

Abstract
There is an urgent need for new and better drugs to treat tuberculosis due to lengthy and complex treatment regimens and a rising problem of drug resistance. Drug discovery efforts have increased over the past few years, with a larger focus on modern high-throughput screening technologies. A combination of target-based approaches, with the traditional empirical means of drug identification, has been complemented by the use of target-based phenotypic screens only recently made possibly with newer genetic tools. Using these approaches, a number of promising compound series have been discovered. However, significant problems remain in developing these into drugs. This review highlights recent advances in TB drug discovery, including an overview of screening campaigns, lessons learned and future directions.
Keywords: drug target, essential genes, high-throughput screening, Mycobacterium tuberculosis, tuberculosis drug discovery
In the last 3 years, over 16,000 peer-reviewed scientific articles pertaining to tuberculosis (TB) research have been published. This number is likely to continue to increase, as the threat of tuberculosis and the emergence of drug resistant strains continues to pose threats to global health. Discovering the means to prevent and eradicate this disease will be at the forefront of many research agendas. There has been resurgence in interest in applied and translational research for vaccines, diagnostics and drugs to combat TB. However, progress has been slower than anticipated and there remains an urgent need for increased resources and investment into these research areas.
The basic problems
There are numerous technical challenges in identifying suitable antitubercular agents. The extremely slow growth rate of Mycobacterium tuberculosis (mean generation time of 24 h) poses a significant, yet inherent challenge, as this determines the rate of progress of discovery research. In addition, M. tuberculosis is a respiratory pathogen, which has to be handled under strict safety conditions (Biosafety Level 3 in the USA) requiring expensive specialist facilities. The cell wall of M. tuberculosis is waxy, making it a barrier that prevents many small molecules from penetrating into the bacteria and thus unable to access the intended target(s). To make matters worse, M. tuberculosis has efflux pumps that have been implicated in resistance to antibiotics [1]. During infection, the bacteria are found in diverse microenvironments, as well as in replicating and nonreplicating states, requiring drugs to be active under several conditions and against multiple physiologically different states. Given the extended therapy required to treat TB, drugs also need to be safe over long periods of time, without significant side effects or drug–drug interactions.
In order to overcome these issues, researchers have turned to a variety of screening methods to identify promising compound series for development. This review will discuss the current state of TB drug discovery research with particular regard to screening campaigns. In addition, we will explore the methods by which drug targets are being identified and/or validated. Lastly, we will provide our thoughts on the future of TB research and drug discovery, as the improved treatment of tuberculosis is of upmost importance. This is not intended to be a comprehensive review, but we have focused on the most recent work (last 5 years) and promising avenues for future developments.
Beyond the golden age of antibiotics
The antibiotics rifampin, isoniazid, ethambutol and pyrazinamide form the core treatment regimen for drug-sensitive M. tuberculosis. Rifampin works by inhibiting the beta subunit of RNA polymerase, blocking transcription [2]. Isoniazid is a pro-drug, which is activated by KatG, and likely has multiple mechanisms of action, including blocking the synthesis of mycolic acids, a key component of the cell wall [3,4]. Ethambutol also targets cell wall biosynthesis by inhibiting arabinogalactan and lipoarabinomannan biosynthesis [5]. Pyrazinamide has a complex mode of action, which involves conversion to pyrazinoic acid, and which likely has multiple effects on the cell.
With the appropriate antibiotic treatment, drug sensitive TB is largely curable; however, it still requires taking the four first line drugs for a minimum of 6 months. Patient compliance with the regimen is an ongoing challenge, to such a degree that directly observed therapy (DOTS) is recommended. The number of people who have multidrug-resistant TB (MDR-TB; resistance to isoniazid and rifampin) or extensively drug-resistant TB (XDR-TB; resistance to isoniazid, rifampin, a fluoroquinolone and one injectabledrug) is increasing. Soon, MDR-TB and XDR-TB will render our core antibiotics ineffective. As such, there is a pressing need for new TB drugs and regimens. Despite the impetus for new TB drugs, the rate of development and deployment in the clinic is slow.
Target-based screening campaigns
Drug discovery in the late 20th century largely focused on target-centered methods and that has been true of TB research. The determination of the entire genome sequence of M. tuberculosis H37Rv, played a critical role in identifying new drug targets [6,7], as well as broadening our understanding of infection biology [8]. The identification of essential proteins led drug discovery researchers to embrace target-based approaches for screening campaigns. However, there are major limitations of this approach, including the difficulty in selecting targets relevant for infection and the lack of translation of in vitro activity against individual proteins into activity against live bacterial cells.
A number of compound classes have been identified via target-based high-throughput screening campaigns, of which some remain under development at the hits-to-leads stage. Several groups have completed high-throughput screens against carefully selected targets (Table 1) [9]. Targets include PanC, FtsZ and FadD32. Furthermore, high-throughput screening campaigns have been launched to identify inhibitors of LeuRS, InhA and isoprenoid biosynthesis [10].
Table 1.
Target-based screens for Mycobacterium tuberculosis.
| Study (year) | Target | Screen | Activity against M. tuberculosis | Ref. |
|---|---|---|---|---|
| Galandrin et al. (2013) | FadD32 | Biochemical | Not tested | [11] |
| Kumar et al. (2013) | PanC | Biochemical | Active | [12] |
| Lin et al. (2014) | FtsZ | Biochemical | Active | [13] |
| Wilsey et al. (2013) | DprE1 | In silico | Not tested | [14] |
| Pauli et al. (2013) | InhA | In silico | Not tested | [15] |
| Abrahams et al. (2012) | PanC | Target-based whole cell | Active | [16] |
| Flipo et al. (2012) | EthR | Target-based whole cell | Active | [17] |
PanC (pantothenate synthetase) is an enzyme required for the synthesis of pantothenate (vitamin B5), which is required for the synthesis of coenzyme A. Several screening campaigns have been conducted against this target [16,18], including one in our own laboratory using 90,000 compounds [12]. From the latter screen, the 3-biphenyl-4-cyanopyrrole-2-carboxylic acid series was identified to have good activity in vitro against PanC and also showed activity against live M. tuberculosis in a PanC-dependent fashion.
FtsZ is an essential protein involved in cell division. Using an assay to test for the GTPase activity of FtsZ, a screening campaign of 20,000 compounds resulted in the discovery of the inhibitor 297F. This inhibitor also shows promise for development, since it has activity against live M. tuberculosis [13].
M. tuberculosis proteins are often difficult to express and purify. Another approach has been to use purified proteins from closely related species. For example M. smegmatis FadD32, a protein involved in mycolic acid synthesis, was used in a screen of 10,000 compounds, using a thermal shift assay [11]. Five chemical classes were identified that bound to FadD32, although it is not clear if these bind to the M. tuberculosis FadD32 homolog, or if they have whole-cell activity.
Virtual screening has become more popular where structural information is available; 3D structures of targets can be utilized to perform in silico screens for potential inhibitors. This approach offers the advantage of minimal bench work, and the ability to screen very large compound libraries. For example, 4 million compounds were virtually screened against DprE1 and 41 compounds were identified as likely inhibitors [14]. Six compounds had activity against M. smegmatis suggesting the validity of the approach, although compounds were not tested against M. tuberculosis.
InhA is a target of isoniazid and is still of interest to many groups. A 3D pharmacophore model was built based on 36 crystal structures of InhA; these included InhA from wild type and drug resistant mutants, the InhA apo form, as well as InhA in complex with either NADH, substrate or ligand [15]. Four docking programs were used in parallel to search for ligands and nearly 1 million compounds screened; 19 molecules were identified as potential inhibitors that did not have cytotoxic effects. Six molecules were tested against purified InhA, and three were found to inhibit the enzyme, although data against live bacteria has not yet been reported.
Target-based whole-cell screens
One of the problems in running target-based screens is the lack of translation of in vitro activity to whole-cell activity, for example, growth inhibition, or bactericidal activity. Many of the biochemical screens have identified inhibitors which have no activity against M. tuberculosis. This could be due to a lack of compound penetration, compound efflux, compound detoxification, or the selection of a target that is not essential or vulnerable to inhibition. In order to circumvent these problems, a number of groups are making use of the increasingly sophisticated genetic tools available to manipulate the mycobacterial genome to run target-based whole-cell screens. The basic premise of these screens is that under-expression of the target will increase the sensitivity of the strain to target-directed inhibitors.
For example, Abrahams et al. constructed a strain of M. tuberculosis that expressed PanC under the control of a tetracycline-inducible promoter and conducted high-throughput screening in both induced and noninduced conditions [16]. A flavone series with differential activity was identified. Interestingly, none of the compounds showed activity against PanC in a biochemical assay, suggesting that they might target another enzyme in the pantothenate bio-synthetic pathway or a coenzyme A-dependent enzyme. This suggests that knockdown strains might also be useful for interrogating entire pathways, as well as individual genes.
Genetically modified M. smegmatis has also been used for a target-based whole-cell approach; in this case by looking for induction of a reporter gene in response to compound exposure. A screen for inhibitors of EthR was carried out using a reporter gene. EthR regulates the expression of EthA, a monooxygenase that activates ethionamide. By placing the EthA promoter/operator upstream of a reporter gene, induction of the reporter could be monitored in M. smegmatis [17]. Using this approach phenylphenoxyacetamide derivatives were identified which interacted with EthR and were able to enhance the activity of ethionamide against M. tuberculosis cultured in macrophages.
Target selection
One major limitation to target-based approaches is that there are very few truly validated targets, and even the definition of target validation is debated in the field. A huge amount of work is required to develop the assays, meaning a reluctance on the part of investigators to take risks. This has led to a limited number of targeted screens and thus limited diversity of chemical inhibitors arising from screening. A number of factors play into target selection, in particular, the ‘druggability’ of the target. The ability to determine where a protein is druggable prior to launching a target-based screening campaign is highly desirable. For example, a target-based campaign launched to identify inhibitors of isocitrate lyase and malate synthase was discontinued as it was determined that isocitrate lyase lacks druggability [9]. New databases may be of assistance in this area. The TuberQ database provides a comprehensive druggability analysis of M. tuberculosis genes [19]. Users can immediately determine whether to pursue their target by first determining if it has a druggable pocket. Similarly, Target Explorer compiles data on essentiality, enzyme function, and druggability, and can aid researchers in prioritizing gene targets [20]. Researchers can also use the recently reannotated M. tuberculosis genome combined with the genomes of clinical strains to identify targets that are conserved across relevant strains [21]. Similarly, the increasing number of solved crystal structures and co-crystal structures better inform in silico screens.
Another problem is that many targets were identified as essential under laboratory conditions but are not required during infection (due to alternate pathways, bacterial scavenging or redundancy) [22]. This requires that researchers test targets individually for essential function in vivo prior to starting target-based screens, which is a labor-intensive and costly process, requiring generation of conditionally expressing strains and animal infection models. Target vulnerability also plays a role, such that those proteins that do not require a significant amount of inhibition in order to block cell growth or cause cell death, such as InhA and RpoB, are better suited for drug discovery efforts than targets that require complete inhibition [23].
Phenotypic (whole-cell) screening
In recent years, there has been a rejuvenation of whole-cell-based phenotypic screening strategies to identify antitubercular agents. The focus of this strategy is to assess specific compound classes for their ability to inhibit growth or kill live mycobacteria directly. The implementation of this strategy has led to the discovery of a number or promising new hit/lead series (Table 2). The advantage of this approach is that molecules exhibit the activity required, in other words, growth inhibition and issues such as compound penetration and target vulnerability are directly addressed. The disadvantage is that the target is not known and substantial work may be required to decipher mode of action.
Table 2.
Compounds series identified by phenotypic screens (whole-cell assays).
| Compound | Target | Mode of action | Clinical development |
|---|---|---|---|
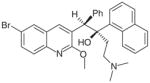 TMC-207 |
ATP synthase | Energy production inhibition | Phase II: MDR-TB FDA approval for pulmonary MDR-TB |
|
SQ109 |
MmpL3 | Cell wall inhibition | Phase II |
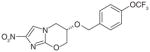 PA-824 |
Unknown | Cell wall inhibition and respiratory poisoning | Phase II |
|
OPC-67683 |
Unknown | Cell wall inhibition | Phase III |
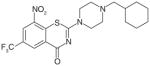 PBTZ169 |
DprE1 | Cell wall inhibition | Preclinical |
A number of technical issues have been solved in order to allow high-throughput screening against live, virulent M. tuberculosis (Table 3). These include the slow growth rate, biosafety containment requirements and notorious potential for clumping in culture [24]. A key parameter is the precise growth medium used, as changes in carbon source and detergent can play major roles in susceptibility to compounds [25]. For example, avoiding glycerol as the primary carbon source is now recommended, as some compounds are only active in environments containing glycerol [26,27]. Similarly, while TMC-207 came from a screen against M. smegmatis, studies suggest that compounds active against M. tuberculosis are best identified by screening against M. tuberculosis, not a model organism [28]. A number of readouts have been used including intracellular ATP or redox-based assays, fluorescent or luminescent strains and multiple detection methods [25].
Table 3.
whole-cell phenotypic screens for Mycobacterium tuberculosis.
| Study (year) | Screening condition | Targets | Ref. |
|---|---|---|---|
| Ananthan et al. (2009) | 7H12 medium (palmitate) | Unknown | |
| Stanley et al. (2012) | 7H9 medium (glucose and glycerol) | DprE1, MmpL3 | [26] |
| Wang et al. (2013) | Biofilm formation | DprE1, MoeW | [29] |
| Gold et al. (2012) | Low pH, low oxygen, butyrate, nitrate | Multifactorial | [30] |
| Christophe et al. (2009) | Inside macrophages | DprE1, QcrB | [31] |
| Ramon-Garcia et al. (2011) | With spectinomycin | Unknown | [32] |
| Stanley et al. (2014) | Inside macrophages | Macrophage targets | [33] |
| Sundaramurthy et al. (2013) | Inside macrophages | Macrophage targets | [34] |
Several whole-cell screens that test compound libraries against replicating M. tuberculosis under typical laboratory conditions have been reported (Table 3). For example, a screen of 100,000 compounds against the well-defined laboratory strain H37Rv used Alamar blue, as a measurement of aerobic respiration, to identify several compound classes with activity [35,36]. Other screens have been conducted using fluorescent strains of H37Rv, for example, 20,000 compounds were screened in medium containing glucose and glycerol and novel inhibitors of DprE1, a cell wall metabolism enzyme, and MmpL3, a protein involved in mycolic acid transport, were found [26].
Since M. tuberculosis likely exists in multiple physiological states during infection, including replicating and nonreplicating, there is increased interest in modifying screening conditions to mimic host-relevant conditions. Mimicking the environment of the host is likely to reveal infection-relevant targets. Thus efforts toward screening compounds under nonreplicating conditions or conditions similar to the host have commenced. For example, the formation of bio-films has been suggested as an antibiotic tolerance or resistance mechanism for M. tuberculosis [37]; a screen against biofilm production identified TCA1 as an inhibitor. Interestingly, this compound appears also to target DprE1, as well as MoeW, an enzyme involved in the synthesis of the enzyme cofactor molybdopterin [29]. A number of models can be used to generate nonreplicating bacteria (low oxygen, persistence, streptomycin dependence and carbon starvation), although there appears to be limited overlap of compound activity between them, suggesting there is not a single nonreplicating model that should be used exclusively [38]. To address this, a screen for compounds active against nonreplicating bacteria has been developed which uses a combination of four conditions: low pH, low oxygen, butyrate as the primary carbon source and nitrate. From a screen of 3600 compounds, oxyphenbutazone, which likely has multiple targets that result in the depletion of thiols and flavins, was identified [30].
To mimic further the host environment, high content screening to identify compounds active against intracellular M. tuberculosis in the macrophage model has been used. By screening 57,000 molecules, the dinitrobenzamides were found, which target DprE1, and Q203 which targets QcrB, a protein involved in electron transport [31].
Since tuberculosis therapy requires multiple drugs and it is highly likely that new drugs will be part of a regimen, rather than a single agent, there has been interest in looking directly for synergistic combinations during screening. In a synergy screen that tested 4900 clinically available drugs that have little or no activity against M. tuberculosis, macrolides, azoles and butyrophenones were identified to enhance the activity of spectinomycin against M. tuberculosis [32].
Another approach to treatment is to identify host-targeted compounds which might act alongside drugs targeting the bacteria. High content screening was used to find bioactive compounds that enhance macrophage-killing properties. Active molecules, such as fluoxetine, were found to enhance autophagy in the infected macrophages or to expedite endosome maturation [33,34].
Other approaches have used surrogate models, such as M. smegmatis. For example, TMC-207 (Sirturo™, Janssen Pharmaceutica), a potent diarylquinoline active against drug-susceptible and drug resistant strains of M. tuberculosis, was identified from a compound library active against M. smegmatis [39]. TMC-207 kills M. tuberculosis via inhibition of ATP synthase.
Targets of phenotypic (whole-cell) screening
Several compound series have been developed based on initial phenotypic activity, including series that contain SQ109, PA824, OPC-67683 and BTZ-043. SQ-109 is a new drug candidate currently in Phase II clinical trials. SQ-109 was identified from a combinatorial library of small-molecules containing a 1,2-ethylenedi-amine core. It demonstrates potent activity in all TB strains including MDR-T and XDR-TB, as well as other mycobacteria. One of its targets is the mycolic acid transporter, MmpL3, which is required for the incorporation of mycolic acids into the M. tuberculosis cell wall [40]. PA-824 (Phase II clinical) is a nitroimidazole pro-drug that requires intracellular activation, in other words, nitro-reduction by the F420-deazaflavin-dependent nitro-reductase (Ddn). PA-824 has in vitro and in vivo activity and is active against replicating and nonreplicating bacteria through a mode of action involving inhibition of cell wall synthesis and respiratory poisoning [41]. Benzothiazinones (BTZ) are active against M. tuberculosis in vitro and in mouse models of infections [42]. BTZ-043 inhibits DprE1 thus preventing the synthesis of cell-wall arabinans. The piperazinobenzothiazinones (PBTZ) derivatives are under development, since they have improved solubility, potency, safety and efficacy in zebrafish and mouse models of TB [43].
One of the drawbacks of the whole-cell screen approach is that identified inhibitors target the same proteins, which are often found in the cell wall. For example, BM212, AU1235, C215, THPP, Spiro, SQ109 and NITD-304 target mmpL3 despite having no structural similarity [33,44–48]. DprE1 is targeted by several compound classes including TCA1 [29], 1,4-azaindoles [49], 4-aminoquinoine piperidine amides and pyrazolopyridones [50,51], as well as novel BTZ analogs consisting of a sulfonamide, reverse-amide or ester [52]. Similarly, Q203 was identified in a screen of infected macrophages while imi-dazopyridines were found in a screen of ATP inhibitors under low oxygen conditions, and both compounds target QcrB, an important component of the electron transport chain [27,53]. Similarly, diarylcoumarin was identified to inhibit FadD32, which is involved in mycolic acid biosynthesis [54] and WecA, an enzyme involved in synthesis of the cell wall, is inhibited by CPZEN-45 [55]. All of the mentioned targets are cell wall-associated. Why cell wall targets are repeatedly discovered, even by screens of disparate conditions, is unclear. One reason may be that as a whole, we are screening the same compound libraries or the targets may be highly susceptible [56]. Alternatively, screening is designed for automation, which may result in missing compounds that act through novel targets and/or the coupling of whole-cell screening with whole genome sequencing of resistant mutants may create a propensity to identify these targets [9,33].
Repurposing: a shortcut to success?
Synthetic chemical libraries are often composed of similar scaffolds and moieties, thus screening these libraries in either target-based or whole-cell-based drug screens tend to identify the same lead compounds repeatedly. The development of new chemical entities has become a laborious, expensive and scientifically difficult endeavor. This has led many pharmaceutical companies to abandon antibiotic development [57]. Along these lines, there has been an increased interest in repurposing known drugs. From a safety and regulatory standpoint, repurposed drugs already have pharmacokinetic and safety data, which are beneficial during the approval process. Furthermore, the semisynthesis of abandoned natural product derivatives introduces a complex, and often stereochemically defined, structural diversity that is frequently missing in synthetic libraries. Oxazolidinones, riminophenazines, rifamycins and fluoroquinolones are all examples of repurposed drugs that were designed to treat other diseases, but are currently being evaluated to treat TB. Examples of repurposed compounds are depicted in Figure 1.
Figure 1.

Discovery timeline of repurposed drugs.
The fluoroquinolones are frequently used as second-line antibiotics for treatment of MDR-TB. A regimen containing gatifloxacin was evaluated in Phase III clinical trials (OFLOTUB trial). Results from this study were presented at the 44th Union World Conference on Lung Health (Paris, France) [58]. Unfortunately, the gatifloxacin-containing regimen failed to shorten treatment time. Both gatifloxacin and moxifloxacin are is currently in Phase III clinical trials for the treatment of drug sensitive TB for shortened treatment duration. However, due to their global usage and availability, there is concern that fluoroquinolone resistant strains of M. tuberculosis will arise.
The clinical success and effectiveness of the 1st generation oxazolidinone, linezolid, has driven the discovery of new generation oxazolidinones. Two analogs, sutezolid and AZD5847, possess improved safety profiles and are currently in Phase II clinical trials [59–61]. Linezolid (as a single agent) and sutezolid (in combination with other antibiotics) are currently being evaluated for efficacy to treat MDR-TB and XDR-TB [60,62–63].
Clofazimine (CFZ), a riminophenazine derivative that was originally used to treat leprosy, demonstrated potent activity against drug-resistant strains. However, clofazimine has poor solubility, is highly lipophilic, has a long terminal half-life and causes skin discoloration. SAR studies of riminophenazine derivatives by Zhang et al. have led to the discovery of C-2 pyridyl bearing derivatives that possess excellent in vitro activity against M. tuberculosis, low acute toxicity in mice, reduced lipophilicity and demonstrates reduced skin discoloration in mice [64]. Two of these derivatives showed equal or better in vivo efficacy against MDR-TB compared with CFZ, and are currently in later stages of preclinical development.
Semisynthetic rifamycins have been the cornerstone of TB treatment. Rifampicin (RMP) is a first-line TB drug. Rifabutin is used as an (expensive) replacement. Rifapentine, which is in Phase II clinical trials for drug-sensitive TB and Phase III clinical trials for latent TB infection, has a longer half-life than RMP and has the potential to shorten treatment length [65]. Rifalazil (RZL) demonstrated good activity in RMP-resistant TB strains, in addition to potent in vivo efficacy and promising pharmacokinetic properties [66,67]. However, the development of RZL was suspended as it proved to be toxic in clinical trials [68]. To this end, Showalter et al. are conducting an extensive SAR study of benzoxazinorifamycins, RZL derivatives, to identify a compound for further preclinical evaluation [69].
Drug-like properties
The search for new antitubercular agents has led to the discovery of a large number of promising chemical series and entities with biological potency. The challenge remains to develop these into drugs, since many of the starting points have poor drug-like properties, in other words, solubility, permeability and bioavailability.
Drug discovery for TB has the same elements as other programs, and the final molecular properties have similar requirements for safety and efficacy. Certain chemical moieties can be incorporated into a chemical scaffold to improve drug-like properties. Thus the incorporation of N-substituted piperazine improves the solubility of PBTZ169 under acidic conditions [43] and riminophenazine bearing a 2-methoxy substituent attached to the C-2 pyridyl moiety has a reduced potential for side effects [64]. However, the normal rules for development may not apply, or may need to be slightly bent. For example, Lipinski’s and Veder’s ‘rules’ are normally employed to improve drug-like properties, but many antibiotics simply break these rules. Discussion of drug development in detail is outside the scope of this review, but it is worth bearing in mind that alternative approaches may be needed.
Drug solubility, stability and delivery can be improved through nanoparticle formulation [70–72], as demonstrated with rifampicin, isoniazid and pyrazinamide encapsulated in a poly(DL-lactide-co-glycolide) (PLG) nanoparticle, which had both improved pharmacokinetics and pharmacodynamics in mice [73]. In addition, nanoparticles could be delivered as nebulized solid lipid particles (SLPs) via the respiratory route in guinea pigs [74] and sustained drug release was maintained for 5 days in plasma and 7 days in organs resulting in complete clearance of infection. Given the risks accompanying TB drug development, further incorporation of nanotechnology may prove beneficial.
One of the leading causes for drugs to be withdrawn from the market are cardiac side effects, normally caused by inhibition of the hERG (human Ether-a-go-go Related Gene) potassium ion channel. Disruption of this channel leads to an increased QT interval which can trigger Torsades de pointes (TdP) arrhythmia and sudden death [75,76]. Current drugs under development, as well as new agents for TB seem to be particularly prone to this potential side effect. For example, clinical doses of fluoroquinolones, such as moxifloxacin and levofloxacin, have been associated with prolonged QT via hERG blockage [75–78]. This blockage has been attributed to the protonated carboxylic acid moiety present on all fluoroquinolones [79,80].
There are a number of structural modifications that can be incorporated to lower hERG blocking without sacrificing potency, such as reducing amine pKa and incorporating oxygen H-bond acceptors and rigidity [81]. These modifications were highlighted in the SAR studies of dofetilide derivatives, where incorporation of permanently positively charged pyridines substantially lowered hERG affinity, and neutral pyridine moieties with rigid (short aliphatic side chains, alkynes) and oxygen-bearing side chains also had reduced hERG affinities [82]. Reducing lipophilicity reduces hERG inhibition, as evidenced by novel N-linked aminopiperidines (inhibitors of bacterial topoisomerase type II) with improved hERG profiles [83]. Optimization of the aminopiperidine core via pKa reduction resulted in the development of a new cis-fluoro aminopiperidine analog with an improved safety profile, which has been advanced into Phase I clinical trials [84]. DNA gyrase inhibitors of the N-linked aminopiperidine class have also been improved with favorable pharmacokinetic properties and significantly reduced hERG liability [85].
Chemical genetics in tuberculosis research
The increase in phenotypic screening means that a large number of chemical entities with activity against M. tuberculosis have been identified within the past 5 years for which there is no known target or mode of action (MOA). The identification of cellular targets and MOA are paramount. As such, recent advances in chemical genetics accompanied by advances in in silico bioinformatics are timely.
The use of whole genome sequencing has been of benefit to target identification [86]. This approach is based on the easy availability of whole genome sequencing, which is used to identify single nucleotide polymorphisms in compound-resistant isolates. Employing this method, we were able to identify resistance-linked genes for eight different compounds with antitubercular activity. Of these genes, four are essential for growth, and three not targeted by existing TB drugs (Pks13, AspS and EccB3).
Garvish et al. used a chemical biology approach to determine the target and MOA of a novel synthetic cyclic peptide, lassomycin [87]. From this study, it was concluded that lassomycin specifically targets ClpC1 ATPase. The Clp complex is essential for in vitro viability of M. tuberculosis. When lassomycin binds ClpC1, ATPase activity is stimulated while simultaneously a loss of proteolytic activity within the Clp complex is observed.
Transcriptome-wide gene expression transcriptional profiling has proven to be a useful chemical genetics method to identify mode of action and affected pathways [88]. Transcriptional profiling of 75 different drugs examined the physiological response in M. tuberculosis [89]. Amides, such as pyrazinamide, nicotinamide and benzamide, induced transcriptional profiles consistent with their effects on intracellular pH homeostasis. Similarly, transcriptional profiles of cell wall synthesis inhibitors gave rise to upregulation of common cell wall gene clusters. The MOA of PA-824 was also illuminated by transcriptomics [41].
With advances in mass spectroscopy and NMR, metabolomics has become an emerging field for MOA studies [90]. Employing an ex vivo stable-isotope metabolomics approach, the molecular target of D-cycloserine was revealed to be D-alanine:D-alanine ligase [91].
Chemical genetics methods employing physical interactions have not been widely applied to M. tuberculosis. For example, affinity chromatography and photoaffinity labeling have been applied to elucidate drug targets elsewhere, but for TB. Newer approaches such as drug affinity responsive target stability (DARTS), as used to identify targets for rapamycin and FK506 may be relevant [92]. However, each of these methods poses challenges, including the amount of labor and need to work with a pathogenic organism.
In silico methods provide a promising complementary approach to in vitro methods. For example, a multicategory naive Bayesian classifier (MCNBC) was built and trained using information from the ChEMBL database, and used to explore the chemogenomic space of 776 compounds which resulted in 1401 compound-target relationships with 84 M. tuberculosis proteins [93]. An in silico domain fishing model for drug target deconvolution which has good prediction accuracy [94] and the TB-drugome, which characterizes all drug-target interactions of the M. tuberculosis proteome [95], are also available.
Conclusion & future perspective
Considerable progress has been made in TB research and drug discovery. Although target-based approaches, both in vitro and in whole cells, have led to the identification of very promising compounds, no drug candidates have yet been advanced to preclinical development. This may, in part, reflect the slow nature and inherent difficulty of working with M. tuberculosis. In contrast, the success from whole-cell screens has led to the identification of several new chemical entities, including the first FDA-approved drug in over 40 years.
Identifying key cellular pathways and proteins is a challenge that must be met. Though nascent in its development, in vitro and in silico chemical genetics look promising in regard to target identification. As our knowledge of the cellular targets of novel compounds and their mechanisms of action increases, it is likely we will find more series which target the same proteins, for example, DprE1, MmpL3, and the ATP synthase. It is not clear why these targets are promiscuous, although it may have something to do with their location in the membrane and the limited chemical diversity of libraries being screened.
We anticipate that a great deal of information from current phenotypic screening campaigns being conducted against multiple compound libraries, under multiple experimental conditions, will become available in the next few years. This will provide a wealth of starting material on which to base medicinal chemistry programs. However, the success of screening campaigns is dependent on the quality and diversity of the chemical libraries available. A deal of effort in sourcing new chemical diversity may be required, as well as continued effort in developing novel screening methods reflective of the infection setting.
The difficulty of working with M. tuberculosis, and the need to develop multiple screens, has led to a more collaborative way of working. Several large consortia have been assembled to focus on drugs; these include the TB Drug Accelerator [96], the Lilly TB Drug Discovery Initiative [97], the EU-funded MM4TB (More Medicines for Tuberculosis) [98], as well as those focused on more basic complementary areas, such as the TB Structural Genomics Consortium [99] or the TB Clinical trials Consortium [100]. We predict that such large consortia will provide the best way forward, as we pool resources and expertise in a common goal, that of effective TB treatment.
EXECUTIVE SUMMARY.
Work with Mycobacterium tuberculosis has inherent difficulties, including its pathogenic nature and slow growth rate.
Many compounds show limited efficacy against M. tuberculosis, due to its thick, waxy cell wall and its efflux systems.
Biochemical screens have been conducted against a small number of targets, with limited success in identifying novel compound hit series with whole-cell activity.
A large increase in phenotypic screening has resulted in multiple new series being identified, but (so far) many series appear to act via a limited number of cell wall-associated targets.
Increased efforts and diversification of methods will be required to identify the targets of phenotypic hits.
Target-based whole-cell screens using genetically modified knockdown expression or reporter strains have great potential to identify new antitubercular agents with different modes of action.
Novel screens to explore nonreplicating, antibiotic tolerant or intracellular bacteria are beginning to generate promising results.
An increasing diversity of screens, as well as chemical matter, will be required to feed the drug pipeline.
Collaboration and cooperation will be key in getting compounds from screens to the clinic.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
The authors were funded by the Bill and Melinda Gates Foundation, under grant OPP1024038 and NIAID of the NIH under award number R01AI099188. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest;
•• of considerable interest
- 1.Rodrigues L, Machado D, Couto I, Amaral L, Viveiros M. Contribution of efflux activity to isoniazid resistance in the Mycobacterium tuberculosis complex. Infect Genet Evol. 2012;12(4):695–700. doi: 10.1016/j.meegid.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 2.Calvori C, Frontali L, Leoni L, Tecce G. Effect of rifamycin on protein synthesis. Nature. 1965;207(995):417–418. doi: 10.1038/207417a0. [DOI] [PubMed] [Google Scholar]
- 3.Takayama K, Wang L, David HL. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1972;2(1):29–35. doi: 10.1128/aac.2.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Timmins GS, Master S, Rusnak F, Deretic V. Nitric oxide generated from isoniazid activation by KatG: source of nitric oxide and activity against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2004;48(8):3006–3009. doi: 10.1128/AAC.48.8.3006-3009.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takayama K, Kilburn JO. Inhibition of synthesis of arabinogalactan by ethambutol in Mycobacterium smegmatis. Antimicrob Agents Chemother. 1989;33(9):1493–1499. doi: 10.1128/aac.33.9.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 7.Fleischmann RD, Alland D, Eisen JA, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184(19):5479–5490. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList-10 years after. Tuberculosis (Edinb) 2011;91(1):1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 9••.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6(1):29–40. doi: 10.1038/nrd2201. Offers a reflection of past target-based screening campaigns, also providing lessons learned and possible direction for future TB drug discovery strategies. [DOI] [PubMed] [Google Scholar]
- 10.Working Group on New TB Drugs. Discovery portfolio. www.newtbdrugs.org/pipeline-discovery.php.
- 11.Galandrin S, Guillet V, Rane RS, et al. Assay development for identifying inhibitors of the mycobacterial FadD32 activity. J Biomol Screen. 2013;18(5):576–587. doi: 10.1177/1087057112474691. [DOI] [PubMed] [Google Scholar]
- 12.Kumar A, Casey A, Odingo J, et al. A high-throughput screen against pantothenate synthetase (PanC) identifies 3-biphenyl-4-cyanopyrrole-2-carboxylic acids as a new class of inhibitor with activity against Mycobacterium tuberculosis. PLoS ONE. 2013;8(11):e72786. doi: 10.1371/journal.pone.0072786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y, Zhu N, Han Y, Jiang J, Si S. Identification of anti-tuberculosis agents that target the cell-division protein FtsZ. J Antibiot (Tokyo) 2014;67 (9):671–676. doi: 10.1038/ja.2014.89. [DOI] [PubMed] [Google Scholar]
- 14.Wilsey C, Gurka J, Toth D, Franco J. A large scale virtual screen of DprE1. Commun Integr Biol. 2013;47:121–125. doi: 10.1016/j.compbiolchem.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Pauli I, Dos Santos RN, Rostirolla DC, et al. Discovery of new inhibitors of Mycobacterium tuberculosis InhA enzyme using virtual screening and a 3D-pharmacophore-based approach. J Chem Inf Model. 2013;53(9):2390–2401. doi: 10.1021/ci400202t. [DOI] [PubMed] [Google Scholar]
- 16••.Abrahams GL, Kumar A, Savvi S, et al. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem Biol. 2012;19(7):844–854. doi: 10.1016/j.chembiol.2012.05.020. Highlights the capabilities of target-based whole-cell screening (TB-WCS) as a tool for tuberculosis drug discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flipo M, Willand N, Lecat-Guillet N, et al. Discovery of novel N-phenylphenoxyacetamide derivatives as EthR inhibitors and ethionamide boosters by combining high-throughput screening and synthesis. J Med Chem. 2012;55(14):6391–6402. doi: 10.1021/jm300377g. [DOI] [PubMed] [Google Scholar]
- 18.White EL, Southwork K, Ross L, et al. A novel inhibitor of Mycobacterium tuberculosis pantothenate synthetase. J Biomol Screen. 2007;12(1):100–105. doi: 10.1177/1087057106296484. [DOI] [PubMed] [Google Scholar]
- 19.Radusky L, Defelipe LA, Lanzarotti E, et al. TuberQ: a Mycobacterium tuberculosis protein druggability database. Database (Oxford) 2014;2014:1–10. doi: 10.1093/database/bau035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pai R, Ioerger TR. Target explorer (prototype) http://saclab.tamu.edu/target_explorer.html.
- 21.Camus JC, Pryor MJ, Médigue C, Cole ST. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology. 2002;148(Pt 10):2967–2973. doi: 10.1099/00221287-148-10-2967. [DOI] [PubMed] [Google Scholar]
- 22.Lechartier B, Rybniker J, Zumla A, Cole ST. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol Med. 2014;6(2):158–168. doi: 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei JR, Krishnamoorthy V, Murphy K, et al. Depletion of antibiotic targets has widely varying effects on growth. Proc Natl Acad Sci USA. 2011;108(10):4176–4181. doi: 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng N, Porter MA, Frick LW, et al. Filtration improves the performance of a high-throughput screen for anti-mycobacterial compounds. PLoS ONE. 2014;9(5):e96348. doi: 10.1371/journal.pone.0096348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franzblau SG, Degroote MA, Cho SH, et al. Comprehensive ana-lysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis (Edinb) 2012;92(6):453–458. doi: 10.1016/j.tube.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Stanley SA, Grant SS, Kawate T, et al. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem Biol. 2012;7(8):1377–1384. doi: 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pethe K, Sequeira PC, Agarwalla S, et al. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat Commun. 2010;1:57. doi: 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altaf M, Miller CH, Bellows DS, O’toole R. Evaluation of the Mycobacterium smegmatis and BCG models for the discovery of Mycobacterium tuberculosis inhibitors. Tuberculosis (Edinb) 2010;90(6):333–337. doi: 10.1016/j.tube.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Wang F, Sambandan D, Halder R, et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc Natl Acad Sci USA. 2013;110(27):E2510–E2517. doi: 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gold B, Pingle M, Brickner SJ, et al. Nonsteroidal anti-inflammatory drug sensitizes Mycobacterium tuberculosis to endogenous and exogenous antimicrobials. Proc Natl Acad Sci USA. 2012;109(40):16004–16011. doi: 10.1073/pnas.1214188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christophe T, Jackson M, Jeon HK, et al. High content screening identifies decaprenyl-phosphoribose 2′ epimerase as a target for intracellular antimycobacterial inhibitors. PLoS Pathog. 2009;5(10):e1000645. doi: 10.1371/journal.ppat.1000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramón-García S, Ng C, Anderson H, et al. Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen. Antimicrob Agents Chemother. 2011;55(8):3861–3869. doi: 10.1128/AAC.00474-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanley SA, Barczak AK, Silvis MR. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog. 2014;10(2):e1003946. doi: 10.1371/journal.ppat.1003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sundaramurthy V, Barsacchi R, Samusik N, et al. Integration of chemical and RNAi multiparametric profiles identifies triggers of intracellular mycobacterial killing. Cell Host Microbe. 2013;13(2):129–142. doi: 10.1016/j.chom.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 35.Ananthan S, Faaleolea ER, Goldman RC, et al. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb) 2009;89(5):334–353. doi: 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maddry JA, Ananthan S, Goldman RC, et al. Antituberculosis activity of the molecular libraries screening center network library. Tuberculosis (Edinb) 2009;89(5):354–363. doi: 10.1016/j.tube.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Islam MS, Richards JP, Ojha AK. Targeting drug tolerance in mycobacteria: a perspective from mycobacterial biofilms. Expert Rev Anti Infect Ther. 2012;10(9):1055–1066. doi: 10.1586/eri.12.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38•.Grant SS, Kawate T, Nag PP, et al. Identification of novel inhibitors of nonreplicating Mycobacterium tuberculosis using a carbon starvation model. ACS Chem Biol. 2013;8(10):2224–2234. doi: 10.1021/cb4004817. The development and application of a novel nonreplicating and secondary assays using a carbon starvation model are presented. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andries K, Verhasselt P, Guillemont J, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:5707. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 40.Sacksteder KA, Protopopova M, Barry CE, III, Andries K, Nacy CA. Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012;7(7):823–837. doi: 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41•.Manjunatha U, Boshoff HI, Barry CE. The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun Integr Biol. 2009;2(3):215–218. doi: 10.4161/cib.2.3.7926. The elucidation of the molecular mechanism of mycobacterial killing demonstrated by PA-824 is discussed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makarov V, Manina G, Mikusova K, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324(5928):801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Makarov V, Lechartier B, Zhang M, et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med. 2014;6(3):372–383. doi: 10.1002/emmm.201303575. Discusses the synthesis of a novel preclinical candidate PBTZ-169, which is an piperazine-containing analog of BTZ-043. Future use in combination therapy is also highlighted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.La Rosa V, Poce G, Canseco JO, et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob Agents Chemother. 2012;56(1):324–331. doi: 10.1128/AAC.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grzegorzewicz AE, Pham H, Gundi VA, et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol. 2012;8(4):334–341. doi: 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Remuiñán MJ, Pérez-Herrán E, Rullás J, et al. Tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamide and N-benzyl-6′,7′-dihydrospiro[piperidine-4,4′-thieno[3,2-c] pyran] analogues with bactericidal efficacy against Mycobacterium tuberculosis targeting MmpL3. PLoS ONE. 2013;8(4):e60933. doi: 10.1371/journal.pone.0060933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tahlan K, Wilson R, Kastrinsky DB, et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56(4):1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao SP, Lakshminarayana SB, Kondreddi RR, et al. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci Transl Med. 2013;5(214):214ra168. doi: 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- 49.Shirude PS, Shandil R, Sadler C, et al. Azaindoles: noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J Med Chem. 2013;56(23):9701–9708. doi: 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- 50.Naik M, Humnabadkar V, Tantry SJ, et al. 4-aminoquinolone piperidine amides: noncovalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J Med Chem. 2014;57(12):5419–5434. doi: 10.1021/jm5005978. [DOI] [PubMed] [Google Scholar]
- 51.Panda M, Ramachandran S, Ramachandran V, et al. Discovery of pyrazolopyridones as a novel class of noncovalent DprE1 inhibitor with potent anti-mycobacterial activity. J Med Chem. 2014;57(11):4761–4771. doi: 10.1021/jm5002937. [DOI] [PubMed] [Google Scholar]
- 52.Tiwari R, Möllmann U, Cho S, Franzblau SG, Miller PA, Miller MJ. Design and syntheses of anti-tuberculosis agents inspired by BTZ043 using a scaffold simplification strategy. ACS Med Chem Lett. 2014;5(5):587–591. doi: 10.1021/ml500039g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mak PA, Rao SP, Ping Tan M, et al. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem Biol. 2012;7(7):1190–1197. doi: 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stanley SA, Kawate T, Iwase N, et al. Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc Natl Acad Sci USA. 2013;110(28):11565–11570. doi: 10.1073/pnas.1302114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ishizaki Y, Hayashi C, Inoue K, et al. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J Biol Chem. 2013;288(42):30309–30319. doi: 10.1074/jbc.M113.492173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldman RC. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis? Tuberculosis (Edinb) 2013;93(6):569–588. doi: 10.1016/j.tube.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 57.Kraus CN. Low hanging fruit in infectious disease drug development. Curr Opin Microbiol. 2008;11(5):434–438. doi: 10.1016/j.mib.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 58.Merle C, Fielding K, Lapujade O, et al. A randomised controlled trial of a 4-month gatifloxacin-containing regimen vs. standard 6-month regimen for treating drug-susceptible pulmonary tuberculosis: main efficacy and safety results of the OFLOTUB trial. Presented at. 44th Union World Conference on Lung Health; Paris, France. 30 October–3 November 2013. [Google Scholar]
- 59.Shaw KJ, Barbachyn MR. The oxazolidinones: past, present, and future. Ann NY Acad Sci. 2011;1241:48–70. doi: 10.1111/j.1749-6632.2011.06330.x. [DOI] [PubMed] [Google Scholar]
- 60.Alffenaar JW, Van Der Laan T, Simons S, et al. Susceptibility of clinical Mycobacterium tuberculosis isolates to a potentially less toxic derivate of linezolid, PNU-100480. Antimicrob Agents Chemother. 2011;55(3):1287–1289. doi: 10.1128/AAC.01297-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balasubramanian V, Solapure S, Iyer H, et al. Bactericidal activity and mechanism of action of AZD5847: a novel oxazolidinone for treatment of tuberculosis. Antimicrob Agents Chemother. 2014;58(1):495–502. doi: 10.1128/AAC.01903-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee M, Lee J, Carroll MW, et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N Engl J Med. 2012;367(17):1508–1518. doi: 10.1056/NEJMoa1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sotgiu G, Centis R, D’ambrosio L, Spanevello A, Migliori GB International Group for the study of Linezolid. Linezolid to treat extensively drug-resistant TB: retrospective data are confirmed by experimental evidence. Eur Respir J. 2013;42(1):288–290. doi: 10.1183/09031936.00191712. [DOI] [PubMed] [Google Scholar]
- 64.Zhang D, Lu Y, Liu K, et al. Identification of less lipophilic riminophenazine derivatives for the treatment of drug-resistant tuberculosis. J Med Chem. 2012;55(19):8409–8417. doi: 10.1021/jm300828h. [DOI] [PubMed] [Google Scholar]
- 65.Rosenthal IM, Zhang M, Williams KN, et al. Daily dosing of rifapentine cures tuberculosis in three months or less in the murine model. PLoS Med. 2007;4(12):e344. doi: 10.1371/journal.pmed.0040344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moghazeh SL, Pan X, Arain T, Stover CK, Musser JM, Kreiswirth BN. Comparative antimycobacterial activities of rifampin, rifapentine, and KRM-1648 against a collection of rifampin-resistant Mycobacterium tuberculosis isolates with known rpoB mutations. Antimicrob Agents Chemother. 1996;40(11):2655–2657. doi: 10.1128/aac.40.11.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang B, Koga H, Ohno H, et al. Relationship between antimycobacterial activities of rifampicin, rifabutin and KRM-1648 and rpoB mutations of Mycobacterium tuberculosis. J Antimicrob Chemother. 1998;42(5):621–628. doi: 10.1093/jac/42.5.621. [DOI] [PubMed] [Google Scholar]
- 68.Dietze R, Teixeira L, Rocha LM, et al. Safety and bactericidal activity of rifalazil in patients with pulmonary tuberculosis. Antimicrob Agents Chemother. 2001;45(7):1972–1976. doi: 10.1128/AAC.45.7.1972-1976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gill SK, Xu H, Kirchhoff PD, et al. Structure-based design of novel benzoxazinorifamycins with potent binding affinity to wild-type and rifampin-resistant mutant Mycobacterium tuberculosis RNA polymerases. J Med Chem. 2012;55(8):3814–3826. doi: 10.1021/jm201716n. [DOI] [PubMed] [Google Scholar]
- 70.Zhang L, Pornpattananangkul D, Hu C-MJ, Huang C-M. Development of nanoparticles for antimicrobial drug delivery. Curr Med Chem. 2010;17(6):585–594. doi: 10.2174/092986710790416290. [DOI] [PubMed] [Google Scholar]
- 71.Pandey R, Ahmad Z. Nanomedicine and experimental tuberculosis: facts, flaws, and future. Nanomedicine. 2011;7(3):259–272. doi: 10.1016/j.nano.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 72.Hauck TS, Giri S, Gao Y, Chan WC. Nanotechnology diagnostics for infectious diseases prevalent in developing countries. Adv Drug Deliv Rev. 2010;62(4–5):438–448. doi: 10.1016/j.addr.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 73.Pandey R, Zahoor A, Sharma S, Khuller GK. Nanoparticle encapsulated antitubercular drugs as a potential oral drug delivery system against murine tuberculosis. Tuberculosis (Edinb) 2003;83(6):373–378. doi: 10.1016/j.tube.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 74.Pandey R, Khuller GK. Solid lipid particle based inhalable sustained drug delivery system against experimental tuberculosis. Tuberculosis (Edinb) 2005;85(4):227–234. doi: 10.1016/j.tube.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 75.Altin T, Ozcan O, Turhan S, et al. Torsade de pointes associated with moxifloxacin: a rare but potentially fatal adverse event. Can J Cardiol. 2007;23(11):907–908. doi: 10.1016/s0828-282x(07)70850-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dale KM, Lertsburapa K, Kluger J, White CM. Moxifloxacin and torsade de pointes. Ann Pharmacother. 2007;41(2):336–340. doi: 10.1345/aph.1H474. [DOI] [PubMed] [Google Scholar]
- 77.Koide T, Shiba M, Tanaka K, et al. Severe QT interval prolongation associated with moxifloxacin: a case report. Cases J. 2008;409:1. doi: 10.1186/1757-1626-1-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kang J, Wang L, Chen XL, Triggle DJ, Rampe D. Interactions of a series of fluoroquinolone antibacterial drugs with the human cardiac K+ channel HERG. Mol Pharmacol. 2001;59(1):122–126. doi: 10.1124/mol.59.1.122. [DOI] [PubMed] [Google Scholar]
- 79.Alexandrou AJ, Duncan RS, Sullivan A, et al. Mechanism of hERG K+ channel blockade by the fluoroquinolone antibiotic moxifloxacin. Br J Pharmacol. 2006;147(8):905–916. doi: 10.1038/sj.bjp.0706678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80•.Ryu S, Imai YN, Oiki S. The synergic modeling for the binding of fluoroquinolone antibiotics to the hERG potassium channel. Bioorg Med Chem Lett. 2013;23(13):3848–3851. doi: 10.1016/j.bmcl.2013.04.074. A proposed mechanism of hERG activity is presented for fluoroquinolone antibiotics. [DOI] [PubMed] [Google Scholar]
- 81.Kerns EH, Di L. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization. 1. Academic Press; MA, USA: 2008. [Google Scholar]
- 82.Carvalho JF, Louvel J, Doornbos ML, et al. Strategies to reduce HERG K+ channel blockade: exploring heteroaromaticity and rigidity in novel pyridine analogues of dofetilide. J Med Chem. 2013;56(7):2828–2840. doi: 10.1021/jm301564f. [DOI] [PubMed] [Google Scholar]
- 83.Reck F, Alm R, Brassil P, et al. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J Med Chem. 2011;54(22):7834–7847. doi: 10.1021/jm2008826. [DOI] [PubMed] [Google Scholar]
- 84.Reck F, Alm RA, Brassil P, et al. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II with reduced pK(a): antibacterial agents with an improved safety profile. J Med Chem. 2012;55(15):6916–6933. doi: 10.1021/jm300690s. [DOI] [PubMed] [Google Scholar]
- 85.Hameed PS, Patil V, Solapure S, et al. Novel N-linked aminopiperidine-based gyrase inhibitors with improved hERG and in vivo efficacy against Mycobacterium tuberculosis. J Med Chem. 2014;57(11):4889–4905. doi: 10.1021/jm500432n. [DOI] [PubMed] [Google Scholar]
- 86.Ioerger TR, O’malley T, Liao R, et al. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS ONEe. 2013;8(9):e75245. doi: 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gavrish E, Sit CS, Cao S, et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol. 2014;21(4):509–528. doi: 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Butcher RA, Schreiber SL. Using genome-wide transcriptional profiling to elucidate small-molecule mechanism. Curr Opin Chem Biol. 2005;9(1):25–30. doi: 10.1016/j.cbpa.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 89.Boshoff HI, Myers TG, Copp BR, Mcneil MR, Wilson MA, Barry CE., 3rd The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem. 2004;279:40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 90.Patti GJ, Yanes O, Siuzdak G. Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;6:263–269. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Prosser GA, De Varvalho LP. Metabolomics deveal D-Alanine:D-Alanine ligase as the target of D-cycloserine in Mycobacterium tuberculosis. ACS Med Chem Lett. 2013;4:1233–1237. doi: 10.1021/ml400349n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lomenick B, Hao R, Jonai N, et al. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci USA. 2009;106(51):21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martínez-Jiménez F, Papadatos G, Yang L, et al. Target prediction for an open access set of compounds active against Mycobacterium tuberculosis. PLoS Comput Biol. 2013;9(10):e1003253. doi: 10.1371/journal.pcbi.1003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prathipati P, Ma NL, Manjunatha UH, Bender A. Fishing the target of antitubercular compounds: in silico target deconvolution model development and validation. J Proteome Res. 2009;8(6):2788–2798. doi: 10.1021/pr8010843. [DOI] [PubMed] [Google Scholar]
- 95.Kinnings SL, Xie L, Fung KH, Jackson RM, Xie L, Bourne PE. The Mycobacterium tuberculosis drugome and its polypharmacological implications. PLoS Comput Biol. 2010;6(11):e1000976. doi: 10.1371/journal.pcbi.1000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seven pharmaceutical companies join academic researchers to speed TB drug discovery. www.idri.org/press-6-25-12.php.
- 97.The Lilly TB drug discovery initiative. www.tbdrugdiscovery.org.
- 98.MM4TB more medicines for tuberculosis. www.mm4tb.org.
- 99.WebTB. www.webtb.org.
- 100.Tuberculosis trials consortium (TBTC) www.cdc.gov.