods
All FTLD patients known to harbor a proven mutation MAPT or GRN were excluded, as were those cases that displayed tau pathology at postmortem. The final study group comprised 460 patients (mean age at onset, 60 years; range, 24–83 years), 244 men (mean age at onset, 60 years; range, 34–81 years), and 181 women, (mean age at onset, 59 years; range, 23–83 years). Control cases were drawn from healthy spouse, 253 being male (mean age, 56 years; range, 30–79 years) and 397 female (mean age, 53 years; range, 26–81 years). The London cohort consisted of 367 cases of FTLD (41% female, average onset of 59). All patients and control subjects were of UK Caucasian ethnic origin and were recruited with ethical committee approval and provided informed consent.
A bacterial artificial chromosome (BAC) library was constructed, from cerebellar tissue from the case of bvFTD described in the introduction, by Bio S&T Inc (Canada) using the pIndigoBAC-5 vector. This was screened to isolate a BAC clone that contained C9orf72, and the expansion. Sanger sequencing was performed using BigDye v3 (Applied Biosystems) run on an ABI 3730 (Applied Biosystems). One microgram of BAC DNA was included in each reaction along with 10 pm of primer, 8 μL of BigDye sequencing master mix v3.1 (Applied Biosystems) in a total volume of 40 μL. When used, DMSO was added, it was at a final concentration of 6% and betaine at 1 M. Ion torrent next generation sequencing was performed according to the manufacturer's instructions (Life Technology). Southern blot and repeat-primed PCR were performed as previously described (Mann et al., 2013; Renton et al., 2011).
3. Results
Our initial thoughts which might explain our inability to detect an expansion in these 2 brothers was that there was an single nucleotide polymorphism under the primer used for repeat-primed PCR that lay outside of the expansion. However, we sequenced this region which was wild type in both patients (not shown). An expansion in C9orf72 was confirmed by Southern blotting in both brothers (Fig. 1). To elucidate as to why the repeat-primed PCR did not detect this, we attempted to sequence the BAC clone of C9orf72 with the expansion we had obtained. When we used standard Sanger sequencing with a primer placed on the telomeric side of the expansion, we obtained good sequence data that read through some of the expansion and confirmed it was the usual GGGGCC hexanucleotide repeat (Supplementary Fig. 2). This also proved that the expansion was not GC clamping the BAC and preventing polymerase read through. However, when we placed a primer on the centromeric side of the repeat and sequenced toward it, the reaction consistently failed to produce any sequence data, even when we placed a primer up to 1 kb away. After ruling out issues of DNA purity, we attempted to use next generation sequencing (Ion Torrent Personal Genome Machine) to sequence the entire BAC. We obtained around a 70-fold read depth for the entire BAC apart from the repeat region where coverage was low, particularly on the centromeric side. Analysis of this data did not reveal any differences to the reference genome, Primary Assembly (GRCh38). Given that Sanger sequencing produced good reads from the telemetric side of the expansion, but not from the centromeric side, we hypothesized that a variation in the sequence in this part of C9orf72 was producing a secondary structure that was preventing sequencing polymerase read through. We therefore attempted a Sanger sequencing reaction with high levels of DNA denaturants (1 M betaine and 6% DMSO). This produced a good sequence and when compared with the reference genome (GRCh38 Primary Assembly), we were able to detect a 10 bp deletion directly adjacent to the start of the repeat (Fig. 2). This 10 bp section contains some AT sequence, and when removed the repeat is then adjacent to more GC rich sequence.
Fig. 1.

Southern blot (as of Mann et al. (2013) for Set A and Beck et al. (2013) for Set B) of C9orf72 of both brothers confirming a repeat expansion is present in both. Set A left to right, marker; negative control, positive control, patient samples. Set (B) left to right; marker; patient samples; positive control; negative control.
Fig. 2.
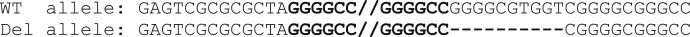
Ten bp deletion adjacent to the GGGGCC repeat region.
Previously, we had been using the assay of Renton et al. (2011) to genotype our FTLD and control cohorts and had identified 37 expansion carriers in the FTLD group. When we repeated the genotyping using the assay of DeJesus-Hernandez et al. (2011) (which should not be affected by the presence of the deletion; Fig. 3) we detected a further 11 cases, an increase of approximately 25%. In addition, we genotyped our ALS cohort (n = 234) and the number of expansion carriers increased from 16 to 20 (Table 1). However, repeated analysis of previously screened London FTLD cohort failed to detect any additional cases.
Fig. 3.

Top–repeat primed PCR showing 6 repeats in C9orf72 using the assay by Renton et al. (2011) in a case with the 10 bp deletion. Bottom–the same case genotyped with the assay by DeJesus-Hernandez et al. (2011) showing the expansion also shown by Southern blot. Abbreviation: PCR, polymerase chain reaction.
Table 1.
rs10967976 genotype analysis in the Manchester FTLD cohort
| rs10967976 | Controls (%) | FTD all (%) | OR | 95% CI | p | FTD and/or MND (%) | OR | 95% CI | p |
|---|---|---|---|---|---|---|---|---|---|
| All cases | |||||||||
| AA | 164 (26.54) | 106 (24.20) | 1.0 | — | 11 (16.92) | 1.0 | — | ||
| GA | 330 (53.40) | 217 (49.54) | 1.01 | 0.75–1.37 | 0.91 | 34 (52.31) | 1.53 | 0.75–3.10 | 0.233 |
| GG | 124 (20.06) | 115 (26.26) | 1.43 | 1.00–2.04 | 0.045 | 20 (30.77) | 2.40 | 1.11–5.20 | 0.026 |
| GA + GG | 454 (73.46) | 332 (75.80) | 1.13 | 0.85–1.50 | 0.86 | 54 (83.08) | 1.77 | 0.90–3.47 | 0.095 |
| AA + GA | 494 (79.94) | 323 (73.74) | 1.0 | — | 45 (69.23) | 1.0 | |||
| GG | 124 (20.06) | 115 (26.26) | 1.42 | 1.06–1.89 | 0.016 | 20 (30.77) | 1.81 | 1.00–3.10 | 0.046 |
| Drop “old” expansion cases | |||||||||
| AA | 164 (26.54) | 103 (25.69) | 1.0 | 11 (21.15) | 1.0 | ||||
| GA | 330 (53.40) | 200 (49.88) | 0.96 | 0.71–1.30 | 0.817 | 25 (48.08) | 1.12 | 0.54–2.35 | 0.745 |
| GG | 124 (20.06) | 98 (24.44) | 1.26 | 0.87–1.80 | 0.213 | 16 (30.77) | 1.92 | 0.86–4.29 | 0.110 |
| GA + GG | 454 (73.46) | 298 (74.31) | 1.04 | 0.78–1.39 | 0.763 | 41 (78.85) | 1.34 | 0.67–2.68 | 0.398 |
| AA + GA | 494 (79.94) | 303 (75.56) | 1.0 | 36 (69.23) | 1.0 | ||||
| GG | 124 (20.06) | 99 (24.44) | 1.28 | 0.95–1.74 | 0.099 | 16 (30.77) | 1.77 | 0.95–3.29 | 0.071 |
| Drop “old” and “new” expansion cases | |||||||||
| AA | 164 (26.54) | 100 (25.64) | 1.0 | 10 (20.41) | 1.0 | ||||
| GA | 330 (53.40) | 198 (50.77) | 0.98 | 0.72–1.33 | 0.917 | 25 (51.02) | 1.24 | 0.58–2.64 | 0.574 |
| GG | 124 (20.06) | 92 (23.59) | 1.21 | 0.84–1.75 | 0.295 | 14 (28.57) | 1.85 | 0.79–4.30 | 0.153 |
| GA + GG | 454 (73.46) | 290 (74.36) | 1.04 | 0.78–1.39 | 0.768 | 39 (79.59) | 1.40 | 0.68–2.88 | 0.349 |
| AA + GA | 494 (79.94) | 298 (76.41) | 1.0 | 35 (71.43) | 1.0 | ||||
| GG | 124 (20.06) | 92 (23.59) | 1.22 | 0.90–1.66 | 0.184 | 14 (28.57) | 1.59 | 0.83–3.20 | 0.160 |
Key: CI, confidence interval; FTD, frontotemporal dementia; FTLD, frontotemporal lobar degeneration; OR, odds ratio.
In a recent study, Jones et al. (2013) reported that residual genetic association to chromosome 9p remained, even when known expansion carriers were removed from their ALS cohort. When we performed this same analysis in our cohort using rs10967976 following removal of old and newly identified expansion carriers, previous residual association was completely abolished (Table 2).
Table 2.
Frequencies of the original and newly identified C9orf72 expansion carriers in the Manchester cohort
| Cases | C9 carriers old | C9 carriers new | C9 carriers total |
|---|---|---|---|
| All FTLD, N = 460 | 37 | 11 | 48 |
| FTD + ALS, N = 74 | 12 | 3 | 15 |
| ALS, N = 234 | 16 | 4 | 20 |
Key: ALS, amyotrophic lateral sclerosis; FTLD, frontotemporal lobar degeneration.
4. Discussion
DRP inclusions are thought to be specific to C9orf72 expansion carriers; however, there has been some controversy as to their role in the neurodegenerative process (Mann, 2014). Nevertheless, they could be considered the equivalent of amyloid plaques in Alzheimer's disease with TDP-43 inclusions being the equivalent neurofibrillary inclusions, and this may account for the discrepancy of DPR distribution and neurodegeneration. Our neuropathologic analyses of the brains of the 2 brothers showing DPR inclusions, when no such expansion had been found using PCR methodology, lead us to reanalyze these cases in depth. Indeed, Southern blotting revealed an expansion that was undetectable using our standard assay (Renton et al., 2011). Subsequently, however, such an expansion became detectable using the assay of DeJesus-Hernandez et al. (2011). These observations highlight to need to consider to what extent genetic variation can affect PCR-based assays and reenforces the utility of relevant neuropathologic investigation. Sequencing of a BAC cloned from genomic DNA from one of the individuals identified a 10 bp deletion adjacent to the repeat region. A 10 base deletion at this region has been reported previously but with a normal nonexpanded GGGGCC repeat (van der Zee et al., 2013). However, the deleted sequence reported by van der Zee et al. (2013) is reversed when compared with the sequence in the current genome Primary Assembly (GRCh38). Investigation of alternative assemblies in the genome databases reveals some heterogeneity at this locus which could explain these differences. Standard Sanger sequencing failed to read through the area containing the deletion in the BAC, and sequence data were only obtained when strong DNA denaturants were included into the reaction. Presumably, the deletion confers a strong secondary structure in the region that prevents polymerase read through. Although denaturants are also included into the repeat-primed PCR assay, the wild-type allele still gets preferentially amplified and dominates over the mutant.
Using the assay of DeJesus-Hernandez et al. (2011), we could bypass issues of the deletion affecting genotyping, and in so doing increased the number of expansion carriers in our cohort by approximately 25%. There were not any robust clinical differences between the newly identified cases and those previously identified. Nonetheless, other previously undetected carriers were not identified in the London cohort, suggesting that the deletion in combinations with the expansion could be geographically specific. The study reported by Jones et al. (2013), reporting residual association in C9orf72 following removal of known expansion carriers, paradoxically claimed to use the assay of DeJesus-Hernandez et al. (2011) and therefore should not have been affected by the 10 bp deletion. However, it is possible there is other genetic variation on the other side of the expansion affecting this assay, with expansion carriers bearing such a variation again being missed, and this explaining the residual association. Clearly, local founder effects do occur as we only find the 10 bp deletion with an expansion on the same haplotype in cases from the North West of England all of which could theoretically be distantly related. However, further genotyping is required to fully establish whether this is the case. Moreover, there has been a recent report of major inconsistencies in correctly genotyping cases of known expansion by different laboratories using local methodologies (Akimoto et al., 2014). This observation has important implications for patients undergoing genetic testing for C9orf72, and we would recommend genotyping samples with both Renton and DeJesus-Hernandez PCR-based methods to ensure that expansion carriers do not go undetected.
Disclosure statement
The authors have no conflicts of interest to disclose.
Acknowledgements
This work was supported by the Medical Research Council (G0701441).
Appendix A. Supplementary data
References
- Akimoto C., Volk A.E., van Blitterswijk M., Van den Broeck M., Leblond C.S., Lumbroso S., Camu W., Neitzel B., Onodera O., van Rheenen W., Pinto S., Weber M., Smith B., Proven M., Talbot K., Keagle P., Chesi A., Ratti A., van der Zee J., Alstermark H., Birve A., Calini D., Nordin A., Tradowsky D.C., Just W., Daoud H., Angerbauer S., DeJesus-Hernandez M., Konno T., Lloyd-Jani A., de Carvalho M., Mouzat K., Landers J.E., Veldink J.H., Silani V., Gitler A.D., Shaw C.E., Rouleau G.A., van den Berg L.H., Van Broeckhoven C., Rademakers R., Andersen P.M., Kubisch C. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet. 2014;51:419–424. doi: 10.1136/jmedgenet-2014-102360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash P.E., Bieniek K.F., Gendron T.F., Caulfield T., Lin W.L., Dejesus-Hernandez M., van Blitterswijk M.M., Jansen-West K., Paul J.W., 3rd, Rademakers R., Boylan K.B., Dickson D.W., Petrucelli L. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M., Mackenzie I.R., Pickering-Brown S.M., Gass J., Rademakers R., Lindholm C., Snowden J., Adamson J., Sadovnick A.D., Rollinson S., Cannon A., Dwosh E., Neary D., Melquist S., Richardson A., Dickson D., Berger Z., Eriksen J., Robinson T., Zehr C., Dickey C.A., Crook R., McGowan E., Mann D., Boeve B., Feldman H., Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Beck J., Poulter M., Hensman D., Rohrer J.D., Mahoney C.J., Adamson G., Campbell T., Uphill J., Borg A., Fratta P., Orrell R.W., Malaspina A., Rowe J., Brown J., Hodges J., Sidle K., Polke J.M., Houlden H., Schott J.M., Fox N.C., Rossor M.N., Tabrizi S.J., Isaacs A.M., Hardy J., Warren J.D., Collinge J., Mead S. Large C9orf72 Hexanucleotide Repeat Expansions Are Seen in Multiple Neurodegenerative Syndromes and Are More Frequent Than Expected in the UK Population. Am J Hum Genet. 2013;92:345–353. doi: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G.Y., Karydas A., Seeley W.W., Josephs K.A., Coppola G., Geschwind D.H., Wszolek Z.K., Feldman H., Knopman D.S., Petersen R.C., Miller B.L., Dickson D.W., Boylan K.B., Graff-Radford N.R., Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M., Lendon C.L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., Petersen R.C., Stevens M., de Graaff E., Wauters E., van Baren J., Hillebrand M., Joosse M., Kwon J.M., Nowotny P., Che L.K., Norton J., Morris J.C., Reed L.A., Trojanowski J., Basun H., Lannfelt L., Neystat M., Fahn S., Dark F., Tannenberg T., Dodd P.R., Hayward N., Kwok J.B., Schofield P.R., Andreadis A., Snowden J., Craufurd D., Neary D., Owen F., Oostra B.A., Hardy J., Goate A., van Swieten J., Mann D., Lynch T., Heutink P. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Jones A.R., Woollacott I., Shatunov A., Cooper-Knock J., Buchman V., Sproviero W., Smith B., Scott K.M., Balendra R., Abel O., McGuffin P., Ellis C.M., Shaw P.J., Morrison K.E., Farmer A., Lewis C.M., Leigh P.N., Shaw C.E., Powell J.F., Al-Chalabi A. Residual association at C9orf72 suggests an alternative amyotrophic lateral sclerosis-causing hexanucleotide repeat. Neurobiol aging. 2013 doi: 10.1016/j.neurobiolaging.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I.R., Neumann M., Baborie A., Sampathu D.M., Du Plessis D., Jaros E., Perry R.H., Trojanowski J.Q., Mann D.M., Lee V.M. A harmonized classification system for FTLD-TDP pathology. Acta neuropathologica. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E., Renton A.E., Mok K., Dopper E.G., Waite A., Rollinson S., Chio A., Restagno G., Nicolaou N., Simon-Sanchez J., van Swieten J.C., Abramzon Y., Johnson J.O., Sendtner M., Pamphlett R., Orrell R.W., Mead S., Sidle K.C., Houlden H., Rohrer J.D., Morrison K.E., Pall H., Talbot K., Ansorge O., Hernandez D.G., Arepalli S., Sabatelli M., Mora G., Corbo M., Giannini F., Calvo A., Englund E., Borghero G., Floris G.L., Remes A.M., Laaksovirta H., McCluskey L., Trojanowski J.Q., Van Deerlin V.M., Schellenberg G.D., Nalls M.A., Drory V.E., Lu C.S., Yeh T.H., Ishiura H., Takahashi Y., Tsuji S., Le Ber I., Brice A., Drepper C., Williams N., Kirby J., Shaw P., Hardy J., Tienari P.J., Heutink P., Morris H.R., Pickering-Brown S., Traynor B.J. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann D.M. Dipeptide repeat protein toxicity in frontotemporal lobar degeneration and in motor neurone disease associated with expansions in C9ORF72-a cautionary note. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Mann D.M., Rollinson S., Robinson A., Bennion Callister J., Thompson J.C., Snowden J.S., Gendron T., Petrucelli L., Masuda-Suzukake M., Hasegawa M., Davidson Y., Pickering-Brown S. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1:68. doi: 10.1186/2051-5960-1-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok K., Traynor B.J., Schymick J., Tienari P.J., Laaksovirta H., Peuralinna T., Myllykangas L., Chio A., Shatunov A., Boeve B.F., Boxer A.L., Dejesus-Hernandez M., Mackenzie I.R., Waite A., Williams N., Morris H.R., Simon-Sanchez J., van Swieten J.C., Heutink P., Restagno G., Mora G., Morrison K.E., Shaw P.J., Rollinson P.S., Al-Chalabi A., Rademakers R., Pickering-Brown S., Orrell R.W., Nalls M.A., Hardy J. The chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol. aging. 2012;33:209.e3–209.e8. doi: 10.1016/j.neurobiolaging.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K., Weng S.M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C., Haass C., Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- Renton A.E., Majounie E., Waite A., Simon-Sanchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A.M., Kaganovich A., Scholz S.W., Duckworth J., Ding J., Harmer D.W., Hernandez D.G., Johnson J.O., Mok K., Ryten M., Trabzuni D., Guerreiro R.J., Orrell R.W., Neal J., Murray A., Pearson J., Jansen I.E., Sondervan D., Seelaar H., Blake D., Young K., Halliwell N., Callister J.B., Toulson G., Richardson A., Gerhard A., Snowden J., Mann D., Neary D., Nalls M.A., Peuralinna T., Jansson L., Isoviita V.M., Kaivorinne A.L., Holtta- Vuori M., Ikonen E., Sulkava R., Benatar M., Wuu J., Chio A., Restagno G., Borghero G., Sabatelli M., Heckerman D., Rogaeva E., Zinman L., Rothstein J.D., Sendtner M., Drepper C., Eichler E.E., Alkan C., Abdullaev Z., Pack S.D., Dutra A., Pak E., Hardy J., Singleton A., Williams N.M., Heutink P., Pickering-Brown S., Morris H.R., Tienari P.J., Traynor B.J. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowden J., Neary D., Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. 2007;114:31–38. doi: 10.1007/s00401-007-0236-3. [DOI] [PubMed] [Google Scholar]
- van der Zee J., Gijselinck I., Dillen L., Van Langenhove T., Theuns J., Engelborghs S., Philtjens S., Vandenbulcke M., Sleegers K., Sieben A., Baumer V., Maes G., Corsmit E., Borroni B., Padovani A., Archetti S., Perneczky R., Diehl-Schmid J., de Mendonca A., Miltenberger-Miltenyi G., Pereira S., Pimentel J., Nacmias B., Bagnoli S., Sorbi S., Graff C., Chiang H.H., Westerlund M., Sanchez-Valle R., Llado A., Gelpi E., Santana I., Almeida M.R., Santiago B., Frisoni G., Zanetti O., Bonvicini C., Synofzik M., Maetzler W., Vom Hagen J.M., Schols L., Heneka M.T., Jessen F., Matej R., Parobkova E., Kovacs G.G., Strobel T., Sarafov S., Tournev I., Jordanova A., Danek A., Arzberger T., Fabrizi G.M., Testi S., Salmon E., Santens P., Martin J.J., Cras P., Vandenberghe R., De Deyn P.P., Cruts M., Van Broeckhoven C., Muller Vom Hagen J., Ramirez A., Kurzwelly D., Sachtleben C., Mairer W., Firmo C., Antonell A., Molinuevo J., Kinhult Stahlbom A., Thonberg H., Nennesmo I., Borjesson-Hanson A., Bessi V., Piaceri I., Helena Ribeiro M., Rosario Almeida M., Oliveira C., Massano J., Garret C., Pires P., Danel A., Maria Fabrizi G., Ferrari S., Cavallaro T. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34:363–373. doi: 10.1002/humu.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.