

Dear Editors,
In a recent paper in JIMD Reports, Al Khallaf et al. present two siblings (ages 4.5 and 2 years, respectively) with infantile Pompe disease (IPD). Despite being CRIM-negative [as determined by CRIM analysis using monoclonal antibodies specific for acid a-glucosidase (GAA)] and treated chronically with Myozyme® (alglucosidase alfa; rhGAA) enzyme replacement therapy (ERT), these patients had “unusually low anti-rhGAA antibody titers and good clinical outcome.” As the authors mention, the favorable outcomes seen in these children could potentially be due to the fact that they are, in fact, CRIM-positive as one of their mutations is a splice mutation. Though not detected by western blot, it is conceivable that sufficient enzyme protein is, nonetheless, detected by the immune system, which tolerizes to a significant extent. In addition, the authors speculate that the lower antibody titers seen in patient 2 could be due to her very early ERT initiation (at age 6 days) compared to her older brother (patient 1), who had commenced ERT at several months of age.
Early initiation of ERT (defined here as ≤ 31 days) is important because of rapid disease progression in IPD. However, early commencement of ERT does not necessarily explain low(er) or no antibody formation, or necessarily preclude the possible need for immune tolerance induction (ITI), ceteris paribus. In two previous studies (Kishnani et al. 2010, Banugaria et al. 2011), some CRIM-negative and CRIM-positive patients developed high and sustained antibody titers despite early (age ≤ 31 days) initiation of ERT. This observation is further supported by data shown in Fig. 1 (Genzyme Corp.) for the 28 patients identified as ≤ 31 days old at the start of Myozyme® treatment (range: 1 to 31 days; mean: 17 days; median: 21 days). Two of 28 patients were CRIM-negative; information regarding CRIM status for the remaining patients was unavailable. In this cohort of 28 patients, 24 patients (86%) had seroconverted. The median peak titer for these 24 patients was 6,400. Five of 24 patients (21%; including the two known CRIM-negative patients) had peak titers ≥ 25,600 sustained for periods of time ranging from 3 months to > 1 year. One of the two documented CRIM-negative patients is also described in Abbott et al. (2011). This patient commenced ERT at day 10 of life [her parents had declined immune tolerance induction (ITI)] and had a peak titer of 25,600 at month 27 of ERT. The second CRIM-negative patient commenced ERT at age 2 weeks and had a peak titer of 409,600. Two of the 28 patients received immunomodulation (CRIM status unknown): one patient had a peak titer of 51,600, which persisted for 6 months post-peak before declining to 200 subsequent to immunomodulation; the second patient received ITI prophylactically and as of the last study time point had not seroconverted. Neutralizing antibody activity, including neutralization of enzyme uptake and catalytic activity, was tested in six of 28 patients. The single patient who tested positively (for inhibition of enzyme uptake) had a high sustained antibody titer of 409,600. While “early ERT initiation” in this analysis was considered as ERT commenced at or before 31 days of age, data from future studies [including those related to newborn screening (NBS)] could lead to a reconceptualization of what is conceived of as “early ERT initiation” and the timescale generally applied.
Fig. 1.
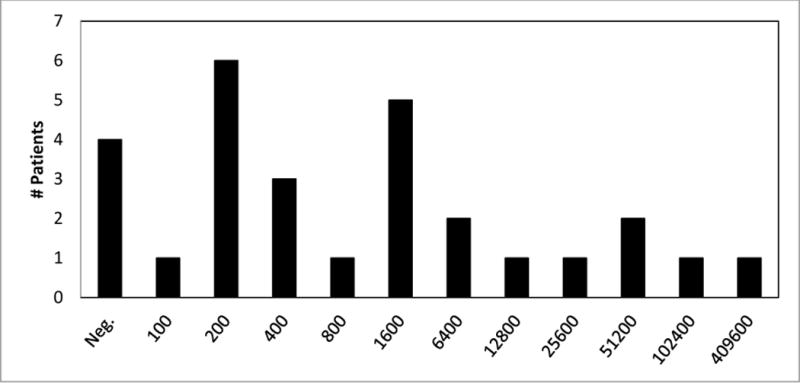
Peak antibody titers for patients ≤ 1 month of age upon initiation of enzyme replacement therapy with Myozyme®.
In a case report by Rohrbach et al. (2010), the authors concluded that an IgE inhibitor, omalizumab, used to mitigate the allergic response, could have played an immunomodulatory role that limited the formation of anti-Myozyme® IgG antibodies in this patient. Based on findings from the subsequent study by Abbott et al. involving a CRIM-negative patient who had only moderately increased, yet persistent, titers (“atypical immune response”), as well as the two new cases from Al Khallaf et al. (none of the 3 patients represented in these two studies received ITI), it is evident that some patients designated as CRIM-negative do not develop high antibody titers with ERT. As we have previously discussed in Abbott et al. (2011), the persistence of titers (not just the presence of high titers per se) could potentially have important clinical implications. In the case by Abbott et al., the patient’s titers peaked at 25,600 after approximately two years of treatment. Titers then fluctuated between 12,800 and 25,600 over the subsequent six months. Although titers ultimately decreased to 6,400 the following year (without immunomodulation, and similar to titers seen for patient 1 in Al Khallaf et al.), her demise was associated with titers sustained in this 6,400 range. This contrasts with what has been seen in long-term infantile survivors, who at most recent follow-up (n=10; time on ERT 23–129 months), had titers of 0 to 1,600 (Prater et al. 2012). The potential clinical impact of titers that persist in low to moderate ranges has yet to be fully characterized and warrants further study.
At present, there is limited information to accurately predict which patients with CRIM-negative IPD are unlikely to develop HSAT or very low titers with regular ERT administration (ERT initiated without ITI). Indeed, most CRIM-negative patients treated in this manner have developed HSAT and have had poor clinical outcomes (e.g., Banugaria et al. 2011). The risk-benefit ratio, therefore, supports the administration of prophylactic short-term ITI (relatively more safe) to CRIM-negative patients at the time of ERT initiation, as opposed to longer-term immune suppression (relatively less safe and less certain efficacy) once HSAT has developed (Messenger et al. 2012; Banugaria et al. 2011).
The report by Al Khallaf et al. builds upon our knowledge that amongst patients designated as CRIM-negative who receive ERT (without ITI), there appears to be a small subset that does not develop high titers per se, but rather a more variable antibody response. However, it is important to remember that the vast majority of CRIM-negative patients (even those diagnosed early, e.g., age ≤ 31 days) do develop high and sustained titers. Consequently, at this time, the risk-benefit ratio justifies the use of ITI in all CRIM-negative patients at the start of ERT. We are continuing to assess the potential clinical impact of antibody titers (low to moderate titers) that remain sustained. Further work exploring the basis for variable immune responsiveness in such patients, including age at start of ERT, potential HLA associations, whether the CRIM assay may miss some CRIM-positive patients, and other genomic factors, is needed.
Acknowledgments
Funding
This study was supported in part by Genzyme, a Sanofi Company (Cambridge, MA), and by the Lysosomal Disease Network, a part of National Institutes of Health Rare Diseases Clinical Research Network (RDCRN). The Lysosomal Disease Network (U54NS065768) is a part of the National Institutes of Health (NIH) Rare Diseases Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS), the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The views expressed in this manuscript represent the opinions of the authors, and do not necessarily represent the official views of the FDA.
Authors’ Contributions
Sean N. Prater contributed toward this work by helping conceive the project, by coordinating data collection, and by writing and revising the manuscript.
Suhrad G. Banugaria helped write and revise the manuscript.
Claire Morgan contributed toward this work through the collection, analysis and interpretation of data, by helping to create the figure, and by writing and revising the manuscript.
Crystal C. Sung contributed toward this work by supporting commercial patient anti-rhGAA antibody testing and providing data interpretation and editing of the manuscript.
Amy S. Rosenberg contributed toward this work by providing critical professional insights and analysis, and by helping write and revise the manuscript.
Priya S. Kishnani contributed toward this project by helping conceive the project, by supervising the project and providing critical professional insights, and by helping write and revise the manuscript.
Article Guarantor
Priya S. Kishnani, MD, accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Competing Interests
Sean N. Prater reports no competing interests.
Suhrad G. Banugaria reports no competing interests.
Claire Morgan works with Genzyme Corporation as a Senior Medical Director, Global Safety Officer for the Pompe program.
Crystal C. Sung works with Genzyme Corporation as Scientific Director, Head of Clinical Specialty Lab.
Amy S. Rosenberg reports no competing interests.
Priya S. Kishnani reports receiving research and grant support from Genzyme. She also receives honoraria and consulting fees from Genzyme and is a member of the Pompe disease and the Gaucher Disease Registry Advisory Boards.
Duke University and the inventors of the method of treatment and precursors of the cell lines used to generate the enzyme (rhGAA) used commercially have received royalties pursuant to the University’s policy on inventions, patents, and technology transfer. This potential conflict for Duke University has been resolved through monetization.
Ethics Approval
Not specifically required for this study.
Patient Consent
Not specifically required for this study.
References
- Abbott MA, Prater SN, Banugaria SG, et al. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol Genet Metab. 2011;104:583–586. doi: 10.1016/j.ymgme.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13:729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banugaria SG, Prater SN, McGann JK, et al. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: lessons learned from Pompe disease. Genet Med. 2013;15:123–131. doi: 10.1038/gim.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger YH, Mendelsohn NJ, Rhead W, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 2012;14:135–142. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myozyme. [Package Insert] Genzyme Corporation; Cambridge, MA: 2006. [Google Scholar]
- Prater SN, Banugaria SG, DeArmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med. 2012;14:800–810. doi: 10.1038/gim.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach M, Klein A, Köhli-Wiesner A, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis. 2010;33:751–757. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]