

Abstract
Prostate cancer (CaP), a commonly diagnosed malignancy, is readily treated by androgen ablation. This treatment temporarily halts the disease, but castration-resistant neoplasms that are refractory to current therapies emerge. While these neoplasms are no longer dependent on physiological levels of androgens, they remain reliant on the expression of the androgen receptor (AR). There are multiple mechanisms by which CaP cells circumvent androgen ablation therapies. These include AR mutations that broaden ligand specificity, AR overexpression, AR activation by growth factors and cytokines, overexpression of AR co-activators, altered steroid metabolism, and a locus-wide histone transcriptional activation of some AR targets. This review focuses on a more recently described mechanism: the expression of low molecular weight AR species that are missing the ligand-binding domain and function independently of ligand to drive proliferation. The etiology, biological activity, unique features, predictive value, and therapeutic implication of these androgen receptor isoforms are discussed in depth.
Keywords: Androgen Receptor, Splice Variant, LNCaP Cell, Enzalutamide, Androgen Receptor Gene
Introduction
Prostate cancer (CaP) remains one of most commonly diagnosed malignancies in the developed world [1]. Normal prostate tissue is dependent on the presence of the androgen receptor (AR) which is essential for mediating androgen signaling [2]. The AR is a steroid hormone receptor that contains four distinct domains: an N-terminal activation domain, a DNA-binding domain, a hinge region, and a C-terminal ligand-binding domain [3]. Activation of AR signaling is dependent on the binding of ligand (testosterone or dihydrotestosterone), which initiates a series of conformational changes that culminate with the translocation of the AR to the nucleus, binding to distinct DNA-binding sites, and an alteration of gene transcription. The dependence on AR for survival and proliferation is retained by most CaPs, hence commonly utilized treatment strategies exploit this feature by targeting the androgen receptor either with therapies that limit the levels of ligand, or with therapeutics that bind the AR to inhibit its activity [4]. These treatments provide a temporary reprieve, since castration-resistant neoplasms inevitably arise. The majority of the castration-resistant cancers continue to express and remain reliant on the AR, which appears to function despite the castration levels of androgen [5–7]. There are multiple mechanisms by which the AR activity becomes independent of ligand binding [8], but this review will focus on the increased expression of low molecular weight (LMW) forms of the AR which are missing the ligand-binding domain hence can function in the absence of ligand to ensure survival and proliferation of prostate tumor cells.
Discovery of the Truncated Forms in CWR22
Truncated isoforms of the androgen receptor (AR) in the context of prostate cancer (CaP) were originally described in castration-resistant CWR22R xenograft tumors and derivative cell lines CWR-R1 [9] and 22Rv1 [10]. The parental CWR22 xenograft was established from a localized tumor and has been extremely valuable for modeling prostate cancer since it faithfully recapitulates the clinical scenario in that its growth in nude athymic mice is highly androgen-sensitive, tumors regress upon castration (i.e., bilateral orchiectomy), and then relapsed variants (designated CWR22R) occur 2–7 months later [11–13]. Importantly, fundamental features of androgen signaling are conserved including the expression of functional AR and secretion of prostate-specific antigen (PSA). In addition to being activated by testosterone and 5α-dihydrotestosterone, the CWR22 AR possesses an H874Y mutation resulting in broadened ligand specificity for estradiol and progesterone, elevated responsiveness to the adrenal androgen dehydroepiandrosterone, and stimulation by the antiandrogen hydroxyflutamide [14]. The findings of truncated ARs in 22Rv1 were particularly exciting since antibody mapping implicated the 75–80-kD truncated AR species as being constitutively active due to their lack of the regulatory ligand-binding domain (LBD), and thereby represented a novel mechanism for evading androgen deprivation [10]. The functionality of the ARΔLBD was validated by its ability to localize to the nucleus, bind to AR consensus DNA-binding sites (e.g., ARE, ARR) with high affinity in the absence of androgen, and activate PSA/KLK3 and probasin promoter-driven reporter constructs under conditions of androgen deprivation. Interestingly, there were two, easily discernible forms of the ARΔLBD, which became more prominent under these conditions. Another noteworthy feature of the 22Rv1 AR repertoire was the expression of an extended full-length AR (114 kD) that was approximately 4 kD greater in mass than LNCaP AR protein. This was derived from the insertion of an additional 39 amino acids caused by an in-frame tandem duplication of exon 3 (117 bp) that encodes the second zinc finger of the DNA-binding domain [10], which resulted from a 35-kb genomic rearrangement around this locus [15]. Screening of a small panel of CWR22/CWR22R tumors demonstrated that the exon 3 duplication mutation (AR-E3DM) originated in the CWR22R-2152 relapsed tumor from which 22Rv1 was derived, which notably was the most aggressive relapse [13]. It was also demonstrated that truncated AR proteins could be derived from the full-length AR by proteolytic processing and that the AR-E3DM mutation sensitized the AR to this process. Although the AR had been demonstrated to be a suitable substrate for caspases [16] and ubiquitin-proteasome processing [17], the treatment of 22Rv1 cells with their respective inhibitors did not diminish the levels of truncated AR. In contrast, it was later shown that calpain-mediated AR cleavage was a prominent mechanism leading to generation of the truncated AR [18]. Since the initial discovery of truncated AR isoforms, there have been intense efforts directed towards characterizing their composition, better defining their functions, elucidating the mechanisms leading to their generation, and therapeutic targeting. These will be discussed below. Following the discovery of the LMW-AR forms, different groups proposed various hypotheses on the etiology of these AR isoforms.
Post-Translation Processing of Full-Length AR into Truncated Isoforms
Calpains, calcium-dependent proteinases, are ubiquitously expressed and proteolyze numerous substrates. In general, calpains cleave proteins at a limited number of sites to generate large polypeptides [19] and proteolysis can serve to alter protein activity or localization. Several members of the steroid hormone receptor superfamily, including the estrogen [20] and glucocorticoid [21] receptors are calpain substrates. Biochemical analyses demonstrated that the AR can be proteolyzed by calpain to fragments ranging from ∼75 to 34/31 kDa [22], with the 75-kDa fragment having retained the N-terminal domain thereby suggesting that proteolysis removed the C-terminal region of the molecule (Fig. 1). Follow-up studies demonstrated that in vivo stimulation of calpain activity in 22Rv1 cells resulted in a decrease in full-length AR (FL-AR) but increase of the LMW-AR[18]. Conversely, treatment of the cells with a calpain inhibitor moderately decreased the level of the LMW-AR. Since calpain cleavage is dependent on protein structure and not simply sequence [23] the insertion of an additional third exon, as found in the 22Rv1 AR, would alter AR structure and may sensitize the mutant molecule to proteolysis. CWR-R1 cells express higher levels of calpain 2 than 22Rv1 cells and higher levels of ERK [24], a kinase that phosphorylates and activates calpain 2 [25]. The expression of LMW-AR in CWR-R1 cells was reduced by treatment with the calpain inhibitor calpeptin, a siRNA targeting calpain 2, or by inhibition of ERK activity. Additionally, expression of the FL-AR cDNA in PC3 cells, which express only trace levels of AR, resulted in the expression of the FL-AR as well as ∼80-kD ARΔLBD forms [24]. Subsequent studies reported that treatment of LNCaP cells, which do not express detectable levels of the ARΔLBD, with the proteasome inhibitor bortezomib resulted in the generation of a C-terminally truncated ARΔLBD [26]. The authors propose that treatment with bortezomib leads to the activation of calpain and calpain-dependent proteolysis of the AR. Studies by Harada [27] found that in a castration-resistant LNCaP derivative, the low levels of the C-terminally truncated AR were enhanced by androgen deprivation, and treatment with a proteasome inhibitor in conjunction with bicalutimide greatly augmented the generation of the truncated AR. It is notable that calpain 2 levels are low in benign prostatic hypertrophy, are elevated in localized prostate cancers, and are highest in metastatic lesions [28]. In total, these studies support the notion that proteolysis of the FL-AR is one mechanism that results in the expression of the castration-resistant ARΔLBD forms and contributes to the development of castration resistance. At this point, it is unclear how much proteolysis contributes to the generation of the ARΔLBD forms in human tumors. While this mechanism does contribute to the expression of the ARΔLBD forms in 22Rv1 cells, the majority of the ARΔLBD forms are generated by alternative splicing.
Fig. 1.
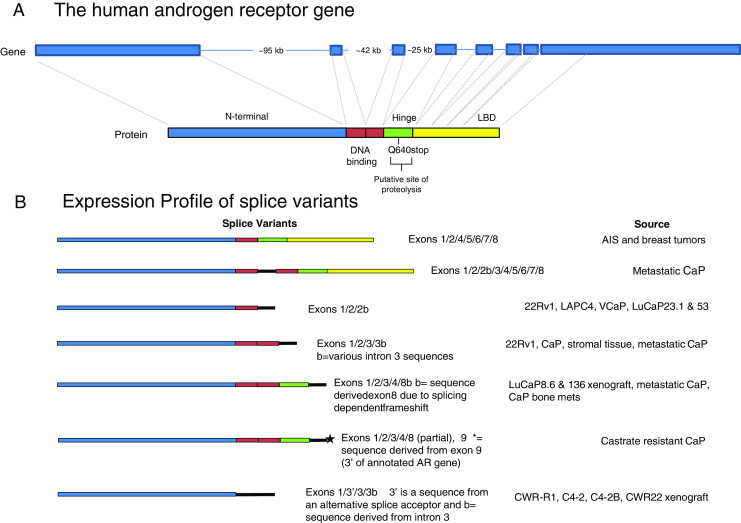
Schematic representation of the AR gene and AR splice variants. a The human AR gene is encoded by eight exons that are separated by large intronic regions. Exon 1 encodes the N-terminal transactivation domain (blue), exons 2 and 3 encode the two zinc fingers of the DNA-binding domain (red), exon 4 encodes the hinge region (green) and exons 5, 6, 7, and 8 encode the ligand-binding domain (yellow). b The various types of splice variants contain specific exons as well are novel sequences encoded by intronic regions. Novel sequences derived from intronic sequences are denoted in black
Premature Chain Termination Generates a C-Terminal Deleted AR
Studies of AR mutations in castration-resistant metastatic cancers led to the identification of a mutation at amino acid position 640 (Q640Stop), which resulted in premature chain termination and the generation of a truncated AR [29]. This mutation mapped to the AR hinge region giving rise to a protein that includes sequences encoded by exons 1, 2, 3, and part of 4 (Fig. 1a). The identical mutation was later identified in a different metastatic prostate cancer [30]. The same study identified additional single basepair mutations, which would result in premature chain termination and the generation of a truncated AR, indicating that such mutations are not rare events. The Q640Stop mutant protein requires phosphorylation [31], could homodimerize [32], and could transactivate expression of certain genes in castrate levels of androgen. However, AR-Q640Stop FL-AR heterodimers were required for activation of additional AR targets under androgen deprivation conditions [32]. An analysis of the interaction of the AR-Q640Stop mutant revealed that CBP and c-Jun were highly recruited by the mutant AR leading to an activation of AP-1, NFAT, and NF-κB transcriptional activity. This property was not observed with the FL-AR, suggesting that it is restricted to the Q640Stop mutant and may be a mechanism by which it contributes to castration resistance [31]. Consistent with the finding that Q640Stop phosphorylation is required for activity, the multikinase inhibitor sorafenib inhibited FL-AR and Q640Stop activity in castration-resistant cells [33].
Truncated AR Variants Resulting from Alternative Splicing
An AR splicing variant in a patient with androgen insensitivity syndrome (AIS) was reported over two decades ago [34]. Shortly afterwards, a study on breast cancers identified an identical AR splice variant that was expressed in a number of breast tumors, but not in normal breast tissue [35]. In both cases, the splice variant had a deletion of exon 3, and encoded a protein that was missing the second zinc finger of the DNA-binding domain (Fig. 1a). The authors of the breast cancer study suggested that given the reduced capacity of such a deletion to bind DNA, the presence of the splice variant in breast tumors would serve to decrease the growth-inhibitory role of the AR in breast tumors and therefore contribute to tumorigenesis. The identification of an identical mutation in AIS strongly supports the hypothesis that the second zinc finger is important for AR function. There are no subsequent follow-up studies on AR splice variants in breast cancer, but several years ago a number of studies detected AR splice variants in prostate tumor-derived cells. The first documented expression of a splice variant in metastatic prostate cancer resulted in an insertion of an intron 2 sequence that encoded 23 amino acids between the two zinc fingers of the DNA-binding domain [36]. This splice variant was shown to localize predominantly to the cytoplasm, but in LNCaP cells could translocate into the nucleus and exhibited partial transcriptional activity. In this case, the insertion of additional sequences between the two zinc fingers compromised AR function.
The majority of studies on AR splice variants utilized the 22Rv1 cell line. This line expresses abundant levels of ARΔLBD forms that differ somewhat in molecular weight. One study reported the expression of a truncated AR form, which was encoded by an mRNA that contained a novel exon (exon 2b) at the 3′-end [37]. This splice variant contained the N-terminal transactivation domain, the first zinc finger, and the novel exon 2b-derived sequence. A siRNA targeting the novel exon was able to reduce the expression of the low molecular weight AR forms. The 1/2/2b splice variant could also be detected in VCaP and LAPC4 cells, although at low levels, as well as in two xenograft cell lines of prostate cancer progression- LuCaP23.1 and LuCaP53.
Additional studies reported that the 22Rv1 cell line expressed a number of splice variants. Several of the variants reported by two groups were identical, and several contained the duplicated exon 3 sequence, hence were specific to the 22Rv1 cell line [38, 39]. However, the majority of variants had only one exon 3 but novel sequences derived from different regions of intron 3 [40, 41]. One particularly abundant transcript, designated as either AR3 [38] or AR-V7 [39] (henceforth referred to as AR3/AR-V7), consisted of exons 1, 2, 3, and a novel cryptic exonic sequence derived from intron 3 which resulted in the addition of 16 unique amino acids at the C-terminus of the protein. The other unique C-terminal extensions derived from various regions of intron 3 differed in sequence and in size. Some novel transcripts contained novel intron 2- derived sequences, but these were less plentiful. These studies indicate that there are two hotspots in the AR gene that give rise to alternative splice variants –intron 2 and intron 3, with the latter being the most common.
Studies utilizing a yeast functional assay led to the identification of two novel transcripts that contained an insertion between first exon 3 and the duplicated exon 3, and between exons 3 and 4 that were derived from intron sequences [42]. Since the insertions resulted in a premature termination codon, the transcripts, while containing sequences corresponding to all of the exons, encoded a protein that contained the N-terminal domain and the DNA-binding domains, as well as unique amino acids derived from the inserted intron 2 or 3 sequences. Hence the repertoire of AR splicing variants expressed in 22Rv1 cells further increased. The protein product generated therefore resembled the previously identified variants in that it consisted of the N-terminal domain, the two zinc fingers, and a novel sequence encoded by intron 3, but the intronic sequences were distinct from those identified in other studies.
Analyses of AR transcripts in two castration-resistant LuCaP xenografts led to the identification of a novel splice variant ARv567es that contained sequences for exons 1, 2, 3, and 4 [43]. The splicing of exon 4 to exon 8 resulted in a frameshift that generated a stop codon after the first 29 nucleotides, thus the exon 8 sequence encoded a novel 10-amino acid sequence at the C-terminus of the variant protein (Fig. 1b). Very similar transcripts were detected in castration-resistant tumor tissue in an independent study [44]. However, these transcripts differed in their 3′- untranslated region where a portion of exon 8 was replaced by sequences mapping 3′ of the gene, which the authors named exon 9. While these transcripts were abundant in castration-resistant tumor tissue, they were less abundant in 22Rv1 cells. These results add an additional layer of complexity and argue that expression of specific splice variants is in part dependent on cellular context.
Biology of AR Variants: Cellular Biology, Activity, Localization, and Expression in Malignancies
The splice variant consisting of exons 1, 2, and 2b was able to transactivate transcription in an androgen-independent manner [37]. While there is no direct evidence that this splice variant can translocate into the nucleus, its ability to transactivate transcription strongly suggests that it does enter the nucleus. In the LuCaP xenograft model, the expression of this splice variant in cells that acquired castration resistance was higher than in the castration sensitive parental lines, indicating that androgen deprivation promotes expression of this splice variant [37].
The AR3/AR-V7 has been particularly well analyzed. Studies revealed that the transcript is expressed in CWR-R1, VCaP cells, and in hormone-refractory prostate tumors. AR3/AR-V can promote transcription in an androgen-independent manner and can function in the absence of FL-AR. Ectopic expression of AR3/AR-V in castration sensitive LNCaP cells confers castration resistance. The expression of this variant is elevated in castration-resistant tumors when compared to hormone naïve cancers [39] and correlates with tumor progression [38]. The latter assertion has been challenged by studies which argue that transcript levels of AR-V1 and AR3/AR-V7 do not predict recurrence in patients [45]. Studies of Guo [38] and Hu [39] reported that AR3/AR-V7 localizes to the nucleus and cytoplasm in cultured cells and in tumor tissue, and nuclear localization is not dependent on the presence of the full-length AR. An additional study conducted in COS1 cells showed that the expression of AR3/AR-V7, ARv567es, as well as several other splice variants is predominantly nuclear, but can be detected in the cytoplasm [46]. Guo and colleagues also observed that antibodies generated against the unique AR3/AR-V7 sequence detected expression in prostatic basal and stomal cells, but not epithelial cells in benign tissue. However, in malignant tissue, the majority of luminal epithelial cells exhibited strong staining, and in castration-resistant tumors there was a redistribution of AR3/AR-V7 protein expression to the nucleus. The splice variant is missing the LBD and as anticipated is refractory to enzalutamide, a second-generation AR antagonist that targets this region [47]. Interestingly, the activity of the AR5/AR-V7 variant has been shown to be regulated by the PTEN-AKT pathway mediated by FOXO1 [48].
The ARv567es splice variant has also been the subject of several inquiries. Like AR3/AR-V7, it is constitutively active [43]. The cellular localization of this variant is predominantly nuclear in both the presence and absence of ligand. Moreover, ectopic expression of ARv567es in LNCaP cells not only conferred castration resistance, but enhanced the expression of the endogenous FL-AR, suggesting that ARv567es can autoregulate endogenous AR transcription. Notably, in this context, ARv567es was more effective at transactivating known AR target genes than the FL-AR. Additional studies suggest that ARv567es can physically interact with the FL-AR and stabilize the FL-AR protein [43].
An analysis of AR3/AR-V7 and ARv567es transcripts in normal and metastatic samples showed that approximately 30 % of the metastatic lesions and 17 % of normal tissue express ARv567es, while 16 % of metastatic lesions and 6 % of normal samples express the AR3/AR-V7 splice variant [43]. An independent study indicated that an ARv567es-like transcript identified in metastatic tumors also was elevated in metastatic lesions, when compared to hormone-naïve tumors [44].
The expression levels of ARv567es, AR3/AR-V7, and AR-V1 were assessed in bone metastases [49]. The AR-V1 and AR3/AR-V7 transcripts were detected in most non-malignant tissue, primary tumors, and metastases, and levels were elevated in metastatic lesions. In contrast, the ARv567es variant was detected in 23 % of the metastases but not in primary tumors. The differences between the above-mentioned study and this analysis may be due to an analysis of different types of metastatic lesions or differences in interpretation and assigning cutoffs for expression at distinct levels. The study of bone metastases also analyzed AR protein expression in a small cohort of tumors using Western immunoblots. Metastatic tumors with the highest levels of AR3/AR-V7 had the highest levels of the ARΔLBD form, while tumors with the lowest level of AR3/AR-V7 had the lowest ARΔLBD levels. However, the levels of ARv567es mRNA did not correlate with levels of the ARΔLBD protein. The Western blot analysis showed that the AR variant proteins constituted ∼32 % of the FL-AR. The authors also noted a discrepancy between protein and RNA ratio of FL-AR and AR3/AR-V7, where the RNA ratio of FL-AR and AR3/AR-V7 (0.4 % of the FL-AR) was almost 2 orders of magnitude lower than the ratios of FL-AR and ARΔLBD protein forms. This led to the suggestion that the difference may be due to possible post-transcriptional stabilization of the splice variants. An alternative explanation is that, as in 22Rv1 cells, there are a number of low molecular weight forms of AR, and the AR3/AR-V7 splice variant encodes a fraction of the observed ARΔLBD protein. However, the correlation of high AR3/AR-V7 mRNA levels and high levels of the ARΔLBD forms suggests that this variant may be a marker of increased aberrant splicing. While elevated expression ARv567es was not indicative of levels of the ARΔLBD protein, it was associated with intense nuclear AR staining.
Some of the proteins encoded by the splice variants localized to the cytoplasm rather than the nucleus. One such variant, AR8, is missing the DNA-binding domain due to the utilization of an alternative exon 3 splice acceptor and a novel intron 3 sequence. The variant is associated with the plasma membrane [41] through palmitoylation of two cysteine residues within its unique C-terminal sequence. As with the other splice variants, the levels of AR8 are elevated in castration-resistant cells. Overexpression of this splice variant promoted the interaction of Src and AR with EGFR in response to EGF and subsequently increased AR tyrosine phosphorylation [41]. Thus the AR8 isoform may promote castration resistance in a nonorthodox manner; by augmenting a response to growth factor signaling.
If LMW-AR forms are associated and possibly predictive of prostate cancer progression, then defining tumors or focal regions of tumors that express these forms would be important in designing therapeutic strategies. The methodologies employed in most studies utilized RT-PCR analysis of mRNA isolated from frozen tumor tissue to detect expression levels of specific splice forms. While these studies have established that castration-resistant tumors express elevated levels of specific variant transcripts, they are detecting AR variants in a tissue that, unless microdissected, contains a heterogeneous population of cells and only one variant transcript is analyzed in a single reaction. An alternative approach is to use Western immunoblot analysis to define the expression of the 80-kDa AR protein. This method, which would detect all LMW-AR forms, is even more problematic, since sufficient amount of frozen tissue is required and as with the mRNA analysis, the assay detects protein expression in a heterogenous mixture of cells. To circumvent these problems, Zhang and co-workers developed an immunohistochemical approach utilizing antibodies that recognize the N-terminal and C-terminal domains of the AR molecule [50]. They reasoned that the ratio of staining intensity obtained with the two antibodies was reflective of the expression levels of ARΔLBD forms. The results obtained showed an overall high frequency of C-terminal truncated AR variants in castration-resistant tumors, leading the authors to argue that this method could be used to stratifying patients for AR targeting therapeutics.
Transcriptional Signature of AR Variants
Several studies sought to identify a transcriptional signature of the AR variants. Tsai et al. generated a recombinant LNCaP subline that inducibly expressed a 22Rv1-derived truncated AR (TC-AR) corresponding to the Q640Stop mutant and containing the exon 3 duplication and found that overexpression of this variant greatly reduced the levels of endogenous AR transcription [51]. This contrasts with the results obtained when ARv567es was ectopically expressed in LNCaP cells [43]. The ability to induce the expression of the truncated receptor allowed for the study of Q640Stop and FL-AR properties in an isogenic background. As previously shown, the variant can translocate into the nucleus and transactivate transcription in castration levels of androgen. The study found that there is a significant overlap in the cohort of genes regulated by FL-AR and the TC-AR, but also identified transcripts that were uniquely transactivated by the Q640Stop, including RHOB, which encodes a protein involved in migration and the morphological changes that are observed when Q640Stop is overexpressed in LNCaP cells.
Ectopic expression of AR3/AR-V7 in LNCaP cells, approximating the levels observed in castration-resistant tumors, identified a set of transcripts that were distinct from the cohort of genes transactivated by FL-AR [44]. The most highly transactivated AR3/AR-V7 gene set consisted predominantly of transcripts that were associated with cell cycle regulation. In contrast, the most highly expressed FL-AR specific cohort consisted of genes that were related to biosynthesis, metabolism, and secretion. The AR3/AR-V7-specific gene set was also transactivated by ectopic expression of the ARv567es variant, suggesting that both splice variants target the same genes. The expression of this cohort of transcripts was further validated in LNCaP95 cells, which express elevated levels of the AR3/AR-V7 variant. Ablation of FL-AR and AR3/AR-V7 variant, but not ablation of FL-AR alone, resulted in decreased expression of the AR3/AR-V7-specific set of transcripts, confirming that this variant is required for transcript expression. An analysis of one AR3/AR-V7-specific transcript, UBE2C, in castration-resistant cancers indicated that UBE2C expression correlated with AR3/AR-V7, but not FL-AR levels.
A siRNA-mediated decrease of AR3/AR-V7 expression in 22Rv1 cells cultured in castration androgen levels identified a set of transcripts that were specifically regulated by AR3/AR-V7, but not FL-AR. One AR3/AR-V7-regulated transcript AKT1, a serine threonine kinase, was further analyzed. Expression of AKT1 was also decreased in CWR-R1 cells following AR3/AR-V7 ablation. Moreover, AKT1 ablation in CWR-R1 cells reduced cellular proliferation.
A similar analysis was conducted in a CWR subline that was enriched for an intron 1 deletion and had elevated levels of the AR variants. However, changes in AKT1 expression were not observed following ablation of the AR3/AR-V7 variant. Additional gene expression profiling studies indicated that many, but not all genes were regulated similarly by ligand-activated FL-AR and the constitutively active splice variant. This study also assessed the transcript profile of LNCaP cells treated with a proliferative dose of DHT (1 mM) and an antiproliferative dose (100 nM) of DHT. These datasets were compared to that of the AR variant-specific transcript dataset. The variant-specific dataset was comparable to that of the LNCaP profile at 1 nM DHT, but not the profile obtained following treatment at 100 nM DHT. Specifically, the M-phase genes identified in other studies as variant enriched or specific, displayed a biphasic response where they were transactivated at low DHT levels, but repressed by 100 nM DHT. The authors argue that this result indicates that the AR variant-specific transcript signature is reflective of a proliferative state, rather than a discrimination between AR variant and FL-AR. This explanation is consistent with the observation that full-length AR and the ligand-independent variants can interchangeably promote growth of the same cells [47].
The gene expression program regulated by the ARv567es variant was independently assessed by ectopic expression in LNCaP cells [43]. The variant-specific set of transcripts was compared to transcripts that were regulated in control LNCaP cells following androgen stimulation. The study revealed the variant could transactivate well-known androgen-regulated transcripts in the absence of ligand as well as ARv567es-specific targets that were not affected by the addition of androgens. Subsets of variant-specific targets that were increased in the absence of androgen were genes whose functions were classified as transcription factors and genes that have roles in steroid biosynthesis and metabolism. It is notable that the ARv567es-dependent regulation of certain androgen-responsive genes was sometimes opposite from that of FL-AR-dependent regulation. The gene expression program of the other splice forms has not been analyzed, so at this time it is unclear which, if any, of the unique gene targets are common to more than one splice variant.
Biological Implications of Splice Variants
While all of the above-cited studies identified splice variant-specific transcripts, there was almost no overlap in the gene sets obtained in the different studies. This could be in part due to the different cellular backgrounds, different specific splice variants, and experimental design (e.g., ectopic expression vs. siRNA-mediated ablation of variant expression). We had previously shown that the AR directed a different transcriptional program in the two related cell lines 22Rv1 and CWR-R1, even though the AR interacted with almost identical DNA sites [52]. AR-dependent transcription is governed by a large number of co-regulators, which are likely to be differentially expressed in distinct cell lines. However, a number of studies utilized the androgen-sensitive LNCaP cell line in which various splice variants were ectopically expressed in the presence or absence of androgens. The individual studies used somewhat different conditions and levels of variant expression, and data sets were evaluated using different criteria. These could all affect the identification of transactivation (or repression) of specific targets and since the studies further stratified on the basis of the most highly elevated or repressed targets, the results obtained would be expected to vary.
Different variants may well have variant-specific unique properties. The ARv567es localizes almost exclusively to the nucleus and has been shown to stabilize FL-AR levels in LNCaP cells. In contrast, the AR3/AR-V7 splice variant localizes to nuclear and cytoplasmic compartments and there is no evidence that it stabilizes the endogenous FL-AR. Furthermore, the Q640Stop variant, when overexpressed in LNCaP cells, represses the expression of the FL-AR. These distinct features could all affect the activity and variant-specific transactivation of gene transcription.
All of the variants used in these studies described above share two features; (1) they can localize to the nucleus in the absence of androgen and (2) they confer castration resistance. This last property indicates that the variants can initiate a cell cycle progression program with an efficacy that is similar to the efficacy of ligand-activated FL-AR. The simplest explanation is that the same critical genes are targeted. However, the critical target genes may not be the same as the most highly transactivated or repressed genes. It is most likely the variants that are missing the LBD and the FL-AR will have unique properties. A number of AR-interacting proteins, which modify AR activity, are dependent on the presence of the LBD (ARA54, ARA55, Hsps, and Tip60); hence, the variants that are missing this domain would not be subject to modulation by these co-regulators. This has been validated by studies which show that AR splice variants are resistant to Hsp90 inhibitors [53]. In contrast, Vav3 an AR co-activator which requires the AR N-terminal domain can effectively interact with and activate AR3/AR-V7 and ARv567es [54].
Genomic Predisposition Associated with Differential Splicing
The commonly used models for the study of AR variant expression, 22Rv1 and CWR-R1, harbor AR genomic aberrations leading to a hypothesis that such aberrations promote alternative splicing. 22Rv1 cells have a 35-kb duplication of exon 3 and flanking sequences. CWR-R1 subclones, which express the ARΔLBD have a 48-kb deletion in AR intron 1. The LuCaP 86.2 xenograft, which expresses only the ARv567es variant has an 8,579-bp intragenic deletion of AR exons 5, 6, and 7 [55]. Another example that supports this hypothesis is the discovery that alternatively spliced AR mRNAs were expressed in a mouse prostate tumor derived from the Myc mouse model of prostate cancer [56]. Notably, a novel sequence contained in the mRNA maps 5′ of the AR gene. Since transcription does not loop back, the most plausible explanation is that in this tumor, the AR has a genomic abnormality, placing the novel sequence 3′ of the transcriptional start site. However, it is unclear if a genomic alteration enhances variant expression or if it is required for variant expression. Given that certain variants are expressed in benign tissue as well as in primary prostate tumors, AR genomic aberrations are either extremely common, or genomic aberrations further potentiate splice variant expression.
Regulation of Alternative Splicing
The information required to define RNA regions of the preRNA that will be included in the mRNA is present in the sequence, but this information is discerned by RNA-binding proteins (RBP), hence the levels and activity of these proteins regulate alternative splicing. These factors interact with exonic and intronic regions of the nascent mRNA, regulating the recruitment and activity of the spliceosome [57]. Some proto-oncogenes, including cyclin D1 and H-Ras, can be alternatively spliced to yield proteins that have distinct properties [58].
Several studies have reported that the levels of AR splice forms increase when cells are placed in castrate levels of androgen. This increase occurs at the transcriptional level within days, and is coincident with a decrease in the levels of the FL-AR suggesting that there is a shift in AR mRNA splicing. In vivo and in vitro studies have indicated that castration induces oxidative stress through redox imbalance by upregulation of ROS-producing and downregulation of ROS-detoxifying enzymes [59]. AR levels and the levels of some AR co-regulators has been reported to be activated by oxidative stress, suggesting that reducing androgen-dependent AR signaling may have an active role in the acquisition of castration resistance.
It is noteworthy that shifts in splicing of certain transcripts have been observed when cells are undergoing a stress response [60]. The p53 regulators MDM2 and MDM4 are alternatively spliced when cells are exposed to irradiation [61]. Moreover, in rhabdomyosarcoma cells, the alternative splicing of MDM2 in response to by UV radiation and cisplatin is dependent on MDM2 intronic elements [62]. Hypoxic stress has been shown to induce alternative splicing of presenilin 2 in human neuroblastoma cells [63], and nutrient starvation inhibits the splicing of G6PD gene transcripts in mouse liver [64].
During a stress response, various splicing factors (hnRNP A1, Slu, DDX5) [65–67] are relocalized to the cytoplasm, where they play a role in translation and stability of mRNA. The splicing factor Sam68 localizes to nuclear stress granules in response to heat shock and TOP2 inhibitors [68]. Sam68has been shown to modulate the splicing of cyclin D, and levels of Sam68 correlate with the levels of cyclin D1b, a splice variant that unlike cyclin D1a can transform cells, promote anchorage-independent growth, and increase cellular invasiveness [69]. A polymorphism at a cyclin D splice site promoted the expression of the cyclin D1b variant [70]. Modification of splicing components has not been well explored in the context of prostate cancer or in the etiology of castration resistance. If androgen deprivation modifies the availability or activity of splicing factors, then such changes may shift the AR splicing pattern in some tumors to favor the expression of the low molecular weight splice variants. The precedent that intronic regions of the MDM2 gene contribute to alternative splicing suggest a unifying hypothesis, where genomic alterations such as intronic deletions or duplications predispose the AR gene to alternative splicing that is further facilitated by a modulation of splicing factors by androgen deprivation.
Biological and Therapeutic Implication
Various studies have shown that AR variants that are missing the LBD are constitutively active and can function independently of the FL-AR. Therefore these variants will be refractory to therapeutics that target the LBD. But most tumors express the FL-AR and the LMW forms, so LBD-targeting therapeutics may have some efficacy in retarding tumor growth. Yet, the benefit from such therapies would be temporary since studies have shown that androgen withdrawal promotes the expression of the LMW-AR. Hence, the tumors would adapt and become more dependent on the variants that are missing the LBD. Targeting the N-terminal domain of the AR would be a more prudent strategy since all the variants and the FL-AR contain this region. The NTD-targeting agent, EPI 001 has been shown to inhibit proliferation of 22Rv1 cells in castrate androgen levels, while the LBD-targeting agent bicalutamide failed to affect cell growth [71]. This agent has promise for treatment of tumors that are reliant on the activity of the ARΔLBD. If the ARΔLBD is generated by increased proteolysis then protease inhibitors may affect proliferation. An alternative strategy is to target critical N-terminal domain interacting proteins. Studies have shown that a disruption of the p160 co-activator interface with the androgen receptor can inhibit the activity of androgen-dependent and castration-resistant prostate tumor cells [72]. ASC-J9, an agent that promotes AR degradation, has been shown to affect FL and ARΔLBD [73], and compounds found in the Kava root have been shown to decrease the levels of FL and ARΔLBD in a xenograft model of prostate tumorigenesis [74]. Future therapies may include the targeting of variants specifically using an RNAi approach, but at this time, these are in an early stage of development.
The presence of ARΔLBD forms is common and has been detected in malignant and non-malignant tissue, but the levels and perhaps the diversity of LMW-AR forms is greater in higher-grade neoplasms, particularly in castration-resistant tumors. Moreover, there are several mechanisms that can give rise to these forms and each is associated with more advanced malignancies. The association of ARΔLBD forms with cancer progression is clear, but this observation is correlative. The studies that demonstrate a causative role for the LMW-AR in the etiology of castration resistance are reliant on studies in the few prostate tumor-derived cell lines. Nevertheless, all of the studies indicate that these forms promote castration resistance. While some of the splice forms do not retain the DNA-binding domain, most do. The deletion of the LBD and retention of the N-terminal transactivation region and DNA-binding domains indicates that they have the potential for promoting transcription. The cell culture studies show that overexpression of the ARΔLBD forms is sufficient to promote proliferation, but in many of these studies, the levels of the ARΔLBD are very high. While the discovery and characterization of truncated AR variants has advanced the field of CaP research, it also has prompted the emergence of numerous questions. So, are ARΔLBD forms the drivers of castration resistance in tumors? In this respect, much more work needs to be done to define the predictive value of the ARΔLBD expression. Do all ARΔLBD forms correlate with castration resistance or are specific splice variants associated with the disease? Some studies suggest the latter, but these results need to be solidified. Do even low levels of the specific ARΔLBD portend a poor clinical outcome, or is there a threshold effect? Do splice variants transactivate different target genes or are they simply able to function in castration levels of androgen to drive cell proliferation? If a splice variant has specific targets, are these responsible for, or contribute to, disease progression? Is there a splice variant signature that can act as a marker for predicting outcome? At this time a specific signature cannot be discerned but with additional data sets a pattern may emerge. Lastly, can the mechanisms that lead to the expression of ARΔLBD be targeted by therapeutics? This necessitates deciphering the molecular details of such mechanisms. Identification of AR variants that are refractory to androgen ablation therapy was a great step in defining a possible cause of castration resistance. The next steps are to solidify these finding and to ultimately design therapies that can circumvent this roadblock to prostate cancer treatment.
Acknowledgments
We thank Dr. Paramita Ghosh and Stephen J. Libertini for critical reading of the manuscript. This material is based on work supported in part by the U.S. Department of Veterans Affairs, Office of Research and Development, Biomedical Laboratory Research Program (VA Merit grant BX001079 to MM), the California Cancer Research Coordinating Committee (MM) and the DOD Prostate Cancer Research Program (W81XWH-09-1-0314; C.G.T.). The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government.
Conflict of Interest
The authors declare that they have no conflict of interest.
Contributor Information
Maria Mudryj, Email: mmudryj@ucdavis.edu.
Clifford G. Tepper, Email: cgtepper@ucdavis.edu
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J Cell Biochem. 2004;91:483–490. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 3.Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991;5:1396–1404. doi: 10.1210/mend-5-10-1396. [DOI] [PubMed] [Google Scholar]
- 4.Javidan J, Deitch AD, Shi XB, de Vere White RW. The androgen receptor and mechanisms for androgen independence in prostate cancer. Cancer Invest. 2005;23:520–528. doi: 10.1080/07357900500202721. [DOI] [PubMed] [Google Scholar]
- 5.Ruizeveld de Winter JA, Trapman J, Vermey M, Mulder E, Zegers ND, van der Kwast TH. Androgen receptor expression in human tissues: an immunohistochemical study. J Histochem Cytochem. 1991;39:927–936. doi: 10.1177/39.7.1865110. [DOI] [PubMed] [Google Scholar]
- 6.Chodak GW, Kranc DM, Puy LA, Takeda H, Johnson K, Chang C. Nuclear localization of androgen receptor in heterogeneous samples of normal, hyperplastic and neoplastic human prostate. J Urol. 1992;147:798–803. doi: 10.1016/s0022-5347(17)37389-5. [DOI] [PubMed] [Google Scholar]
- 7.Sadi MV, Walsh PC, Barrack ER. Immunohistochemical study of androgen receptors in metastatic prostate cancer. Comparison of receptor content and response to hormonal therapy. Cancer. 1991;67:3057–3064. doi: 10.1002/1097-0142(19910615)67:12<3057::AID-CNCR2820671221>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 8.Devlin HL, Mudryj M. Progression of prostate cancer: multiple pathways to androgen independence. Cancer Lett. 2009;274:177–186. doi: 10.1016/j.canlet.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, French FS. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718–5724. [PubMed] [Google Scholar]
- 10.Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF, Pretlow TG, Kung HJ. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606–6614. [PubMed] [Google Scholar]
- 11.Pretlow TG, et al. Xenografts of primary human prostatic carcinoma. J Natl Cancer Inst. 1993;85:394–398. doi: 10.1093/jnci/85.5.394. [DOI] [PubMed] [Google Scholar]
- 12.Wainstein MA, et al. CWR22: androgen-dependent xenograft model derived from a primary human prostatic carcinoma. Cancer Res. 1994;54:6049–6052. [PubMed] [Google Scholar]
- 13.Nagabhushan M, et al. CWR22: the first human prostate cancer xenograft with strongly androgen-dependent and relapsed strains both in vivo and in soft agar. Cancer Res. 1996;56:3042–3046. [PubMed] [Google Scholar]
- 14.Tan J, et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450–459. doi: 10.1210/me.11.4.450. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011;71:2108–2117. doi: 10.1158/0008-5472.CAN-10-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wellington CL, et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–9167. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- 17.Sheflin L, Keegan B, Zhang W, Spaulding SW. Inhibiting proteasomes in human HepG2 and LNCaP cells increases endogenous androgen receptor levels. Biochem Biophys Res Commun. 2000;276:144–150. doi: 10.1006/bbrc.2000.3424. [DOI] [PubMed] [Google Scholar]
- 18.Libertini SJ, Tepper CG, Rodriguez V, Asmuth DM, Kung HJ, Mudryj M. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res. 2007;67:9001–9005. doi: 10.1158/0008-5472.CAN-07-1072. [DOI] [PubMed] [Google Scholar]
- 19.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 20.Murayama A, Fukai F, Murachi T. Action of calpain on the basic estrogen receptor molecule of porcine uterus. J Biochem. 1984;95:1697–1704. doi: 10.1093/oxfordjournals.jbchem.a134783. [DOI] [PubMed] [Google Scholar]
- 21.Kim YS, Kim J, Kim Y, Lee YH, Kim JH, Lee SJ, Shin SY, Ko J. The role of calpains in ligand-induced degradation of the glucocorticoid receptor. Biochem Biophys Res Commun. 2008;374:373–377. doi: 10.1016/j.bbrc.2008.07.040. [DOI] [PubMed] [Google Scholar]
- 22.Pelley RP, Chinnakannu K, Murthy S, Strickland FM, Menon M, Dou QP, Barrack ER, Reddy GP. Calmodulin-androgen receptor (AR) interaction: calcium-dependent, calpain-mediated breakdown of AR in LNCaP prostate cancer cells. Cancer Res. 2006;66:11754–11762. doi: 10.1158/0008-5472.CAN-06-2918. [DOI] [PubMed] [Google Scholar]
- 23.Tompa P, Buzder-Lantos P, Tantos A, Farkas A, Szilagyi A, Banoczi Z, Hudecz F, Friedrich P. On the sequential determinants of calpain cleavage. J Biol Chem. 2004;279:20775–20785. doi: 10.1074/jbc.M313873200. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Libertini SJ, Wang Y, Kung HJ, Ghosh P, Mudryj M. ERK regulates calpain 2-induced androgen receptor proteolysis in CWR22 relapsed prostate tumor cell lines. J Biol Chem. 2010;285:2368–2374. doi: 10.1074/jbc.M109.049379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glading A, Chang P, Lauffenburger DA, Wells A. Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J Biol Chem. 2000;275:2390–2398. doi: 10.1074/jbc.275.4.2390. [DOI] [PubMed] [Google Scholar]
- 26.Frezza M, Yang H, Dou QP. Modulation of the tumor cell death pathway by androgen receptor in response to cytotoxic stimuli. J Cell Physiol. 2011;226:2731–2739. doi: 10.1002/jcp.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harada N, Inoue K, Yamaji R, Nakano Y, Inui H. Androgen deprivation causes truncation of the C-terminal region of androgen receptor in human prostate cancer LNCaP cells. Cancer Sci. 2012;103:1022–1027. doi: 10.1111/j.1349-7006.2012.02250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rios-Doria J, Day KC, Kuefer R, Rashid MG, Chinnaiyan AM, Rubin MA, Day ML. The role of calpain in the proteolytic cleavage of E-cadherin in prostate and mammary epithelial cells. J Biol Chem. 2003;278:1372–1379. doi: 10.1074/jbc.M208772200. [DOI] [PubMed] [Google Scholar]
- 29.Ceraline J, et al. Constitutive activation of the androgen receptor by a point mutation in the hinge region: a new mechanism for androgen-independent growth in prostate cancer. Int J Cancer. 2004;108:152–157. doi: 10.1002/ijc.11404. [DOI] [PubMed] [Google Scholar]
- 30.Lapouge G, et al. Unexpected paracrine action of prostate cancer cells harboring a new class of androgen receptor mutation—a new paradigm for cooperation among prostate tumor cells. Int J Cancer. 2007;121:1238–1244. doi: 10.1002/ijc.22830. [DOI] [PubMed] [Google Scholar]
- 31.Lapouge G, Marcias G, Erdmann E, Kessler P, Cruchant M, Serra S, Bergerat JP, Ceraline J. Specific properties of a C-terminal truncated androgen receptor detected in hormone refractory prostate cancer. Adv Exp Med Biol. 2008;617:529–534. doi: 10.1007/978-0-387-69080-3_53. [DOI] [PubMed] [Google Scholar]
- 32.Streicher W, et al. AR-Q640X, a model to study the effects of constitutively active C-terminally truncated AR variants in prostate cancer cells. World J Urol. 2012;30:333–339. doi: 10.1007/s00345-012-0842-0. [DOI] [PubMed] [Google Scholar]
- 33.Zengerling F, Streicher W, Schrader AJ, Schrader M, Nitzsche B, Cronauer MV, Hopfner M. Effects of sorafenib on C-terminally truncated androgen receptor variants in human prostate cancer cells. Int J Mol Sci. 2012;13:11530–11542. doi: 10.3390/ijms130911530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quigley CA, et al. Complete androgen insensitivity due to deletion of exon C of the androgen receptor gene highlights the functional importance of the second zinc finger of the androgen receptor in vivo. Mol Endocrinol. 1992;6:1103–1112. doi: 10.1210/me.6.7.1103. [DOI] [PubMed] [Google Scholar]
- 35.Zhu X, Daffada AA, Chan CM, Dowsett M. Identification of an exon 3 deletion splice variant androgen receptor mRNA in human breast cancer. Int J Cancer. 1997;72:574–580. doi: 10.1002/(SICI)1097-0215(19970807)72:4<574::AID-IJC4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 36.Jagla M, Feve M, Kessler P, Lapouge G, Erdmann E, Serra S, Bergerat JP, Ceraline J. A splicing variant of the androgen receptor detected in a metastatic prostate cancer exhibits exclusively cytoplasmic actions. Endocrinology. 2007;148:4334–4343. doi: 10.1210/en.2007-0446. [DOI] [PubMed] [Google Scholar]
- 37.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Z, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu R, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate. 2011;71:1656–1667. doi: 10.1002/pros.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang X, et al. Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells. J Biol Chem. 2011;286:36152–36160. doi: 10.1074/jbc.M111.265124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marcias G, et al. Identification of novel truncated androgen receptor (AR) mutants including unreported pre-mRNA splicing variants in the 22Rv1 hormone-refractory prostate cancer (PCa) cell line. Hum Mutat. 2010;31:74–80. doi: 10.1002/humu.21138. [DOI] [PubMed] [Google Scholar]
- 43.Sun S, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu R, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao H, Coram MA, Nolley R, Reese SW, Young SR, Peehl DM. Transcript levels of androgen receptor variant AR-V1 or AR-V7 do not predict recurrence in patients with prostate cancer at indeterminate risk for progression. J Urol. 2012;188:2158–2164. doi: 10.1016/j.juro.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 46.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287:19736–19749. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mediwala SN, et al. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate. 2013;73:267–277. doi: 10.1002/pros.22566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, Wikstrom P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai HC, Boucher DL, Martinez A, Tepper CG, Kung HJ. Modeling truncated AR expression in a natural androgen responsive environment and identification of RHOB as a direct transcriptional target. PLoS One. 2012;7:e49887. doi: 10.1371/journal.pone.0049887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen H, et al. Genome-wide analysis of androgen receptor binding and gene regulation in two CWR22-derived prostate cancer cell lines. Endocr Relat Cancer. 2010;17:857–873. doi: 10.1677/ERC-10-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shafi AA, Cox MB, Weigel NL. Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression. Steroids. 2013;78(6):548–554. doi: 10.1016/j.steroids.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peacock SO, Fahrenholtz CD, Burnstein KL. Vav3 enhances androgen receptor splice variant activity and is critical for castration-resistant prostate cancer growth and survival. Mol Endocrinol. 2012;26:1967–1979. doi: 10.1210/me.2012-1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31:4759–4767. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cammas A, Lewis SM, Vagner S, Holcik M. Post-transcriptional control of gene expression through subcellular relocalization of mRNA binding proteins. Biochem Pharmacol. 2008;76:1395–1403. doi: 10.1016/j.bcp.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 58.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shiota M, Yokomizo A, Naito S. Pro-survival and anti-apoptotic properties of androgen receptor signaling by oxidative stress promote treatment resistance in prostate cancer. Endocr Relat Cancer. 2012;19:R243–R253. doi: 10.1530/ERC-12-0232. [DOI] [PubMed] [Google Scholar]
- 60.Dutertre M, Sanchez G, Barbier J, Corcos L, Auboeuf D. The emerging role of pre-messenger RNA splicing in stress responses: sending alternative messages and silent messengers. RNA Biol. 2011;8:740–747. doi: 10.4161/rna.8.5.16016. [DOI] [PubMed] [Google Scholar]
- 61.Chandler DS, Singh RK, Caldwell LC, Bitler JL, Lozano G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res. 2006;66:9502–9508. doi: 10.1158/0008-5472.CAN-05-4271. [DOI] [PubMed] [Google Scholar]
- 62.Singh RK, Tapia-Santos A, Bebee TW, Chandler DS. Conserved sequences in the final intron of MDM2 are essential for the regulation of alternative splicing of MDM2 in response to stress. Exp Cell Res. 2009;315:3419–3432. doi: 10.1016/j.yexcr.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 63.Higashide S, et al. Identification of regulatory cis-acting elements for alternative splicing of presenilin 2 exon 5 under hypoxic stress conditions. J Neurochem. 2004;91:1191–1198. doi: 10.1111/j.1471-4159.2004.02798.x. [DOI] [PubMed] [Google Scholar]
- 64.Amir-Ahmady B, Salati LM. Regulation of the processing of glucose-6-phosphate dehydrogenase mRNA by nutritional status. J Biol Chem. 2001;276:10514–10523. doi: 10.1074/jbc.M010535200. [DOI] [PubMed] [Google Scholar]
- 65.van der Houven van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, Caceres JF. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J Cell Biol. 2000;149:307–316. doi: 10.1083/jcb.149.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shomron N, Alberstein M, Reznik M, Ast G. Stress alters the subcellular distribution of hSlu7 and thus modulates alternative splicing. J Cell Sci. 2005;118:1151–1159. doi: 10.1242/jcs.01720. [DOI] [PubMed] [Google Scholar]
- 67.Cohen AA, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–1516. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 68.Busa R, Geremia R, Sette C. Genotoxic stress causes the accumulation of the splicing regulator Sam68 in nuclear foci of transcriptionally active chromatin. Nucleic Acids Res. 2010;38:3005–3018. doi: 10.1093/nar/gkq004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paronetto MP, et al. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 2010;70:229–239. doi: 10.1158/0008-5472.CAN-09-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Comstock CE, et al. Cyclin D1 splice variants: polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin Cancer Res. 2009;15:5338–5349. doi: 10.1158/1078-0432.CCR-08-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andersen RJ, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 72.Nakka M, Agoulnik IU, Weigel NL. Targeted disruption of the p160 coactivator interface of androgen receptor (AR) selectively inhibits AR activity in both androgen-dependent and castration-resistant AR-expressing prostate cancer cells. Int J Biochem Cell Biol. 2013;45:763–772. doi: 10.1016/j.biocel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamashita S, et al. ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia. 2012;14:74–83. doi: 10.1593/neo.111436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li X, Liu Z, Xu X, Blair CA, Sun Z, Xie J, Lilly MB, Zi X. Kava components down-regulate expression of AR and AR splice variants and reduce growth in patient-derived prostate cancer xenografts in mice. PLoS One. 2012;7:e31213. doi: 10.1371/journal.pone.0031213. [DOI] [PMC free article] [PubMed] [Google Scholar]