

Abstract
The procedure to efficiently and reproducibly differentiate retinal cells from human pluripotent stem cells (hPSCs) is described below. Cells are taken through a stepwise protocol to direct them toward a neural fate by treatment with neural induction medium (NIM), then to a retinal fate by exposure to retinal differentiation medium (RDM). Undifferentiated hPSCs are enzymatically lifted from matrigel-coated plates and exposed to NIM in suspension. Differentiation in suspension allows the cells to form 3 dimensional aggregates. At 7 days of differentiation, aggregates are plated and attach to 6 well plates, where a neuroepithelial fate begins to be established. Upon 16 days of differentiation, neurospheres are lifted and maintained in RDM to create a three-dimensional optic vesicle-like structure. This procedure allows for the efficient and timely generation of a variety of retinal cell types, including ganglion cells, retinal pigment epithelium, as well as cone and rod photoreceptors. The use of this protocol to generate a myriad of retinal cell types facilitates in vitro studies of human retinogenesis, and will enable retinal dysfunction to be more easily studied in vitro, as well as providing a large population of cells with which to aid in drug development and patient specific therapies.
Keywords: Human pluripotent stem cells (hPSCs), retina, development, differentiation, optic vesicle
INTRODUCTION
In recent years, several groups have described the ability to direct human pluripotent stem cells (hPSCs) to a retinal fate (Buchholz et al., 2013; Carr et al., 2009; Hirami et al., 2009; Lamba et al., 2006; Lamba et al., 2010; Nakano et al., 2012; Osakada et al., 2008). In order to serve as an effective in vitro model for human retinogenesis, as well as provide a foundation for translational applications of these cells, the stepwise differentiation of hPSCs through all of the major stages of retinogenesis helps to ensure the proper differentiation and prospective identification of hPSC-derived retinal progeny (Gamm and Meyer, 2010; Meyer et al., 2011; Meyer et al., 2009; Sridhar et al., 2013). The procedure to efficiently and reproducibly differentiate retinal cells from hPSCs is described below (Figure 1). Cells are taken through a stepwise protocol to direct them toward a neural fate by treatment with neural induction medium (NIM), then to a retinal fate by exposure to retinal differentiation medium (RDM). Undifferentiated hPSCs are enzymatically lifted from matrigel-coated plates and exposed to NIM in suspension. Differentiation in suspension allows the cells to form three-dimensional aggregates. At 7 days of differentiation, aggregates are plated and attached to 6 well plates, where a neuroepithelial fate is established. Upon 16 days of differentiation, neurospheres are lifted and maintained in RDM to establish a three-dimensional optic vesicle-like fate. This procedure allows for the efficient and timely generation of a variety of retinal cell types, including ganglion cells, retinal pigment epithelum, as well as cone and rod photoreceptors. The use of this protocol to generate a myriad of retinal cell types facilitates in vitro studies of human retinogenesis (Meyer et al., 2011; Meyer et al., 2009; Zhong et al., 2014), and will enable studies of retinal dysfunction (Jin et al., 2012; Meyer et al., 2011; Singh et al., 2013b; Wahlin et al., 2014; Wright et al., 2014), as well as provides a large population of cells with which to aid in drug development in addition to patient specific therapies (Al-Shamekh and Goldberg, 2014; Carr et al., 2009; Lamba et al., 2010; Stern and Temple, 2014).
Figure 1. Overview of retinal differentiation protocol.

hPSCs can be directed to differentiate into all cell types of the retina in a step-wise process. From cultures of undifferentiated hPSCs, cells are directed to differentiate by the generation of embryoid bodies. By 7 total days of differentiation, embryoid bodies are plated and adherent cultures maintained for a total of 16 days. At this point, neurospheres are generated and retinal progenitor populations may be enriched by 20 days of differentiation. Maintained culture of these retinal neurospheres will yield all of the major cell types of the neural retina within the first 70 days of differentiation. Alternatively, optic vesicle-like cultures at day 16 of differentiation may be utilized to generate retinal pigmented epithelium through the maintenance of adherent cultures.
NOTE: All medium and solutions added directly to cells must be warm. It is recommended that reagents be heated in a 37° C water bath prior to use.
NOTE: All protocols below should be performed in a Class II biological culture hood to prevent contamination of cells.
NOTE: Standard incubation temperature is 37° C with 5% CO2.
ENZYMATIC PASSAGING OF HUMAN PLURIPOTENT STEM CELLS (BASIC PROTOCOL 1)
The following procedure can be used to maintain and passage hPSCs for long-term use (Ludwig et al., 2006; Meyer et al., 2011; Meyer et al., 2009; Park et al., 2008; Sridhar et al., 2013; Takahashi et al., 2007; Thomson et al., 1998; Yu et al., 2007). The protocol detailed below (Figure 1) focuses on the use of mTeSR1 medium and matrigel to maintain hPSCs, although previous reports have demonstrated the ability to maintain hPSCs in alternate systems such as fibroblast feeder cells (Meyer et al., 2011; Meyer et al., 2009; Sridhar et al., 2013). Cells are maintained on Matrigel-coated 6-well culture plates and are split when a confluency of approximately 70% is reached. This will aid in preventing spontaneous differentiation of cells due to overgrowth and subsequently ensures that an abundant amount of cells can be collected for directed differentiation. Typically, hPSCs are expanded at a ratio of 1:6, with a single well of cells capable of seeding an entire six well plate. A starting population of hPSCs should display a tightly clustered and bright morphology as well as exhibit immunoreactivity to pluripotency markers (Figure 2).
Figure 2. Characterization of undifferentiated hPSCs.

hPSCs displayed a typical undifferentiated morphology, including tightly packed colonies of cells and clearly defined edges (A). RT-PCR analysis demonstrated the expression of characteristic pluripotency makers in hPSCs, while lacking mesodermal, endodermal and ectodermal markers (B). Immunocytochemistry further demonstrated widespread expression of pluripotency-associated transcription factors (C–E) as well as cell surface markers (F–H). See Tables 1 and 2 for a listing of primers and antibodies used for RT-PCR and immunocytochemistry.
Materials
hPSCs plated on matrigel coated 6 well plates
Matrigel, hESC-qualified (BD Biosciences, see Reagents and Solutions unit)
mTeSR1 (StemCell Technologies)
Neural Induction Medium (NIM, see Reagents and Solutions unit)
DMEM-F/12, 1:1 (Life Technologies)
Dispase (Life Technologies, see Reagents and Solutions unit)
6-well culture plates (Falcon)
Inverted light microscope
T75 Flask (Falcon)
Passaging Undifferentiated Cells
-
1
Matrigel-coat 6-well plates (1 mL/well) and transfer to incubator for a minimum of 1 hour.
-
2
Aspirate excess matrigel from plates then add 2 ml mTeSR1 medium to each well and set aside until step 13.
-
3
Prepare two 15 mL conical tubes, one to collect undifferentiated cells to expand and another to collect cells that will be directed to differentiate (see section below regarding the generation of embryoid bodies).
-
4
Using an inverted light microscope, mark areas of spontaneous differentiation in wells that will be used for expansion.
-
5
Transfer plate to biological safety cabinet and using a 1 ml pipette tip, scrape away any cells from the marked areas of differentiation.
-
6
Aspirate medium from wells, add 1 ml of dispase to each well and transfer to incubator for 10 minutes.
-
7
Monitor the plates every few minutes thereafter to ensure the dispase is allowing for the start of cell detachment from the culture surface. If dispase is sufficiently warmed to 37°C, this process should not take more than 15 minutes. Once a majority of the cell clusters display curled edges, dispase should be removed immediately by aspiration.
-
8
Wash each well once with 1 ml DMEM/F12, ensuring that the medium is added to the side of the well, not directly onto the cells. Doing so will ensure that colonies are not prematurely detached from the culture surface during the wash step.
-
9
Aspirate DMEM/F12 from wells and add an additional 1 ml of DMEM/F12 in each well, this time added with force directly onto the cells to detach colonies from the plate by pipetting. Note that it is better to forcefully dislodge colonies by pipetting 3–4 times than to gently agitate any more than this. Minimizing the amount the cells are broken up is key to ensure maximum survival.
-
10
Transfer the cells from marked wells that will be expanded in one 15 ml conical tube and remaining wells for retinal differentiation to another 15 ml conical tube.
Expansion of Undifferentiated hPSCs
-
11
Allow cells to settle to the bottom of 15 ml conical tube (either by gravity or centrifuge for 1 minute at 800 rpm) and aspirate supernatant, taking care to avoid aspirating the cell pellet.
-
12
Re-suspend cells in mTeSR1 medium so that each new well will receive 500 μL of cell suspension per well. For example, if one well of hPSCs is being passaged to six new wells, 3 ml of mTeSR1 should be used. Break up clusters by pipetting forcefully 4–5 times with a 5 ml serological pipette. Note that each cell line can vary in ease of breaking up cell clusters. If cell clusters are not broken up sufficiently, increase pipetting during the next passage. It is better that clusters remain large than to break them up too much.
-
13
Pipet 500 μL of undifferentiated cell suspension at a 90 degree angle into each new well from step 2.
-
14
Transfer to the incubator and agitate plates in side-to-side followed by front-to-back motions to ensure even distribution of cells. Be sure to pause briefly between each series of agitations to ensure that cells are evenly dispersed across the well rather than accumulated in the middle.
-
15
Medium should be changed daily (2 mL/well) until the next passage, typically within 4–5 days.
Generation of Embryoid Bodies for Differentiation
To begin the differentiation process, cells must be slowly transitioned out of mTeSR1 medium, into neural induction medium (NIM), and maintained in a T75 flask. Day 0 is defined as the day cells are lifted from the Matrigel plate.
-
16
After cells have settled by gravity in the 15 ml conical tube (from step 10), aspirate the supernatant. Gently resuspend the cell pellet in a 3:1 mixture of mTeSR1:NIM and transfer to a T75 flask, and place flask in the incubator overnight.
-
17
Over the first few days of differentiation, embryoid bodies should be gradually transitioned from mTeSR1 to Neural Induction Medium (NIM). To accomplish this, transition cells with the following ratios of mTeSR1 to NIM.
Day 0–3:1
Day 1–1:1
Day 2–1:3
Day 3- Complete NIM
-
18
After day 3 of differentiation, change NIM every other day until day 7 is reached.
INDUCTION TO A PRIMITIVE ANTERIOR NEUROEPITHELIAL FATE (BASIC PROTOCOL 2)
As retinal cells are derived from a pluripotent source through a stepwise process in vivo (Livesey and Cepko, 2001; Marquardt and Gruss, 2002; Oliver and Gruss, 1997; Zhang et al., 2002), likewise hPSCs should be differentiated through analogous stages of differentiation, including a primitive anterior neural fate, an optic vesicle stage, and eventually a retinal and/or RPE fate (Meyer et al., 2011; Meyer et al., 2009; Sridhar et al., 2013; Zhong et al., 2014). To initiate this stepwise process, embryoid bodies are kept in suspension to begin differentiation for the first 7 days, at which point they are allowed to adhere to 6-well plates and maintained until day 16. By day 10 of differentiation, cells can be characterized by a larger, more uniform appearance as well as the expression of typical neural and eye field transcription factors (Figure 3).
Figure 3. Induction of hPSCs to a Neural Progenitor Fate.

After ten days of differentiation and plating (A), differentiating hPSCs were analyzed by RT-PCR and demonstrated expression of the neural markers PAX6 and SOX1. An anterior neural, eye-field fate was further indicated by the expression of OTX2. RAX, SIX3, and LHX2 (B). Immunocytochemistry demonstrated the widespread expression of many of these transcription factors (C–E).
Materials
hPSC-derived embryoid bodies in T75 flask (from Basic Protocol 1)
NIM (see Reagents and Solutions unit)
Fetal Bovine Serum
P200 pipetman
Plating Embryoid Bodies
After 7 total days of differentiation, cells should be plated onto 6-well culture plates to allow for further neural differentiation. This can be accomplished by the addition of 10% Fetal Bovine Serum (FBS) for the first 24 hours of the plating process to ensure cells adhere to the wells. In general, EBs derived from 5 wells of undifferentiated cells will plate on 1 six well plate, approximately 30–40 EBs per well.
Collect EBs in a 15 mL conical tube and allow them to settle by gravity, typically within 5 minutes.
Add 2 mL of NIM to each well of a 6 well plate.
Aspirate the supernatant from the EBs, being careful to avoid removing the cell pellet. Resuspsend the EBs in NIM so that each well will receive 200 μL of the cell suspension, and add cells to 6-well plate(s) accordingly. For one plate, cells would thus be resuspended in 1.2 ml of NIM.
Add 250 μL (~10%) FBS to each well to allow for cell attachment to the culture plate.
Transfer plates to the incubator, agitating plates in a side-to-side and then front-to-back manner to ensure an even distribution of cells within each well.
The following day, aspirate medium containing FBS and add fresh NIM to each well (2 mL/well) without FBS.
Medium should be changed every other day until day 16 of differentiation is reached, by which point these cells will have acquired a primitive anterior neuroepithelial fate.
DIFFERENTIATION OF PRIMITIVE ANTERIOR NEUROEPITHELIAL CELLS TO A RETINAL PIGMENTED EPITHELIAL FATE (BASIC PROTOCOL 3)
During normal development, the retinal pigmented epithelium (RPE) is the first retinal cell type to be specified from a more primitive source. The RPE layer develops in a manner that is distinctly separate from the neural retinal populations of cells, and is known to be specified in the absence of factors instrumental in directing a neural retinal fate (Fuhrmann et al., 2000; Martinez-Morales et al., 2003; Shibahara et al., 2000). Likewise, RPE cells generated from hPSCs are also found to differentiate through a similar process in which RPE cells are often found in close proximity to, although distinctly separate from, neural retinal populations (Capowski et al., 2014; Zhong et al., 2014). These hPSC-derived RPE cells can be readily identified by their accumulation of pigmentation and their distinct hexagonal morphology (Figure 4), and have been successfully generated by many groups in recent years (Buchholz et al., 2009; Buchholz et al., 2013; Capowski et al., 2014; Carr et al., 2009; Ferrer et al., 2014; Liao et al., 2010; Maruotti et al., 2013; Meyer et al., 2011; Meyer et al., 2009; Rowland et al., 2013; Singh et al., 2013a; Sridhar et al., 2013; Vugler et al., 2008).
Figure 4. Differentiation of hPSCs to Retinal Pigmented Epithelium.
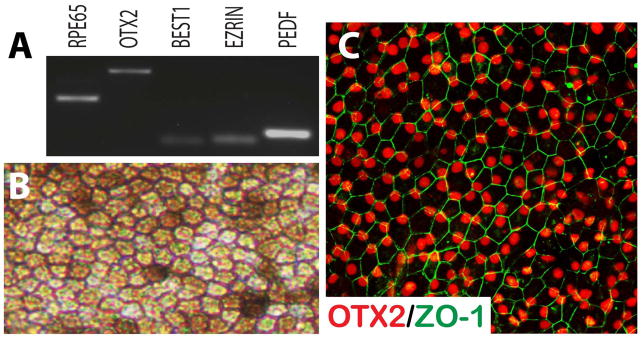
hPSC-derived RPE-like cells expressed typical RPE associated markers when screened by RT-PCR (A). Under brightfield microscopy, these cells displayed proper morphological features distinct to RPE, including a hexagonal shape and areas of pigmentation (B). Immunocytochemical analysis revealed the features typical of the retinal pigmented epithelium (C).
Materials
hPSC-derived neuroepithelial cells plated on 6-well plates (from Basic Protocol 2)
Retinal Differentiation Medium (see Reagents and Solutions unit)
Poly D-Ornithine/Laminin-coated coverslips in a 4 or 24 well plate (see Support protocols)
P20 pipetman
Inverted light microscope
FGF2 (working concentration of 20 ng/ml)
EGF (working concentration of 20 ng/ml)
Heparin (working concentration of 2 μg/ml)
hPSC-derived primitive anterior neuroepithelial cells on 6-well plates can also be used to generate a highly pure population of RPE, characterized by a cobblestone-like morphology as well as dark pigmentation. Continued growth and differentiation of these cells should be maintained until an RPE morphology emerges, typically within the first 60 days of differentiation.
Once day 16 of differentiation is reached, medium should be switched from NIM to RDM and medium should be changed every 2–3 days until distinct populations of RPE cells are readily observed, typically within 60 total days of differentiation.
At day 60 of differentiation, RPE can be microdissected and isolated. To microdissect RPE, identify an area of RPE cells of suitable purity by pigmentation and morphology under the microscope.
Using a pointed object (e.g. tungsten needle, pipette tip, etc), gently scratch away an area around the region of cells to be microdissected, freeing up the RPE cells.
Using a P100 pipette, transfer this cluster of freed RPE cells with 50 μl of medium to a laminin/poly-ornithine-coated coverslip in a 4 or 24 well plate, and repeat this process for as many coverslips as needed (see support protocols for coating coverslips). Typically, one cluster of RPE cells should be sufficient per coverslip.
Transfer the plate to incubator and allow RPE to attach to coverslip overnight.
The next day, RPE cell clusters should have adhered to the coverslips. Add 500 μl of RDM supplemented with EGF (20 ng/ml), FGF2 (20 ng/ml), and heparin to allow for proliferation of RPE cells. Replace medium and growth factors every 2 days.
Within 7–10 days, RPE cells should have lost most pigmentation and their hexagonal shape, and will have occupied most of the coverslip.
To allow for maturation and reacquisition of RPE morphology, remove medium containing EGF, FGF2, and heparin and replace with RDM. Maintain in this state for 2–3 weeks, or until a desired stage of RPE maturation is reached.
DIFFERENTIATION AND LONG-TERM MAINTENANCE OF RETINAL PROGENITOR CELLS (BASIC PROTOCOL 4)
During in vivo development, after cells have adopted a primitive anterior neural phenotype, a subset of cells are known to acquire a retinal fate and are characterized by numerous retinal-associated features that distinguish these cells from other neural lineages (Belecky-Adams et al., 1997; Bharti et al., 2008; Horsford et al., 2005; Rowan et al., 2004). Once this retinal identity has been established, all mature retinal cell types (cones, rods, retinal ganglion cells, etc.) will eventually arise. Likewise, hPSCs can acquire a retinal progenitor fate (Figure 5), eventually yielding all of the major cell types of the retina (Capowski et al., 2014; Meyer et al., 2011; Meyer et al., 2009; Phillips et al., 2014; Sridhar et al., 2013). To accomplish this differentiation event, cells are lifted and maintained in floating suspension with retinal differentiation medium (RDM) after approximately 16 days total days of differentiation. Retinal and non-retinal cells can then be manually separated and maintained until the desired stage of differentiation is reached.
Figure 5. Identification, Enrichment, and Characterization of Retinal Progenitor Cells.

After 30 days of differentiation, hPSCs were isolated into two morphologically distinct and readily identifiable populations (A). Retinal neurospheres, characterized by a bright ring surrounding the outer layer (B) and a non-retinal neural population displaying a larger, more uniform appearance (C). RT-PCR analysis revealed striking differences between these two neurosphere populations. While both populations expressed neural associated transcription factors (PAX6, NeuroD1, and FABP7), retinal neurospheres expressed CHX10, RAX, and SIX6, characteristic of retinal progenitors which were absent from non-retinal neural populations. Conversely, the non-retinal neural cells expressed forebrain-associated transcription factors including SOX1, DLX1, and EMX1, which were absent from retinal neurospheres (D). Immunocytochemical analysis revealed that retinal neurospheres widely expressed retinal progenitor markers such as CHX10 and PAX6 (E), but largely lacked the expression of the forebrain-associated marker SOX1 (F). hPSC-derived retinal progenitors also remained highly proliferative within the first 30 days of differentiation (G). Non-retinal neural populations displayed typical features of emerging forebrain neurons, including the expression of βIII-Tubulin and OTX2 (H), as well as the forebrain-associated DLX5, but lacked the retinal progenitor marker CHX10 (I). Non-retinal neural cells also retained the expression of both PAX6 and SOX1 (J).
Materials
Primitive neuroepithelial cells maintained in 6 well plates (from Basic Protocol 2)
Retinal Differentiation Medium (see Reagents and Solutions unit)
60 mm × 15 mm polystyrene petri dishes
6 well plates (Falcon)
P1000 pipetman
Generation of neurospheres from primitive anterior neuroepithelial cells
At day 16 of differentiation (see Figure 3), cells should be lifted from culture plates to allow for the development of a three-dimensional optic vesicle-like structure.
-
1
Using a P1000 pipetman, draw up 1 ml of medium from a well of cells and dislodge the center of each aggregate from the plate by vigorously pipetting the medium directly at the center of each aggregate. The center of each aggregate will dislodge, leaving behind a ring of peripheral cells possessing a flattened appearance. It is better to pipette forcefully 5–6 times instead of gently pipetting any more than this, as excessive pipetting results in cell death and reduced yield of neurospheres.
-
2
After dislodging the cells from the plates, transfer aggregates to a 15 ml conical tube. Allow the cell aggregates to settle by gravity or by centrifugation for 1 minute at 800 rpm.
-
3
Aspirate the supernatant and resuspend the cells in 5 ml of RDM.
-
4
Transfer the cell suspension to a 60 mm dish and return cells to the incubator. Cells at this stage should have their medium changed every 2–3 days.
Manual Enrichment of Retinal Neurospheres
By day 20–25 of differentiation, two populations of neurospheres will begin to emerge from the primitive anterior neuroepithelial cells - those possessing a golden ring around the outside (retinal neurospheres) and those with a darker appearance lacking this golden ring (non-retinal forebrain neurospheres). These morphological differences can easily be observed with an inverted microscope under a 4x objective. By 30 total days of differentiation, distinct transcriptional profiles emerge that distinguish these two populations (Figure 5).
-
5
Enrichment of retinal neurospheres should be performed by day 25 of differentiation. While viewing the cells under an inverted light microscope with a 4x objective, swirl the plate gently in a circular motion to collect cells in the middle of the dish.
-
6
Looking at the neurospheres through the microscope, gently gather retinal neurospheres based on their bright outer ring appearance using a P20 pipetman and transfer these neurospheres to a 15 ml conical tube containing 5 ml of RDM. Repeat until all retinal neurospheres have been collected in the same 15 ml conical tube.
-
7
After collecting retinal neurospheres, transfer this population along with the medium to a 60 mm dish and return cells to the incubator. Non-retinal neurospheres may be similarly maintained for neuronal cultures if desired, or discarded at this stage.
Maintenance of Retinal Neurospheres
From this point forward, medium will need to be replaced every other day until the desired stage of retinal differentiation is reached.
-
8
Tilt the plate towards you to allow the medium and neurospheres to collect on the bottom half of the plate.
-
9
Using a P1000 pipetman, transfer 1 ml of medium containing as many neurospheres as possible to a 15 ml conical tube while still maintaining the tilt of the plate.
-
10
Collect the remaining medium and rinse over the entire surface of the plate to collect any remaining neurospheres that may remain in the plate.
-
11
Transfer this remaining medium to the 15 ml conical tube.
-
12
Allow neurospheres to settle to the bottom of the tube and then aspirate the supernatant.
-
13
Resuspend neurospheres in 5 ml of fresh RDM and transfer this cell suspension back to the dish. The dish should be stored in the incubator for further use.
INDUCTION OF RETINAL PROGENITORS TO SPECIFIC RETINAL SUBTYPES (BASIC PROTOCOL 5)
Previous studies have demonstrated the ability to yield all of the major classes of retinal cells from hPSC-derived retinal progenitor cells, including photoreceptors (Gonzalez-Cordero et al., 2013; Lamba et al., 2006; Lamba et al., 2010; Mellough et al., 2012; Meyer et al., 2011; Meyer et al., 2009; Osakada et al., 2008; Reichman et al., 2014; Tucker et al., 2013a; Tucker et al., 2013b; Zhong et al., 2014) and retinal ganglion cells (Lamba et al., 2010; Meyer et al., 2011; Sridhar et al., 2013; Zhong et al., 2014). In order to derive these various types of retinal cells, retinal neurospheres must be maintained in differentiating cultures for extended periods of time. Within 90 days of total differentiation, neural retinal cell types including photoreceptors and retinal ganglion cells can be identified (Figure 6). In order to analyze cells by immunocytochemistry, neurospheres should be dissociated with accutase and then plated onto Poly-D-Ornithine and laminin-coated coverslips (See Support Protocol 1 for coating coverslips). At this point, cells demonstrate the presence of a wide variety of retinal specific transcription factors and distinct neuroretinal morphologies such as neurite outgrowth and/or axonal/dendritic arborization typical of retinal ganglion cell or photoreceptor morphologies (Figure 6).
Figure 6. Differentiation of hPSCs to Retinal Neurons.

Within 90 total days of differentiation, hPSC-derived retinal cells displayed typical neuronal morphologies under DIC microscopy (A). Analysis by RT-PCR illustrated an array of retinal-associated transcription factors including those associated with ganglion cells (BRN3 and Islet1), as well as those associated with photoreceptors (CRX and NeuroD4). Furthermore, proteins associated with phototransduction could also be identified, including Red/Green Opsin, Arrestin, and Transducin (B). Immunocytochemistry analysis confirmed the expression of BRN3-positive retinal ganglion cells extending Map2-positive dendrites (C) as well as photoreceptor-like phenotypes including the expression of CRX, OTX2 and Recoverin (D, E).
Materials
Retinal Neurospheres at 40 days total differentiation (from Basic Protocol 4)
Retinal Differentiation Medium (see Reagents and Solutions unit)
Poly-D-Ornithine and laminin-coated coverslips (see Support protocols)
Accutase (BD Biosciences)
P100 pipetman
Inverted light microscope
Dissociation and Plating of Retinal Neurospheres
Place the plate of retinal neurospheres in suspension under a microscope and swirl in a circular motion to gather cells towards the center of the field of view.
-
Gather neurospheres that will be used for dissociation and plating in a 1.5 mL tube.
For plating coverslips, 2–3 neurospheres/coverslip typically provide enough density for sufficient plating and microscopy.
Allow neurospheres to settle at the bottom and gently remove excess medium with a pipette.
Add 100 μL of accutase.
Transfer to 37°C water bath for 10 minutes.
Every ten minutes, remove tube from water bath and forcefully agitate cells about 4 or 5 times with a P100 (50 μL setting) pipetman to break up the cells.
Repeat steps 5 and 6 if clusters need further dissociation
Once retinal neurospheres have been dissociated to aggregates of a desired size, centrifuge cell suspension for 1 minute at 800 rpm. Viability is greatly increased if cells are dissociated to yield small aggregrates of cells rather than a single cell suspension.
Gently remove supernatant with a pipet and resuspend in enough RDM to ensure each coverslip receives 50 μL of cell suspension.
Pipet 50 μL of cell suspension onto each poly-D-ornithine and laminin-coated coverslip (see support protocols).
Transfer to incubator and allow cells to adhere overnight.
If cells are to be maintained on coverslips for further differentiation, add 500 μL of RDM the following day and every other day thereafter until the desired stage of differentiation is reached. If cells are to be fixed immediately for immunocytochemistry, no additional medium should be added.
SUPPORT PROTOCOLS
Support Protocol 1: Coating coverslips with Poly-D-ornithine and Laminin
This brief protocol will explain how to coat the coverslips for use in Basic Protocols 3 and 5. While the Poly-D-ornithine increases adhesion of cells to the coverslip, the laminin promotes cell growth. Cells grown on Poly-D-Ornithine and Laminin-coated coverslips can easily be utilized for immunocytochemical analysis and readily transferred to slides for visualization by microscopy.
24- and/or 4-well plates
EtOH washed and subsequently autoclaved glass coverslips (12 mm)
Stock of poly-D-ornithine (see Reagents and Solutions)
Transfer one coverslip to each well and ensure it lies flat on the bottom of the well.
Pipet 100 μL Poly-D-Ornithine into the center of each coverslip. Let sit at room temperature for 30 minutes, ensuring the Poly-D-Ornithine remains on the coverslip.
Remove Poly-D-Ornithine and wash each well with 1 mL of sterile water. Repeat two more times.
After removing the third wash, check coverslips under microscope to make sure any precipitate is washed away. Continue rinsing if any residue remains.
Remove last wash and allow coverslips to air dry in the biological safety cabinet overnight. Be sure to leave the hood fan on and the sash slightly open.
The following day, remove dry coverslips from hood and store at room temperature for future use.
Laminin-Coating of Coverslips
Add 50 μL of laminin (20 μg/ml in DMEM) directly to the center of each coverslip previously coated with Poly-D-Ornithine.
Transfer plates to the incubator and let stand for at least 4 hours or overnight.
After incubation to allow for thorough coating of coverslips with laminin, aspirate excess laminin just before addition of cell suspension.
REAGENTS AND SOLUTIONS
All solutions should be made in a biological safety cabinet and filtered through a steriflip or bottle top filter to ensure solutions are sterile.
Dispase
DMEMF/12 containing:
2 mg/mL dispase powder
Ensure DMEMF/12 is warm and dissolve dispase completely into solution.
Combine reagents and filter
Store up to 2 weeks at 4°C
NIM (Neural Induction Medium)
489.5 mL DMEMF/12
5 mL N2 supplement
5 mL MEM NEAA
0.5 mL Heparin (2 mg/ml)
Combine reagents and filter
Store up to 1 month at 4°C
RDM (Retinal Differentiation Medium)
240 mL DMEMF/12
240 mL DMEM
10 mL B27 Supplement
5 mL MEM Non-essential amino acids
5 mL antibiotics
Combine reagents and filter
Store up to 1 month at 4°C
Laminin
Staring with a 1 mg/mL stock, dilute laminin 1:50 in cold DMEM to a final concentration of 20 μg/mL.
Store up to 1 month at 4°C
Matrigel
Dilute according to the manufacturer’s specifications in DMEM
Poly-D-ornithine
Poly-D-ornithine (10 mg Sigma)
Sterile H20
1 mL pipetman
250 mL autoclaved beaker
50 mL conical tubes
Steriflip 0.2 mm filtering device
Store up to 6 months at 4°C
Tap the bottle of poly-D-ornithine on the surface of the hood and open carefully as to not lose any powder.
Slowly pipet 1 mL of sterile water into the bottle, put the top back on, and shake vigorously. Remove cap carefully and transfer to the beaker.
Repeat process about 4 more times to remove trace amounts from the bottle.
To the beaker, add sterile water up to a volume of 100 mL. Pipet up and down to mix thoroughly.
Transfer 50 mL into each conical tube. Sterile filter each using a Steriflip 0.2 μm filtering device.
COMMENTARY
The ability to direct the differentiation of hPSCs to a retinal fate represents a limitless source of retinal cells for in vitro studies of human retinogenesis, as well as a unique and exciting tool with which to study retinal disease progression, screen compounds for potential therapeutic efficacy, and even provide a source of replacement cells for transplantation purposes. For the above reasons, several groups have explored the ability to differentiate hPSCs to a retinal fate (Buchholz et al., 2009; Carr et al., 2009; Hirami et al., 2009; Lamba et al., 2006; Liao et al., 2010; Maruotti et al., 2013; Mellough et al., 2012; Meyer et al., 2011; Meyer et al., 2009; Nakano et al., 2012; Osakada et al., 2008; Sridhar et al., 2013; Zhong et al., 2014), with the traditional focus upon the differentiation of photoreceptors and RPE cells due to the availability of unique and specific characteristics with which to identify these cells.
The protocol described within this manuscript not only details a method with which to differentiate retinal cells from a pluripotent stem cell population, but this protocol is significant due to the ability to faithfully identify and enrich for cells at all of the major stages of retinal development (Meyer et al., 2011; Meyer et al., 2009; Sridhar et al., 2013). Starting from an undifferentiated population of hPSCs, cells are efficiently differentiated to a primitive anterior neuroepithelial fate in high purity after as little as 10 days of differentiation (Figure 2). From this point, differentiating cells yield neurosphere populations representing either forebrain progenitor cells or optic vesicle-like retinal progenitor cells, the latter of which exclusively gives rise to more mature retinal phenotypes including photoreceptor cells and retinal ganglion cells (Meyer et al., 2011; Sridhar et al., 2013). The ability to readily identify cells at each of the major stages of retinal development is unique to the method outlined in this manuscript to date, and helps to establish hPSCs as a valuable in vitro model of human retinogenesis.
The ability to morphologically identify and isolate retinal progenitor neurospheres within the first 20–25 days of differentiation is noteworthy for a variety of reasons (Figure 4). First, the use of morphological cues to identify and isolate retinal progenitor neurospheres represents a novel method of enrichment of retinal cells apart from other cellular lineages that may be found in such cultures, effectively yielding highly enriched populations of retinal progenitor cells that can readily differentiate into all of the major cell types of the retina (Meyer et al., 2011; Zhong et al., 2014). Furthermore, this ability to highly enrich for retinal progenitor cells allows for the more definitive identification of many more mature retinal cell types, including retinal ganglion cells. Within the retina itself, retinal ganglion cells are often identified by the expression of the transcription factor BRN3 (Badea and Nathans, 2011; Bryant et al., 2002; Shi et al., 2013). However, this transcription factor is also expressed in other neural cell types, including some auditory neurons (Bryant et al., 2002; Weir et al., 2000) as well as many somatosensory neurons (Badea et al., 2012; Chambers et al., 2012). Thus, when starting with a pluripotent stem cell source that has the potential to give rise to any cell type of the body, the expression of BRN3 by itself is not sufficient to definitively identify cells as retinal ganglion cells. However, due to the ability to enrich for retinal progenitor cells from hPSCs as described by the current protocol, traditional markers such as BRN3 can be utilized to identify these retinal cells as other non-retinal cell types will be effectively eliminated from the culture system. The ability to identify BRN3-positive cells from a highly enriched retinal progenitor population allows for the more definitive identification of these cells as retinal ganglion cells.
When working with a pluripotent stem cell population, the definitive and conclusive identification of a particular differentiated cell type may often be difficult. Many markers traditionally utilized to identify differentiated cell types, including many markers used to identify specific retinal cell types such as retinal ganglion cells, are often expressed in other areas of the body. Thus, when working with a pluripotent cell population that has the potential to give rise to any cell type of the body, the simple expression of many of these cell type-associated markers may not be sufficient to definitively identify a differentiated cell as a particular cell type. Retinal ganglion cells represent such a cell type that would ordinarily be difficult to conclusively identify from pluripotent stem cells, as there are no markers used to identify retinal ganglion cells that are not expressed elsewhere in the nervous system.
Due to the ease of differentiation of hPSCs described in this protocol, including the minimal culture conditions required to achieve this differentiation event, this procedure allows for the application of hPSCs as a novel model for in vitro studies of human retinogenesis (Capowski et al., 2014; Meyer et al., 2011; Meyer et al., 2009; Phillips et al., 2014; Sridhar et al., 2013; Zhong et al., 2014). The ability to identify each of the major stages of retinal development has led to the recent use of this method as a means of studying the molecular basis for cell fate determination in the retina between those cells of the neural retina and the retinal pigmented epithelium (Capowski et al., 2014; Phillips et al., 2014). Not only are these results significant due to the ability to study these events in human cells, but these cell fate determination events are known to occur at stages of development that would otherwise be inaccessible to experimental investigation. Furthermore, recent studies have demonstrated the ability to expand upon this method to generate three-dimensional stratified retinal-like structures in vitro (Phillips et al., 2012; Zhong et al., 2014), allowing for future possibilities not only for studies of cell fate determination, but also for the maturation of these cells.
The ability to derive a full complement of retinal cell types from hPSCs also allows for the ability to study human inherited diseases of the retina, particularly those leading to blindness such as age-related macular degeneration (AMD), retinitis pigmentosa, and optic neuropathies including glaucoma, among others. Owing to the ability to enrich for retinal progenitor cells with this protocol, large numbers of retinal cells are readily obtainable from patient-derived induced pluripotent stem cells, and this ability has recently been utilized as an effective in vitro model for a variety of blinding disorders, including gyrate atrophy and Best disease (Meyer et al., 2011; Singh et al., 2013b), with the potential for studies of numerous other retinal degenerative disorders. Beyond the use of these cells as an in vitro model of retinal disease, the potential exists for pharmacological screening of novel compounds for therapeutic efficacy using patient-derived cells. This was originally described in disorders affecting the RPE(Meyer et al., 2011; Singh et al., 2013b), and has been expanded to disorders affecting other retinal cell types such as photoreceptors (Jin et al., 2011). It is expected that hPSCs will likely be an effective complement to traditional model systems and will also have the potential to add to current methods to study human samples, typically from post-mortem tissue or from readily accessible cell sources that are often unaffected by disease processes, particularly in the case of the retina. Thus, it is believed that hPSCs bridge an important gap for both basic and translational research between traditional model systems and the existing human condition.
Critical Parameters
Passaging hPSCs
It is crucial to minimize the amount of pipetting as even a little excessive pipetting can result in a dramatic decrease in the yield of EBs. Cells that have been treated with dispase should lift off with little effort.
When cells have been collected into their respective tubes (expansion or differentiation), allow at least 5 minutes for cells to fully settle at the bottom to ensure no cells are being aspirated with the supernatant.
Plating Embryoid Bodies with 10% FBS
EBs must be plated at a critical density to ensure the proper differentiation into neuroepithelium. Without close proximity to neighboring cells, cell survival decreases. When plated at too high of a density, cells will lack the space they require to develop properly. Agitation of plates when placed into the incubator also helps in achieving a uniform distribution of cells within the well.
Manually Separating Retinal Neurospheres
Separation of retinal neurospheres by day 25 is critical to ensure that morphology can be definitively used to separate retinal and non-retinal cells. After day 25, the retinal morphology may begin to disappear as the neurospheres continue proliferating.
Maintenance of Retinal Neurospheres
When changing medium of cells in suspension, unnecessary cell loss can be avoided by rinsing the well or dish with extra medium. This ensures all cells are collected and receive fresh medium in a timely manner.
Troubleshooting
| Problem | Possible Cause | Solution |
|---|---|---|
| EB survival is low after passaging | Pipetting too vigorously to remove cells from plates Cells are being aspirated with the supernatant |
Leave dispase on hPSCs longer to ensure easy detachment of cells Allow cells to settle for at least 5 minutes by gravity or centrifuge for 1 minute at 800 RPM |
| Neurosphere yield is low | Cells were not plated densely enough with FBS | Increase density upon next plating |
| Cells are not adhering to plates with 10% FBS | Too much NIM was added to the well resulting in less than 10% FBS in the final volume | Instead of changing the medium, add 5% more FBS to each well and return to incubator overnight. Change medium the following day and add fresh NIM to each well |
| Undifferentiated cells growing too slowly | Cells were broken up too much during passaging | At next passage, pipet cells fewer times to break up cell aggregates and increase this number with each subsequent passage |
| Colonies are taking 15–20 minutes to detach from plate | Dispase is not warm Dispase powder did not completely dissolve when solution was made |
Allow dispase to warm in 37°C water bath for 15 min When making dispase, add powder to warm DMEMF/12 and allow enzyme to dissolve at 37° for at least 20 minutes |
| Recently plated undifferentiated hPSCs or EBs are not evenly distributed across the well | Plates were not agitated when placed in the incubator or were disturbed by opening/closing of incubator door | Agitate plates front/back and side/side in short, quick movements to ensure an even distribution of cells on the well surface. Place plates near the back of the incubator to minimize disturbance by opening/closing of incubator door |
Anticipated Results
The use of this procedure yields a 90% pure population of CHX10-positive retinal progenitor cells that can be further differentiated into every retinal cell type.
Time Considerations
With practice, passaging of hPSCs can take as little as 20 minutes once the necessary supplies and reagents have been prepared. When EBs are ready to be plated down, this process should take about 10 minutes for three plates. Manually separating retinal neurospheres from a mixed population can take anywhere from 30 minutes to an hour, depending on the yield of neurospheres and the experience of the researcher. Using accutase to disassociate neurospheres can take anywhere from 10 minutes to 40 minutes, with varying neurosphere size and the degree to which spheres are meant to be disassociated.
Table 1.
Primary antibodies used for immunocytochemistry
| Antibody | Source | Catalog Number | Dilution |
|---|---|---|---|
| βIII tubulin | Covance | PRB-435P | 1:100 |
| Brn3 | Santa Cruz | SC-6026 | 1:200 |
| Chx10 | Santa Cruz | SC-21690 | 1:200 |
| Crx | Abnova | H00001406-M02 | 1:100 |
| Dlx5 | Abcam | Ab64827 | 1:200 |
| Ki-67 | BD Biosciences | 556003 | 1:500 |
| Lhx2 | Santa Cruz | SC-19344 | 1:200 |
| Map2 | Santa Cruz | SC-20172 | 1:200 |
| Nanog | R&D Systems | AF1997 | 1:100 |
| Oct4 | Stemgent | 09-0023 | 1:200 |
| Otx2 | R & D Systems | AF1979 | 1:2000 |
| Pax6 | Developmental Studies Hybridoma Bank | PAX6 | 1:50 |
| Recoverin | Chemicon | AB5585 | 1:2000 |
| Sox1 | R & D Systems | AF3369 | 1:1000 |
| Sox2 | R & D Systems | AF2018 | 1:1000 |
| SSEA-4 | Chemicon | 09-0006 | 1:500 |
| Tra-1-60 | Chemicon | 09-0010 | 1:1000 |
| Tra-1-81 | Chemicon | 09-0011 | 1:1000 |
| ZO-1 | Zymed | 61-7300 | 1:100 |
Table 2.
Primers used for RT-PCR and quantitative RT-PCR.
| Gene amplified | Forward | Reverse | Size (bp) |
|---|---|---|---|
| α-Fetoprotein | AGA ACC TGT CAC AAG CTG TG | GAC AGC AAG CTG AGG ATG TC | 676 |
| ARRESTIN | ACA AGC TAG GGG ACA ATG CC | TTG TGC TAG AGG CCA GGT TG | 597 |
| BEST1 | GGT GTG GTT TGC CAA CCT GTC AAT | TGT TCA TCT CGT TCA GCA GGC TCT | 92 |
| BRACHYURY | ACC CAG TTC ATA GCG GTG AC | CAA TTG TCA TGG GAT TGC AG | 218 |
| BRN3 | CTC ACA CTG TCC CAC AAT AAT A | CCG GCG GAA TAT TTC ATT CT | 311 |
| CHX10 | ATT CAA CGA AGC CCA CTA CCC AGA | ATC CTT GGC TGA CTT GAG GAT GGA | 229 |
| CRX | TAT TCT GTC AAC GCC TTG GCC CTA | TGC ATT TAG CCC TCC GGT TCT TGA | 253 |
| DLX1 | CAA CCA GCA AAT GTC TCC TTC TC | CGC ACT TCA CCG CCT TCC | 282 |
| EMX1 | AGA CGC AGG TGA AGG TGT GG | CAG GCA GGC AGG CTC TCC | 403 |
| EZRIN | ACC ACC ATG GAT GCA GAG CTG GA | ACA CTT CCC GGA GGC CGA TAG T | 100 |
| FABP7 | AGG CAG GTG GGA AAT GTG AC | CAT AGT GGC GAA CAG CAA CC | 298 |
| ISLET1 | GTG TGA TCC GGG TCT GGT TT | AAT TAG AGC CCG GTC CTC CT | 300 |
| KLF4 | AGT CCC GCC GCT CCA TTA CCA A | TGC TCG GTC GCA TTT TTG GCA C | 316 |
| LHX2 | CAA GAT CTC GGA CCG CTA CT | CCG TGG TCA GCA TCT TGT TA | 284 |
| LIN28 | AGT GGT TCA ACG TGC GCA TGG G | AGG TCC GGT GAC ACG GAT GGA T | 203 |
| NANOG | CAA AGG CAA ACA ACC CAC TT | TCT GCT GGA GGC TGA GGT AT | 158 |
| NEUROD1 | TAC TGC TGC AAA GTG CAA ATA C | AAG TGC TAA GGC AAC ACA ATA AC | 539 |
| NEUROD4 | AGG TCT GGG CTC CCA AAA TG | GCC CCG GAG ACT GAT AGT TG | 557 |
| OCT4 | CGA GCA ATT TGC CAA GCT CCT GAA | TTC GGG CAC TGC AGG AAC AAA TTC | 324 |
| OPSIN | GAA GTT CAA GAA GCT GCG CC | TCT CAC ATT GCC AAA GGG CT | 253 |
| OTX2 | CAA CAG CAG AAT GGA GGT CA | CTG GGT GGA AAG AGA GAA GC TG | 429 |
| PAX6 | CGG AGT GAA TCA GCT CGG TG | CCG CTT ATA CTG GGC TAT TTT GC | 300 (+5a) 258 (−5a) |
| PEDF | AGA TCT CAG CTG CAA GAT TGC CCA | ATG AAT GAA CTC GGA GGT GAG GCT | 127 |
| RAX | GAA TCT CGA AAT CTC AGC CC | CTT CAC TAA TTT GCT CAG GAC | 279 |
| RPE65 | TAC CAC AGA AGG TTC ATC CGC ACT | GGG AAA GCA CAG GTG CCA AAT TCT | 92 |
| SIX3 | CGA GCA GAA GAC GCA TTG CTT CAA | CGG CCT TGG CTA TCA TAC ATC ACA | 394 |
| SIX6 | ATT TGG GAC GGC GAA CAG AAG ACA | ATC CTG GAT GGG CAA CTC AGA TGT | 385 |
| SOX1 | CAA TGC GGG GAG GAG AAG TC | CTC TGG ACC AAA CTG TGG CG | 464 |
| SOX2 | CCC CCG GCG GCA ATA GCA | TCG GCG CCG GGG AGA TAC AT | 448 |
| TRANSDUCIN | CAC GAT GCC CAA GGA GAT GT | GGT GGT TGC AGA TGC TGT TG | 419 |
Acknowledgments
This work was supported by funding from the BrightFocus Foundation (Grant #G2012027), a grant from the Indiana University Collaborative Research Grant fund of the Office of the Vice President for Research, startup funds from the School of Science at IUPUI, as well as funds from a Project Development Team within the ICTSI NIH/NCRR Grant Number UL1TR001108.
References
- Al-Shamekh S, Goldberg JL. Retinal repair with induced pluripotent stem cells. Transl Res. 2014;163:377–386. doi: 10.1016/j.trsl.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea TC, Nathans J. Morphologies of mouse retinal ganglion cells expressing transcription factors Brn3a, Brn3b, and Brn3c: analysis of wild type and mutant cells using genetically-directed sparse labeling. Vision Res. 2011;51:269–279. doi: 10.1016/j.visres.2010.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea TC, Williams J, Smallwood P, Shi M, Motajo O, Nathans J. Combinatorial expression of Brn3 transcription factors in somatosensory neurons: genetic and morphologic analysis. J Neurosci. 2012;32:995–1007. doi: 10.1523/JNEUROSCI.4755-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belecky-Adams T, Tomarev S, Li HS, Ploder L, McInnes RR, Sundin O, Adler R. Pax-6, Prox 1, and Chx10 homeobox gene expression correlates with phenotypic fate of retinal precursor cells. Invest Ophthalmol Vis Sci. 1997;38:1293–1303. [PubMed] [Google Scholar]
- Bharti K, Liu W, Csermely T, Bertuzzi S, Arnheiter H. Alternative promoter use in eye development: the complex role and regulation of the transcription factor MITF. Development. 2008;135:1169–1178. doi: 10.1242/dev.014142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant J, Goodyear RJ, Richardson GP. Sensory organ development in the inner ear: molecular and cellular mechanisms. Br Med Bull. 2002;63:39–57. doi: 10.1093/bmb/63.1.39. [DOI] [PubMed] [Google Scholar]
- Buchholz DE, Hikita ST, Rowland TJ, Friedrich AM, Hinman CR, Johnson LV, Clegg DO. Derivation of functional retinal pigmented epithelium from induced pluripotent stem cells. Stem Cells. 2009;27:2427–2434. doi: 10.1002/stem.189. [DOI] [PubMed] [Google Scholar]
- Buchholz DE, Pennington BO, Croze RH, Hinman CR, Coffey PJ, Clegg DO. Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem Cells Transl Med. 2013;2:384–393. doi: 10.5966/sctm.2012-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capowski EE, Simonett JM, Clark EM, Wright LS, Howden SE, Wallace KA, Petelinsek AM, Pinilla I, Phillips MJ, Meyer JS, Schneider BL, Thomson JA, Gamm DM. Loss of MITF expression during human embryonic stem cell differentiation disrupts retinal pigment epithelium development and optic vesicle cell proliferation. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AJ, Vugler AA, Hikita ST, Lawrence JM, Gias C, Chen LL, Buchholz DE, Ahmado A, Semo M, Smart MJ, Hasan S, da Cruz L, Johnson LV, Clegg DO, Coffey PJ. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS One. 2009;4:e8152. doi: 10.1371/journal.pone.0008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, Bilsland J, Cao L, Stevens E, Whiting P, Shi SH, Studer L. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. 2012;30:715–720. doi: 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M, Corneo B, Davis J, Wan Q, Miyagishima KJ, King R, Maminishkis A, Marugan J, Sharma R, Shure M, Temple S, Miller S, Bharti K. A Multiplex High-Throughput Gene Expression Assay to Simultaneously Detect Disease and Functional Markers in Induced Pluripotent Stem Cell-Derived Retinal Pigment Epithelium. Stem Cells Transl Med. 2014;3:911–922. doi: 10.5966/sctm.2013-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann S, Levine EM, Reh TA. Extraocular mesenchyme patterns the optic vesicle during early eye development in the embryonic chick. Development. 2000;127:4599–4609. doi: 10.1242/dev.127.21.4599. [DOI] [PubMed] [Google Scholar]
- Gamm DM, Meyer JS. Directed differentiation of human induced pluripotent stem cells: a retina perspective. Regen Med. 2010;5:315–317. doi: 10.2217/rme.10.28. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cordero A, West EL, Pearson RA, Duran Y, Carvalho LS, Chu CJ, Naeem A, Blackford SJ, Georgiadis A, Lakowski J, Hubank M, Smith AJ, Bainbridge JW, Sowden JC, Ali RR. Photoreceptor precursors derived from three-dimensional embryonic stem cell cultures integrate and mature within adult degenerate retina. Nat Biotechnol. 2013;31:741–747. doi: 10.1038/nbt.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirami Y, Osakada F, Takahashi K, Okita K, Yamanaka S, Ikeda H, Yoshimura N, Takahashi M. Generation of retinal cells from mouse and human induced pluripotent stem cells. Neurosci Lett. 2009;458:126–131. doi: 10.1016/j.neulet.2009.04.035. [DOI] [PubMed] [Google Scholar]
- Horsford DJ, Nguyen MT, Sellar GC, Kothary R, Arnheiter H, McInnes RR. Chx10 repression of Mitf is required for the maintenance of mammalian neuroretinal identity. Development. 2005;132:177–187. doi: 10.1242/dev.01571. [DOI] [PubMed] [Google Scholar]
- Jin ZB, Okamoto S, Osakada F, Homma K, Assawachananont J, Hirami Y, Iwata T, Takahashi M. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS One. 2011;6:e17084. doi: 10.1371/journal.pone.0017084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin ZB, Okamoto S, Xiang P, Takahashi M. Integration-free induced pluripotent stem cells derived from retinitis pigmentosa patient for disease modeling. Stem Cells Transl Med. 2012;1:503–509. doi: 10.5966/sctm.2012-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba DA, Karl MO, Ware CB, Reh TA. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A. 2006;103:12769–12774. doi: 10.1073/pnas.0601990103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba DA, McUsic A, Hirata RK, Wang PR, Russell D, Reh TA. Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PLoS One. 2010;5:e8763. doi: 10.1371/journal.pone.0008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JL, Yu J, Huang K, Hu J, Diemer T, Ma Z, Dvash T, Yang XJ, Travis GH, Williams DS, Bok D, Fan G. Molecular signature of primary retinal pigment epithelium and stem-cell-derived RPE cells. Hum Mol Genet. 2010;19:4229–4238. doi: 10.1093/hmg/ddq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey FJ, Cepko CL. Vertebrate neural cell-fate determination: lessons from the retina. Nat Rev Neurosci. 2001;2:109–118. doi: 10.1038/35053522. [DOI] [PubMed] [Google Scholar]
- Ludwig TE, Bergendahl V, Levenstein ME, Yu J, Probasco MD, Thomson JA. Feeder-independent culture of human embryonic stem cells. Nat Methods. 2006;3:637–646. doi: 10.1038/nmeth902. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Gruss P. Generating neuronal diversity in the retina: one for nearly all. Trends Neurosci. 2002;25:32–38. doi: 10.1016/s0166-2236(00)02028-2. [DOI] [PubMed] [Google Scholar]
- Martinez-Morales JR, Dolez V, Rodrigo I, Zaccarini R, Leconte L, Bovolenta P, Saule S. OTX2 activates the molecular network underlying retina pigment epithelium differentiation. J Biol Chem. 2003;278:21721–21731. doi: 10.1074/jbc.M301708200. [DOI] [PubMed] [Google Scholar]
- Maruotti J, Wahlin K, Gorrell D, Bhutto I, Lutty G, Zack DJ. A simple and scalable process for the differentiation of retinal pigment epithelium from human pluripotent stem cells. Stem Cells Transl Med. 2013;2:341–354. doi: 10.5966/sctm.2012-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellough CB, Sernagor E, Moreno-Gimeno I, Steel DH, Lako M. Efficient stage-specific differentiation of human pluripotent stem cells toward retinal photoreceptor cells. Stem Cells. 2012;30:673–686. doi: 10.1002/stem.1037. [DOI] [PubMed] [Google Scholar]
- Meyer JS, Howden SE, Wallace KA, Verhoeven AD, Wright LS, Capowski EE, Pinilla I, Martin JM, Tian S, Stewart R, Pattnaik B, Thomson JA, Gamm DM. Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells. 2011;29:1206–1218. doi: 10.1002/stem.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JS, Shearer RL, Capowski EE, Wright LS, Wallace KA, McMillan EL, Zhang SC, Gamm DM. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2009;106:16698–16703. doi: 10.1073/pnas.0905245106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, Saito K, Yonemura S, Eiraku M, Sasai Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 2012;10:771–785. doi: 10.1016/j.stem.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Oliver G, Gruss P. Current views on eye development. Trends Neurosci. 1997;20:415–421. doi: 10.1016/s0166-2236(97)01082-5. [DOI] [PubMed] [Google Scholar]
- Osakada F, Ikeda H, Mandai M, Wataya T, Watanabe K, Yoshimura N, Akaike A, Sasai Y, Takahashi M. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol. 2008;26:215–224. doi: 10.1038/nbt1384. [DOI] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Phillips MJ, Perez ET, Martin JM, Reshel ST, Wallace KA, Capowski EE, Singh R, Wright LS, Clark EM, Barney PM, Stewart R, Dickerson SJ, Miller MJ, Percin EF, Thomson JA, Gamm DM. Modeling human retinal development with patient-specific induced pluripotent stem cells reveals multiple roles for visual system homeobox 2. Stem Cells. 2014;32:1480–1492. doi: 10.1002/stem.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ, Wallace KA, Dickerson SJ, Miller MJ, Verhoeven AD, Martin JM, Wright LS, Shen W, Capowski EE, Percin EF, Perez ET, Zhong X, Canto-Soler MV, Gamm DM. Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Invest Ophthalmol Vis Sci. 2012;53:2007–2019. doi: 10.1167/iovs.11-9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichman S, Terray A, Slembrouck A, Nanteau C, Orieux G, Habeler W, Nandrot EF, Sahel JA, Monville C, Goureau O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc Natl Acad Sci U S A. 2014;111:8518–8523. doi: 10.1073/pnas.1324212111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, Chen CM, Young TL, Fisher DE, Cepko CL. Transdifferentiation of the retina into pigmented cells in ocular retardation mice defines a new function of the homeodomain gene Chx10. Development. 2004;131:5139–5152. doi: 10.1242/dev.01300. [DOI] [PubMed] [Google Scholar]
- Rowland TJ, Blaschke AJ, Buchholz DE, Hikita ST, Johnson LV, Clegg DO. Differentiation of human pluripotent stem cells to retinal pigmented epithelium in defined conditions using purified extracellular matrix proteins. J Tissue Eng Regen Med. 2013;7:642–653. doi: 10.1002/term.1458. [DOI] [PubMed] [Google Scholar]
- Shi M, Kumar SR, Motajo O, Kretschmer F, Mu X, Badea TC. Genetic interactions between Brn3 transcription factors in retinal ganglion cell type specification. PLoS One. 2013;8:e76347. doi: 10.1371/journal.pone.0076347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara S, Yasumoto K, Amae S, Udono T, Watanabe K, Saito H, Takeda K. Regulation of pigment cell-specific gene expression by MITF. Pigment Cell Res. 2000;13(Suppl 8):98–102. doi: 10.1034/j.1600-0749.13.s8.18.x. [DOI] [PubMed] [Google Scholar]
- Singh R, Phillips MJ, Kuai D, Meyer J, Martin JM, Smith MA, Perez ET, Shen W, Wallace KA, Capowski EE, Wright LS, Gamm DM. Functional analysis of serially expanded human iPS cell-derived RPE cultures. Invest Ophthalmol Vis Sci. 2013a;54:6767–6778. doi: 10.1167/iovs.13-11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Shen W, Kuai D, Martin JM, Guo X, Smith MA, Perez ET, Phillips MJ, Simonett JM, Wallace KA, Verhoeven AD, Capowski EE, Zhang X, Yin Y, Halbach PJ, Fishman GA, Wright LS, Pattnaik BR, Gamm DM. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet. 2013b;22:593–607. doi: 10.1093/hmg/dds469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar A, Steward MM, Meyer JS. Nonxenogeneic growth and retinal differentiation of human induced pluripotent stem cells. Stem Cells Transl Med. 2013;2:255–264. doi: 10.5966/sctm.2012-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern J, Temple S. Stem cells for retinal repair. Dev Ophthalmol. 2014;53:70–80. doi: 10.1159/000357328. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Tucker BA, Anfinson KR, Mullins RF, Stone EM, Young MJ. Use of a synthetic xeno-free culture substrate for induced pluripotent stem cell induction and retinal differentiation. Stem Cells Transl Med. 2013a;2:16–24. doi: 10.5966/sctm.2012-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker BA, Mullins RF, Streb LM, Anfinson K, Eyestone ME, Kaalberg E, Riker MJ, Drack AV, Braun TA, Stone EM. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. Elife (Cambridge) 2013b;2:e00824. doi: 10.7554/eLife.00824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vugler A, Carr AJ, Lawrence J, Chen LL, Burrell K, Wright A, Lundh P, Semo M, Ahmado A, Gias C, da Cruz L, Moore H, Andrews P, Walsh J, Coffey P. Elucidating the phenomenon of HESC-derived RPE: anatomy of cell genesis, expansion and retinal transplantation. Exp Neurol. 2008;214:347–361. doi: 10.1016/j.expneurol.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Wahlin KJ, Maruotti J, Zack DJ. Modeling retinal dystrophies using patient-derived induced pluripotent stem cells. Adv Exp Med Biol. 2014;801:157–164. doi: 10.1007/978-1-4614-3209-8_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir J, Rivolta M, Holley M. Identification of differentiating cochlear hair cells in vitro. Am J Otol. 2000;21:130–134. doi: 10.1016/s0196-0709(00)80087-3. [DOI] [PubMed] [Google Scholar]
- Wright LS, Phillips MJ, Pinilla I, Hei D, Gamm DM. Induced pluripotent stem cells as custom therapeutics for retinal repair: progress and rationale. Exp Eye Res. 2014;123:161–172. doi: 10.1016/j.exer.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhang SS, Fu XY, Barnstable CJ. Molecular aspects of vertebrate retinal development. Mol Neurobiol. 2002;26:137–152. doi: 10.1385/MN:26:2-3:137. [DOI] [PubMed] [Google Scholar]
- Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao LH, Peters A, Park TS, Zambidis ET, Meyer JS, Gamm DM, Yau KW, Canto-Soler MV. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun. 2014;5:4047. doi: 10.1038/ncomms5047. [DOI] [PMC free article] [PubMed] [Google Scholar]