

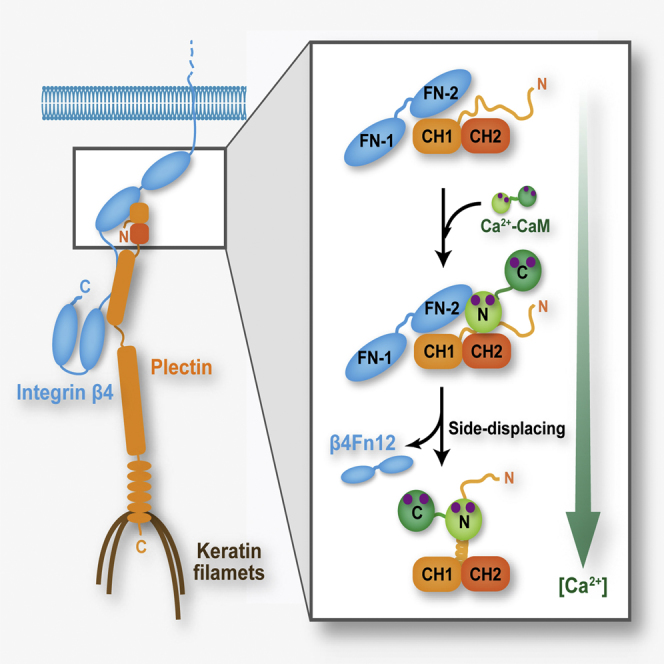
Summary
The mechanical stability of epithelial cells, which protect organisms from harmful external factors, is maintained by hemidesmosomes via the interaction between plectin 1a (P1a) and integrin α6β4. Binding of calcium-calmodulin (Ca2+-CaM) to P1a together with phosphorylation of integrin β4 disrupts this complex, resulting in disassembly of hemidesmosomes. We present structures of the P1a actin binding domain either in complex with the N-ter lobe of Ca2+-CaM or with the first pair of integrin β4 fibronectin domains. Ca2+-CaM binds to the N-ter isoform-specific tail of P1a in a unique manner, via its N-ter lobe in an extended conformation. Structural, cell biology, and biochemical studies suggest the following model: binding of Ca2+-CaM to an intrinsically disordered N-ter segment of plectin converts it to an α helix, which repositions calmodulin to displace integrin β4 by steric repulsion. This model could serve as a blueprint for studies aimed at understanding how Ca2+-CaM or EF-hand motifs regulate F-actin-based cytoskeleton.
Graphical Abstract
Highlights
-
•
Calmodulin binds to plectin 1a via its N-terminal lobe in an extended conformation
-
•
The disordered N-ter tail of plectin 1a folds in an α helix upon calmodulin binding
-
•
Suitably positioned calmodulin displaces integrin β4 from complex with plectin 1a
Hemidesmosomes attach the top level of skin to plasma membrane via the assembly between integrin α6β4, plectin, and intermediate filaments. Hemidesmosome disassembly and remodeling are regulated by phosphorylation and Ca2+. We provide a structural mechanism of how Ca2+/CaM shunts integrin α6β4 and F-actin from the assembly with plectin.
Introduction
The skin forms a barrier against the environment and protects us from mechanical trauma, pathogens, radiation, dehydration, and perilous temperature fluctuations. It is composed of an epidermal and a dermal layer, which are separated by a basement membrane. The epithelium of the skin, the epidermis, is made primarily of keratinocytes, while the dermis contains different cell types, including fibroblasts, endothelial cells, and macrophages, as well as the extracellular matrix. Integrity of epithelial cells is maintained by multiprotein complexes termed hemidesmosomes (HDs), which attach the top level of the skin to plasma membrane (Green and Jones, 1996). HDs are dynamic structures, which can be quickly disassembled if required, for example, during wound healing, differentiation, or carcinoma invasion (Litjens et al., 2006). Integrin α6β4 is the principal player in the interaction between the intermediate filament (IF) cytoskeleton and extracellular matrix at the site of HDs, which traverses the plasma membrane (Borradori and Sonnenberg, 1999; Green and Jones, 1996). The cytoplasmic part of the β4 subunit is unusually long and consists of two pairs of fibronectin type III (FnIII) domains separated by the connecting segment (Tamura et al., 1990). On the cytoplasmic side, integrin α6β4 interacts with plectin, which mediates association between the HDs and the keratin cytoskeleton. Binding of plectin to the β4 subunit of integrin α6β4 is the critical step in the formation of HDs (Litjens et al., 2006). The importance of integrin α6β4 and plectin for stability of HDs is substantiated by in vivo studies in mice, which display reduced levels or complete absence of HDs when deficient in either plectin or integrin subunits α6 and β4 (Andra et al., 1997; Georges-Labouesse et al., 1996; van der Neut et al., 1996). In fact, inherited or acquired diseases in which integrin α6β4 or plectin are missing or are structurally perturbed result in tissue fragility and blistering (Walko et al., 2014).
Plectin, a member of the plakin family, connects different elements of the cytoskeleton and is expressed in a wide variety of mammalian cells (Castanon et al., 2013). At its N terminus there is an actin binding domain composed of two calponin homology (CH) domains, which is followed by a plakin domain, the coiled-coil rod of over 1,000 residues, six plectin repeat domains, and a short terminal tail. Due to alternative splicing, plectin is expressed as 11 isoforms with diverse N-ter sequences that dictate its differential subcellular targeting (Fuchs et al., 1999). Plectin 1a (P1a) and 1c (P1c) are major isoforms of plectin expressed in basal keratinocytes. However, P1a is HD specific, while P1c colocalizes with microtubules (Andra et al., 2003; Walko et al., 2011).
Most of the efforts to understand the regulation of HD disassembly have focused on the main organizer of the HD, the α6β4 integrin. Multiple binding sites mediate the interactions between plectin and integrin α6β4, most of which are regulated by several phosphorylation events at the interaction interfaces (Frijns et al., 2010, 2012; Rabinovitz et al., 2004; Wilhelmsen et al., 2007). One site that is not regulated by phosphorylation is between the ABD of plectin and the first pair of the FnIII domains and the connecting segments of integrin β4 (Frijns et al., 2010; Geerts et al., 1999; Niessen et al., 1997; Rabinovitz et al., 2004; Wilhelmsen et al., 2007). Besides phosphorylation, Ca2+ also serves as a modulator in keratinocyte proliferation and differentiation. In particular, integrin α6β4 is downregulated during Ca2+-induced differentiation of cultured keratinocytes (Kostan et al., 2009; Tennenbaum et al., 1996). Interestingly, Ca2+/calmodulin (Ca2+-CaM) was shown to reduce the interaction between integrin α6β4 and HD-specific P1a and to inhibit the interaction of P1a with F-actin in a Ca2+-dependent manner (Kostan et al., 2009), thus contributing to the regulation of HD disassembly.
The aim of this study is to address the question of how Ca2+-CaM regulates the P1a interaction with integrin β4 and F-actin, and whether the interaction mode with integrin β4 is isoform specific. Our results revealed the molecular mechanism underlying the Ca2+-CaM regulation of the P1a/integrin β4 and P1a/F-actin interaction via shunting the integrin β4 and F-actin from the complex with P1a. Comparison with the structure of the complex between integrin β4 and the P1c isoform (de Pereda et al., 2009) shows that the plectin-integrin β4 interaction mode is not isoform specific, while Ca2+-CaM binds to plectin in an isoform-specific manner. Based on these results, we suggested the model for Ca2+-CaM-induced disruption of the P1aABD-integrin β4 complex. Finally, the related proteins α/β-spectrin, dystrophin, and utrophin (members of the spectrin superfamily), as well as F-actin binding protein filamin A, may be regulated in related ways by Ca2+-CaM or by their own EF hands motifs.
Results
Interaction of P1a and CaM in Epithelial Cells
Previous work has shown that Ca2+-CaM interacts specifically with the ABD of isoform P1a (P1aABD) (Kostan et al., 2009). To further characterize the role of the N-ter isoform-specific sequence of P1a in binding to Ca2+-CaM, we prepared several N-terminally truncated mutants of P1aABD. Pull-down assays were used in the presence of either Ca2+ or EDTA. While full-length P1aABD and mutant versions lacking the first 11 or 22 N-ter residues (P1aABDΔ11 and P1aABDΔ22) bound to CaM in a calcium-dependent manner, P1aABDΔ32 lacking 32 N-ter residues, and P1aABDΔ37 lacking the entire isoform-specific sequence failed to bind to Ca2+-CaM (Figure S1). This suggested that interaction of Ca2+-CaM with P1a is restricted to the N-ter region spanning residues 23–32.
To analyze CaM association with P1a in vivo, we performed Förster resonance energy transfer (FRET) analysis of the P1a-CaM interaction in 804G epithelial cells. The cells were cotransfected with cDNAs encoding C-terminally CFP-tagged P1aABD (P1aABD-CFP) or its mutant P1aABDmut (P1aABDmut-CFP), and with C-terminally yellow fluorescence protein (YFP)-tagged CaM (CaM-YFP) (Figure 1A). The mutant P1aABDmut construct encoded two point mutations in the N-ter segment of P1a (Leu25Asp and Val29Asp) and was designed to be deficient in the interaction (see the section on “Crystal Structure of the P1a ABD in Complex with CaM N-ter Lobe”). The acceptor photobleaching method (Novotny et al., 2011) gave a positive FRET signal for the P1aABD-CFP/CaM-YFP pair (19% ± 0.9%). The P1aABDmut-CFP fusion protein, however, showed a significantly lower FRET signal (6.2% ± 0.5%) despite a similar expression level, reaching the mean FRET efficiency of the P1aABD-CFP/empty YFP plasmid (8.0% ± 0.4%) used as a negative control (Figures 1A and 1B). These results confirmed that P1aABD interacts with CaM in vivo and that CaM binds to the isoform-specific N-ter segment of P1a.
Figure 1.

The P1aABD and CaM Interact In Vivo
(A) 804G cells ectopically expressing combinations of P1aABD-CFP and P1aABDmut-CFP with CaM-YFP or YFP alone. FRET was measured by acceptor photobleaching. Pseudocolor pre- and postbleach images of acceptor (CaM-YFP or YFP alone) and FRET images (FRETeff) are shown. FRET efficiency was determined as a relative increase of donor fluorescence in the cytoplasmic region of interest shown in boxed area, and enlarged in insets (bottom right corners). Bar, 10 μm.
(B) Box and whisker plots indicate the median FRET (middle line in the box), 25th percentile (bottom line of the box), 75th percentile (top line of the box), and minimum and maximum values (whiskers). A CFP-YFP fusion was used as a positive control for FRET (first column); cotransfection of P1aABD-CFP and empty YFP vector was used as a negative control (last column). The number of assessed cells/individual bleach events (n) obtained from three or more independent experiments is indicated.
P1a Interacts with the N-ter Lobe of CaM
In order to identify proximity residue pairs in the P1aABD/CaM complex, we applied chemical crosslinking combined with mass spectrometry (XL-MS) and bioinformatics analysis. We used the zero-length crosslinker 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) in the presence of N-hydroxysulfosuccinimide (sulfo-NHS). A band corresponding to ∼50 kDa that appeared on SDS-PAGE upon one- or two-step crosslinking (Figure 2A) was excised, trypsinized, and analyzed by high-resolution liquid chromatography (LC)-MS/MS. In total, 12 crosslinks were identified for the P1aABD/CaM complex (Table S1). The major crosslinking products involve the adjacent K residues 36 and 37 at the border between the sequence-specific N-ter segment of P1aABD, and E14 residing in the A helix of the first CaM EF-hand. A representative MS/MS spectrum of a specific crosslinking product identifying a linkage between E14 of CaM and K37 of P1aABD is shown in Figure 2B. Other less prominent crosslinking products involve several sites of the C-lobe of CaM (CaMCL). These data showed that both lobes of CaM have the capacity to interact with P1aABD. However, crosslinked peptides connecting the N-lobe of CaM (CaMNL) to the N-ter tail of P1aABD were more prominent and more frequently detected (Table S1), suggesting a higher binding affinity of CaMNL.
Figure 2.

P1aABD Associates Preferentially with the N-ter Lobe of CaM
(A) SDS-PAGE analysis of the P1aABD/CaM complex crosslinked by EDC and sulfo-NHS. Proteins were incubated in the binding buffer either in the absence of crosslinker (-), or subjected to EDC and sulfo-NHS in a one-step (+/1) or two-step (+/2) reaction (for details see Supplemental Materials and Methods). Complexes of crosslinked P1aABD and CaM are readily visible as a higher-molecular weight band (arrow).
(B) MS/MS spectrum identifying a pair of crosslinked peptides after in-gel proteolysis of the complex band obtained by two-step crosslinking of CaM and P1aABD. Fragment ions are annotated for the α-peptide (red) and the β-peptide (green). Insets show mapping of the fragment ions onto the crosslinked peptide sequences (left) and the corresponding high-resolution MS spectrum displaying the isotopic distribution of the crosslinking product (right).
see also Table S1.
To quantify the preferential binding of CaMNL to P1a, we determined and compared the binding affinities of CaM, CaMNL, and CaMCL with P1aABD by isothermal titration calorimetry (ITC). Schematic illustrations of plectin and CaM depicting the boundaries of the protein fragments used in this study are shown in Figure 3A. CaM binds to P1aABD more strongly (Kd 4.2 ± 0.4 μM) than CaMNL alone (Kd 10.5 ± 1.1 μM), while CaMCL displays even weaker affinity (Kd 27.7 ± 3.9 μM) (Figures 3B and 3C; Figure 7C), corroborating the XL-MS results. In a competitive experiment, CaMCL was displaced when the P1aABD/CaMCL complex was titrated with CaMNL (Figure 3D). As expected, CaMCL could not displace CaMNL from the P1aABD/CaMNL complex (Figure 3E). These results indicate that CaMNL and CaMCL bind to the same site on P1aABD, to which CaMNL binds with higher affinity. Furthermore, full-length CaM has a higher affinity for P1aABD than CaMNL alone, suggesting a possible auxiliary role of CaMCL in the binding event. In summary, XL-MS data combined with ITC conclusively showed that CaM binds to P1a preferentially via its N-ter lobe.
Figure 3.

N-ter Lobe of CaM Binds to P1aABD with Higher Affinity Than the C-ter Lobe
(A) Schematic illustrations of plectin and CaM. CH, calponin homology domain; PD, plakin domain; ROD, coiled-coil rod domain; CTD, C-terminal domain. Arrows indicate positions of the first N-ter residue of P1aABD and P1aABDΔ22 constructs, respectively. Schematic illustration of the P1a fragment used in the assays is shown in boxed area.
(B–E) ITC assays showing preferential binding of CaMNL to P1aABD. (B) P1aABD (40 μM) was titrated with CaMNL (400 μM). (C) P1aABD (40 μM) was titrated with CaMCL (400 μM). (D) P1aABD/CaMCL complex (40/60 μM) was titrated with CaMNL (400 μM). (E) P1aABD/CaMNL complex (40/60 μM) was titrated with CaMCL (400 μM). Data are expressed as mean values ± SD. ND, not determined.
Figure 7.

Displacement of β4Fn12 from the β4Fn12/P1aABD Complex by Ca2+-CaM
(A) The SAXS-derived tentative model of the CaM/P1aABD complex (for detail see Figure 4E), shown as surface, was superimposed on the ABD of the plectin of the P1aABDΔ22/β4Fn12 complex, shown as ribbon. Steric clashes of Fn2 domain with CaM/P1aABD are indicated with a purple dashed line.
(B–D) Displacement of β4Fn12 by Ca2+-CaM as analyzed by ITC. (B) P1aABD (0.08 mM) was titrated with β4Fn12 (0.8 mM) and exhibited entropy-driven binding. (C) P1aABD (0.1 mM) was titrated with CaM (1 mM). CaM bound to P1aABD with a higher affinity than β4Fn12. (D) For the displacement assay, 1 mM CaM was injected into the sample cell containing 0.1 mM P1aABD and 0.1 mM β4Fn12. Data are expressed as mean values ± SD.
see also Figure S6.
Crystal Structure of the P1a ABD in Complex with CaM N-ter Lobe
To understand the structural basis of the Ca2+-CaM–P1a interaction, we set out to determine the crystal structure. Prediction of disordered protein regions (Ishida and Kinoshita, 2007) suggested that the N-ter extension of P1a is intrinsically disordered (Figure S2) and hence its presence is unfavorable for crystallization studies. As we found the minimal region of P1a sufficient for the interaction with CaM to span N-ter residues 22–32, we first focused on crystallization of the P1aABDΔ22/CaM complex, which was unsuccessful. We therefore turned our attention to the P1aABDΔ22/CaMNL complex, since we showed that CaMNL binds to P1a with higher affinity than CaMCL (Figures 3B–3E). The structure of the P1aABDΔ22/CaMNL complex was determined to 1.8 Å resolution, with final R and Rfree factors of 0.151 and 0.187, respectively. Data collection and refinement statistics are summarized in Table 1.
Table 1.
Data Collection and Refinement Statistics
| P1aABDΔ22/CaMNL | P1aABDΔ22/β4Fn12 | P1aABDΔ22 | |
|---|---|---|---|
|
Data Collection | |||
| Source | ID14-1 (ESRF) | ID23-2 (ESRF) | ID14-4 (ESRF) |
| Wavelength (Å) | 0.933 | 0.873 | 0.939 |
| Resolution (Å) | 48.93–1.8 (1.9–1.8)a | 48.16–4.0 (4.47–4.0) | 60.37–2.30 (2.38–2.30) |
| Space group | P212121 | P65 | C2221 |
| Unit cell (Å, °) | a = 59.08, b = 65.38, c = 87.3 α = β = γ = 90 |
a = 96.32, b = 96.32, c = 207.8 α = 90, β = 90, γ = 120 |
a = 41.60, b = 159.39, c = 183.83 α = 90, β = 90, γ = 90 |
| Molecules/a.u. | 2 | 4 | 2 |
| Unique reflections | 31805 (4301) | 9188 (2594) | 28020 (2698) |
| Completeness (%) | 99.2 (94.7) | 99.1 (99.5) | 99.7 (99.8) |
| Rmergeb | 0.095 (0.488) | 0.272 (0.793) | 0.125 (0.424) |
| Rmeasc | 0.101 (0.527) | 0.300 (0.879) | 0.143 (0.483) |
| Rpimd | 0.033 (0.193) | 0.123 (0.364) | 0.067 (0.224) |
| Multiplicity | 9.4 (7.2) | 5.7 (5.6) | 4.4 (4.2) |
| Mean I/sig(I) | 17.5 (3.7) | 7.4 (3.9) | 6.9 (2.2) |
| CC (1/2)e | 0.998 (0.877) | 0.977(0.751) | 0.945 (0.856) |
| Refinement | |||
| Rworkf/Rfreeg | 0.151/0.187 | 0.224/0.287 | 0.183/0.245 |
| Rmsd bonds (Å) | 0.007 | 0.004 | 0.009 |
| Rmsd angles (°) | 0.963 | 0.892 | 1.246 |
| Ramachandran outliers | 0 | 0 | 0 |
see also Figure S5.
Values in parentheses are for the highest-resolution shell.
, where Ii is the intensity of the ith observation, and is the mean intensity of the reflection.
.
, where Ii(hkl) is the observed intensity, and is the average intensity of multiple observations of symmetry-related reflections.
CC(1/2) = the Pearson correlation coefficient of random half data sets.
, calculated from working data set.
Rfree is calculated from 5% of data randomly chosen and not included in refinement.
In the complex, CaMNL binds to the N-ter tail of P1a, which forms an α helix. This α helix extends the A helix of the first CH domain and protrudes away from the body of the ABD (Figure 4A). Each EF-hand of CaMNL coordinates one calcium ion (Figure 4A). Comparison of CaMNL with the crystal structure of unbound Ca2+-CaM (Protein Data Bank [PDB], 3CLN) (Babu et al., 1988) (root-mean-square deviation (rmsd), 0.40 Å over 62 equivalent Cα atoms in N-lobe) showed that CaMNL did not change the conformation upon binding to plectin.
Figure 4.

Structure of the P1aABD/CaM Complex
(A) Crystal structure of the P1aABDΔ22/CaMNL complex. The complex is displayed in two orientations (90° rotated along the y axis). CH1 and CH2 in the ABD are colored respectively in bronze and orange, the N-ter tail of P1a in yellow and CaMNL in green. Ca2+ is depicted as a violet sphere.
(B) The binding interface of the P1aABDΔ22/CaMNL complex. CaMNL is shown according to electrostatic surface potential, with blue and red depicting positive and negative electrostatic potentials, respectively. Two residues of P1a (L25 and V29) important for interaction with CaM are buried in the hydrophobic cleft of CaMNL.
(C) A salt bridge between E14 of CaM and R40 of plectin (2.7 Å apart). XL-MS analysis shows that CaM E14 is crosslinked with K36 (5.7 Å apart) and K37 of plectin (7.0 Å).
(D) Experimental SAXS data of the P1aABD/CaM complex is shown in red; calculated scattering curves from SAXS models are individually presented in green (rigid-body modeling by CORAL) and blue (ab initio modeling by DAMMIF) lines.
(E) The ab initio molecular shape of the P1aABD/CaM complex (shown in transparent beads) was superimposed over the SAXS-derived rigid-body model of the complex. Flexible residues (1–21 residues of P1aABD and 74–82 residues of CaM) were modeled as dummy residues (colored spheres).
CaMNL binds to the N-ter extension of P1a mainly via hydrophobic interactions. Three hydrophobic residues of P1aABDΔ22 (L25, V29, and A32) are buried in the hydrophobic cleft of CaMNL (Figure 4B), consistent with the known binding motif of Ca2+-CaM, termed 1-5-8, which bears hydrophobic residues at these positions (Rhoads and Friedberg, 1997). To assess the role of these residues in binding to Ca2+-CaM, we mutated amino acids at motif positions 1 and 5 to negatively charged residues (L25D and V29D; P1aABDmut). We showed by ITC, size exclusion chromatography (SEC), and FRET analysis that the P1aABDmut does not interact with Ca2+-CaM, confirming that the hydrophobic positions 25 and 29 are essential for binding (Figure 1; Figure S3).
Two polar contacts further stabilize the complex: an interaction between Q41 (in the loop between helices B and C of CaMNL) and R31 of P1a (in the N-ter tail), and a salt bridge between E14 (in the A helix of EF-hand 1) and R40 in P1a (in the A helix of the CH1 domain). The latter seems less important for CaM binding, as other plectin isoforms that do not bind to Ca2+-CaM also host an R at this position (Figure 4C). Finally, mapping the crosslinked residues on the three-dimensional structure of the P1aABDΔ22/CaMNL complex showed that the carboxylate group of E14 in CaMNL is 5.7 Å and 7.0 Å away from the amino groups of plectin residues K36 and K37 (Figure 4C), respectively, which is in good agreement with the XL-MS results (Figure 2B; Table S1).
CaM Binds to P1a in an Extended Conformation
To determine the low-resolution shape of the nontruncated P1aABD in complex with full-length Ca2+-CaM, small-angle X-ray scattering (SAXS) experiments were conducted (Table S2). The Rg of Ca2+-CaM in the complex with P1aABD (20.9 Å; CRYSOL [Svergun et al., 1995]) was found to be similar to the Rg of extended Ca2+-CaM in solution (21.3 ± 0.2 Å) (Heidorn and Trewhella, 1988). This indicated that the Ca2+-CaM in complex with P1aABD exhibits an extended conformation in which the two lobes are connected with an interlobe linker, modeled here with dummy residues (Figures 4D and 4E). Furthermore, in the P1aABD/CaM complex, neither the CaMCL nor the first 22 residues of the N-ter tail of P1a participate in the interaction.
The CaM binding motif of P1aABD (L25-V29-A32) represents a subgroup of 1-5-8-14 motifs in which hydrophobic residues at position 1 and 14 are the primary requirements for Ca2+-CaM binding and anchoring the interaction partner to the two lobes of CaM (Figure S2) (Rhoads and Friedberg, 1997). P1a contains D38 at position 14, suggesting that the C-terminal lobe of CaM cannot bind due to the absence of a hydrophobic residue at this site. In addition, D38 is the first residue of helix A of the CH1 domain, and binding of the C-terminal lobe to this site would lead to a steric clash of helix C of CaM with the ABD, as shown by superposition of CaMNL in complex with P1aABDΔ22 on Ca2+-CaM in complex with skeletal muscle light chain kinase (PDB, 1CDL) (Chattopadhyaya et al., 1992) (rmsd, 0.684 Å over 57 Cα) (Figure S4). In summary, our results showed that Ca2+-CaM binds with CaMNL to a 1-5-8-type recognition site on the P1aABD N-ter tail in an extended conformation. This is to our best knowledge the first example of a complex in which Ca2+-CaM binds to the interaction partner via its N-ter lobe in an extended conformation. Namely, a search of PDB for complexes in which CaM binds to the interaction partner in extended conformation gave seven hits, but in all of them, only the C-ter lobe or both lobes are involved in binding (Table S3).
N-ter Tail of P1a Folds upon Binding to CaM
Although the N-ter segment of P1aABD is predicted to be intrinsically disordered (Figure S2B), the amino acid residues 23–37 form an α helix in the P1aABDΔ22/CaMNL complex (Figures 4A and 4B), a feature often observed by intrinsically disordered proteins (Dyson and Wright, 2005). To validate the bioinformatics prediction that the entire N-ter tail is structurally disordered in the absence of Ca2+-CaM, we solved the crystal structure of P1aABDΔ22 and performed SAXS analysis of P1ABD.
The electron density of the first 15 residues (aa 23–37) preceding the CH1 domain was missing in the crystal structure of P1aABDΔ22 (Figure 5A; Table 1), suggesting that this part of the protein is structurally disordered. SDS-PAGE analysis of the crystals confirmed that the N-ter tail was intact and was not proteolytically removed in the process of crystallization. In addition, no significant conformational changes took place in the ABD upon Ca2+-CaM binding, as shown by comparison of P1aABDΔ22 in complex with CaMNL with the structure of P1aABDΔ22 alone (Figure S5) (rmsd, 0.69 Å over 209 equivalent Cα atoms).
Figure 5.

The N-ter Tail of P1a Is Intrinsically Disordered
(A) The crystal structure of P1aABDΔ22 is displayed in ribbon; CH domains 1 and 2 (CH1 and CH2) are shown in bronze and orange, respectively. The electron density corresponding to 15 residues of N-ter tail (amino acid residues 23–37) is absent in the structure. The electron density map (2Fo-Fc) is contoured at 1.5 σ.
(B) SAXS analysis of the P1aABD. The frequency distributions of Rg generated from EOM compared with the pool (red curve) and the selected ensemble (black curve) are shown. The inset shows the experimental scattering curve (red) and the simulated scattering curve of the selected ensemble by EOM (black).
(C) Eight superimposed models from the selected ensemble, shown in two different orientations (rotated 90° along the x axis). The N-ter tail adopts random conformations.
In the SAXS analysis of P1ABD, the N-ter extension was modeled as an ensemble of structurally variable moieties using the Ensemble optimization method (EOM) (Bernado et al., 2007), while the crystal structure of P1aABDΔ22 was used as a rigid-body constraint. This yielded a fit to the experimental data with χ = 1.09 (Figure 5B, inset). Data collection and structural parameters derived from SAXS analysis on P1aABD are reported in Table S2. EOM analysis showed a broad Rg distribution, typical of extended and flexible structures (Figure 5B). Eight models from the selected ensemble (50 models) superimposed on ABD show that the N-ter tails of plectin isoform 1a adopt extended and variable conformations (Figure 5C). These data show that the N-ter segment of P1a is structurally disordered in the unbound state; however, it folds into an α helix upon binding to Ca2+-CaM, undergoing a coupled folding and binding process (Dyson and Wright, 2005).
N-ter Tail of P1a Is Not Involved in Interaction with Integrin β4
Our structural and biochemical data showed that Ca2+-CaM interacts with the N-ter isoform-specific sequence of P1a via its N-ter lobe. In vivo, this interaction contributes to the disassembly of the P1a-integrin β4 complex. In the published structure of the P1cABD/integrin β4 complex, the N-ter tail of P1c binds to integrin β4 mainly through the polypeptide backbone (de Pereda et al., 2009). In order to address the question whether this interaction mode is isoform specific and whether the coupled folding and binding mechanism observed for the interaction with Ca2+-CaM applies also for the interaction of P1a and integrin β4, we determined the crystal structure of P1aABD in complex with the first pair of the FnIII domains of integrin β4. A construct encompassing the first two FnIII domains of integrin β4 (β4Fn12; 1126–1355) was used for crystallization with P1aABDΔ22 (Figure 6A). The asymmetric unit contained one P1aABDΔ22/β4Fn12 complex and one free copy of ABD and β4Fn12 each. The structure was determined to 4.0 Å resolution and refined to the final Rwork and Rfree factors of 0.224 and 0.287, respectively (Figure 6B; Table 1).
Figure 6.

Structure of P1aABD/β4Fn12 Complex
(A) Schematic illustrations of integrin β4 and P1a fragments used in the experiments (boxed areas).
(B) The crystal structure of the P1aABDΔ22/β4Fn12 complex is shown in two orientations. β4Fn12 is displayed in cyan, and the CH1 and CH2 of the P1aABDΔ22 are displayed in bronze and orange, respectively. The binding interface comprises three salt bridges between P1a and integrin β4; D123/R1225 (2.60 Å), R71/E1242 (3.04 Å), and E68/R1281 (2.57 Å).
see also Figures S7 and S8; Table S2.
Structural comparison of our P1aABDΔ22/β4Fn12 complex with the complex of P1cABD/β4Fn12 (PDB, 3F7P; de Pereda et al., 2009) showed that no substantial conformational differences occurred and that the overall architecture was maintained in both complexes (rmsd, 0.87 Å over 367 equivalent Cα atoms). The first 15 residues of the P1aABDΔ22 construct were not visible in the electron density of the P1aABDΔ22/β4Fn12 crystal structure, suggesting that it is structurally disordered in this complex and most likely not involved in the interaction with integrin β4. This was further confirmed by data showing that the P1a N-ter segment encompassing residues (1–60 aa) does not bind to β4Fn12 in ITC assays (Figure S6A).
To further characterize and validate the architecture of the P1aABD/β4Fn12 complex in solution, we performed SAXS analysis. Data collection and structural parameters are summarized in Table S2. The ab initio molecular envelope yielded a fit of χ2 = 1.01 (Figure S7A) and agreed well with the crystal structure (Figure S7B). Since the concentration of the sample used (41.3 μM) was similar to the Kd (41.7 μM) of the complex determined by ITC (Figure 7B), a polydisperse solution (calculated 38.0% of complex) was expected. To account for this, we used the program OLIGOMER (Konarev et al., 2003) to determine the volume fractions of the complex and its subunits in solution. The best fit to the experimental data (χ2 = 0.90) corresponded to a mixture containing 54% ± 3% of the complex, 25% ± 3% of P1aABD, and 21% ± 3% of β4Fn12, in line with the Kd-based estimation. To further validate the structures of the P1aABD/β4Fn12 complex, we used XL-MS. Mapping the crosslinked residues on the three-dimensional structure of the complex further corroborated the structural results (Figures S8A and S8B): four crosslinked peptides cluster to one site in the P1aABD/β4Fn12 complex (Table S1). Both plectin K36 and K37, located on the border between helix A of the CH1 domain and the N-terminal segment, were crosslinked to integrin β4 residue E1286 residing on the loop between two β strands in the FnIII-2 domain. These lysine residues of plectin are in contact with CaM in the P1aABDΔ22/CaMNL complex (Figure 2B; Figure 4C), indicating that interaction with CaM should exclude that with integrin β4. In the crystal structure, the first residue of the CH1 domain visible in the electron density is D38, located 12.8 Å from integrin β4 Glu1286 (Cα–Cα distance, Figure S8C), showing that the two adjacent lysine residues (K36 and K37) are close to E1286, as also shown by XL-MS data.
Molecular Determinants of Integrin β4 Displacement from the Complex with P1a by CaM
In order to enlighten the regulation mechanism of the integrin β4-P1a complex by Ca2+-CaM at the molecular level, we mimicked binding of Ca2+-CaM to the P1aABDΔ22/β4Fn12 complex by superimposing the SAXS-derived tentative model of P1aABD/CaM on the crystal structure of the P1aABDΔ22/β4Fn12 complex (rmsd, 0.77 Å over 206 Cα equivalent atoms). As shown in Figure 7A, binding of Ca2+-CaM to the N-ter extension of P1a results in steric clashes with the second FnIII domain of the integrin β4, implying a disruption of the P1aABDΔ22/β4Fn12 interaction.
To obtain quantitative understanding of interactions and dissociations of CaM, β4Fn12, and P1aABD, we carried out ITC assays. The binding of β4Fn12 to P1aABD is an entropy-driven reaction with weak affinity (Kd, 41.7 ± 4.2 μM, Figure 7B). Conversely, the binding of Ca2+-CaM to P1aABD is an enthalpy-favored process and more than one order of magnitude stronger (Kd, 4.2 ± 0.4 μM, Figure 7C). No interaction was observed between Ca2+-CaM and β4Fn12 (Figure S6B). In the displacement experiment, in which the P1aABD/β4Fn12 complex was titrated with Ca2+-CaM, the apparent binding affinity of Ca2+-CaM was reduced due to the competitive binding of β4Fn12 to P1aABD (Kd, 7.7 ± 0.7 μM) (Figure 7D), while the enthalpy and entropy changes (ΔH and ΔS) increase as a result of the replacement of an entropic binding with an enthalpic one. The measured binding affinity of Ca2+-CaM P1aABD (Kd, 4.2 ± 0.4 μM) is consistent with the calculated one using the competitive binding model (Kd, 2.3 μM) (Sigurskjold, 2000). Moreover, the binding affinity of Ca2+-CaM to P1aABDΔ22 (Kd, 2.6 ± 1.0 μM) is comparable with that of P1aABD (Figure S6C). Due to the higher binding affinity of P1aABD to CaMNL (Kd, 10.5 ± 1.1 μM), compared with its affinity to β4Fn12 (Kd, 41.7 μM ± 4.2), CaMNL alone expectedly displaced β4Fn12 from the P1aABD/β4Fn12 complex (Figure S6D).
In addition, we compared the thermodynamics of the β4Fn12 and Ca2+-CaM binding to P1aABD. The formation of the P1aABD/β4Fn12 complex is not enthalpy but entropy driven (ΔH, 3.69 kcal/mol, Figure 7B). This indicates that ordered water molecules solvating the binding partners are released upon complex formation (Dunitz, 1994). For the P1aABD/CaM interaction, we showed that the folding of the structurally disordered N-ter tail into an α helix is coupled with binding to Ca2+-CaM. This reaction is entropy unfavorable, as shown by ITC (Figure 7C). However, the enthalpy contribution compensates for the entropy cost to instigate the binding reaction (Figures 7C and 7D) (Dyson and Wright, 2005), which is inferred from the fact that the binding affinity of Ca2+-CaM to P1aABD (Kd, 4.2 μM) is lower than that reported for other binding partners (Kd, 10−7–10−11 M) (Crivici and Ikura, 1995).
To explore structural determinants of the preferential affinity of CaMNL for P1a compared with CaMCL, we generated a model of the P1aABDΔ22/CaMCL complex, by superimposing CaMCL (82–146 aa; PDB, 3CLN [Babu et al., 1988]) on the crystal structure of the P1aABDΔ22/CaMNL complex (rmsd, 0.50 Å over 53 equivalent Cα atoms out of 65). Amino acid residues involved in the interaction interfaces were analyzed by PISA (Krissinel and Henrick, 2007) (Figure S9A). Comparison of the interaction interfaces showed differences in three positions involved in critical stabilizing interactions, which are at the basis of higher affinity of the N-ter lobe: CaMNL Q41/CaMCL E114, CaMNL L18/CaMCL V91, and CaMNL V55/CaMCL A128 (Figure S9B). Although E114 in the C-lobe can potentially form a stronger interaction with the P1aABD R31 residue compared with the corresponding Q41 in the N-lobe, the two bulkier hydrophobic residues in the N-lobe (L18 and V55) contribute to the establishment of a larger hydrophobic interface with plectin than the equivalent C-lobe residues (V91 and A128) (Figure S9B). The total interface area of CaMNL (635 Å2, 14.9% of the solvent accessible surface area) is larger than that of CaMCL (618 Å2, 13.4% of the solvent accessible surface area) (Table S4).
We further compared the intermolecular interfaces of the P1aABDΔ22/CaMNL and P1aABD/β4Fn12 complexes. Probability measures PΔG,IF of specific interfaces were derived from the gain in solvation energy upon complex formation, with PΔG,IF > 0.5 pointing to hydrophilic/unspecific and PΔG,IF < 0.5 to hydrophobic/specific interfaces using PISA. In the case of P1aABDΔ22/CaMNL, the PΔG,IF values (<0.15) are in the range of probabilities derived from typical protein interfaces (0.1–0.4). In the case of P1aABD/β4Fn12, however, the PΔG,IF value close to 0.6 suggests a less specific interaction, with concomitantly smaller solvation energy gain (Table S4). These data collectively show that CaMNL is the high-affinity ligand of P1aABD. The finely tuned hydrophobic interactions of CaMNL with the N-ter tail of P1a lead to its capacity to disrupt the weaker and less specific interaction between P1a and integrin β4.
Discussion
Specific subcellular localizations have been identified for several plectin isoforms (Castanon et al., 2013). Nevertheless, few isoform-specific binding partners of these variants have been reported. Our results showed that Ca2+-CaM binds specifically to the P1a isoform, in particular to the ten residues of the isoform-specific N-ter tail, directly preceding the isoform-conserved ABD. The role of the 22 isoform-specific amino acid residues preceding the CaM binding site is still unknown. However, the specific localization of P1a to HDs suggests that this part of the isoform-specific sequence most likely harbors a signal for targeting of P1a into HDs. Plectin isoform diversity suggests that the function and nature of the plectin interactions with binding partners will vary, depending on both interaction partners and specific N-ter extensions.
Obviously, the strength of the interactions as measured in vitro may well vary from those in vivo, in which the interaction of P1a with integrin β4 is further stabilized by other interaction sites (Walko et al., 2014), contributing to localization of P1aABD in the vicinity of β4Fn2, thus increasing the probability of these domains to interact. In any case, the interaction of P1aABD with Ca2+-CaM displays affinity that is an order of magnitude higher than that with β4Fn12, and these differential affinities are crucial for the regulation of HD disassembly. Moreover, dimer and higher oligomeric forms of P1a contribute to the stability of HDs (Walko et al., 2011) and might have additional impact on HD disassembly regulated by Ca2+-CaM.
In addition to its role in regulating the interaction of P1a and integrin β4, Kostan et al showed that Ca2+-CaM also prevents plectin ABD from binding to F-actin (Kostan et al., 2009). To understand the molecular mechanism of this process, we superimposed the SAXS structure of the P1aABD/CaM complex on the homologous CH1 domain of α-actinin bound to F-actin (PDB, 3LUE) (Galkin et al., 2010) (rmsd, 0.54 Å over 98 equivalent Cα atoms). Superimposition of the P1aABD/CaM model onto the ABD of α-actinin bound to F-actin (Galkin et al., 2010) showed that CaM does not directly affect the actin binding sites of CH1 (Bresnick et al., 1990; Levine et al., 1990) but rather produces steric clashes between Ca2+-CaM and F-actin (Figure 8), which likely explains the inability of this complex to bind F-actin.
Figure 8.

Ca2+-CaM Regulates Binding of P1a to F-actin
The SAXS-derived tentative model of the P1aABD/CaM complex was superimposed to the CH1 domain of α-actinin (red), which is bound to F-actin (light gray, PDB: 3LUE). The steric clash between the P1aABD/CaM complex and F-actin is shown by purple arrows.
The results presented here support and extend the previously proposed model of HD disassembly (Kostan et al., 2009) and explain isoform specificity of P1a binding to Ca2+-CaM. In our extended model (Figure 9), the P1a/integrin β4 complex is maintained at low cytosolic calcium concentrations in HDs. In this complex, P1a and integrin β4 interact via the isoform-independent interface. The N-ter isoform-specific tail of P1a is disordered in this complex and is not involved in the interaction. Upon increased cytosolic Ca2+ concentrations during keratinocyte differentiation or wound healing, activated CaM binds to the N-ter tail of P1a via its N-lobe, while the C-lobe plays an auxiliary role in this interaction. The N-ter tail folds into an α helix upon CaM binding to P1a, leading to a steric clash between both the N- and C-terminal lobes of CaM and β4Fn12, which in turn instigates the displacement of integrin β4 from P1a.
Figure 9.

Model for the CaM-Driven Disruption of the P1aABD/β4Fn12 Complex
(A) At low cytosolic calcium concentrations, the P1a/integrin α6β4 complex is maintained in HDs.
(B) Increased cytosolic calcium concentration, during differentiation or wound healing, leads to activation of CaM (Ca2+-CaM). CaM in its active form binds to the structurally disordered N-ter tail of P1a via its N-ter lobe.
(C) CaM binding leads to the folding of the N-ter tail of P1a into an α helix. The steric clash caused by CaM bound to the α helix results in shunting of integrin β4 from the complex, contributing to HD disassembly.
For HD dynamics, being structurally disordered is an essential property of the P1a N-ter tail for three main reasons. First, although the N-ter extension does not contribute to the complex formation with integrin β4, an α helix in place of a structurally disordered peptide would generate a steric clash with integrin β4. Second, being structurally disordered in complex with integrin β4 enables exploration of a large conformational space, facilitating the recruitment of Ca2+-CaM upon increased cytosolic calcium levels. Intrinsically disordered regions provide conformational fluctuations, which can facilitate intermolecular interactions, forming complexes with high specificity and relatively low affinity. This is critical for processes in which not only specific association but also subsequent dissociation of binding partners is required (Tompa and Csermely, 2004). Third, binding to Ca2+-CaM leads to the formation of an α helix, which suitably positions Ca2+-CaM to displace integrin β4 and F-actin from the complex with plectin.
This study contributes to the understanding of molecular mechanisms regulating the functional states of HDs. Using a combination of structural, mutational, biophysical and in vivo studies, we show that the binding of Ca2+-CaM to the N-ter extension of P1a is involved in disruption of the plectin/integrin β4 and plectin/F-actin complexes by shunting integrin β4 and F-actin, without directly competing for the binding site. We further showed that the interaction of plectin with Ca2+-CaM is isoform specific, while the interaction with integrin β4 is not, implying that the main function of the N-ter tail is regulation of P1a interaction with integrin β4 and F-actin, and not direct binding to these partners.
Finally, several cytoskeletal proteins of the spectrin superfamily (spectrin, utrophin, dystrophin, β-actinin) harbor EF-hand motifs, which may play important roles in regulating cytoskeletal interactions near the plasma membranes (Bennett and Healy, 2008). The studies on titin-α-actinin (Young and Gautel, 2000) and recent studies on spectrin-ankyrin, actin, and protein 4.2 (Korsgren and Lux, 2010; Korsgren et al., 2010) suggest that these interactions are governed by a pseudoligand mechanism. Furthermore, Ca2+-CaM was reported to regulate filamin A and utrophin interactions with F-actin (Nakamura et al., 2005; Winder and Kendrick-Jones, 1995). Our study provides the first structural insight into a mechanism of Ca2+-CaM-regulated interactions of an actin binding protein. Although the regulatory mechanics depicted above might differ, this analysis could serve as a blueprint for future studies aiming at understanding how Ca2+/CaM or EF-hand motifs regulate interactions in actin-associated proteins.
Experimental Procedures
Protein Cloning, Expression, Purification, and Crystallization
Proteins were expressed as His6-fusions in Escherichia coli and purified via Ni-NTA agarose and SEC as described in the Supplemental Materials and Methods.
Structure Determination of the P1aABDΔ22/CaMNL and P1aABDΔ22/β4Fn12 Complex
Equimolar mixture of P1aABDΔ22 and CaMNL was applied to an SEC column and elution peak containing the complex concentrated to 11 mg/ml. Crystals were grown by the hanging-drop vapor diffusion method at 4°C from a solution containing 0.1 M Bis-Tris (pH 6.5), 0.2 M MgCl2, and 13% PEG 8000. The structure was solved by molecular replacement using human plectin ABD (PDB, 1MB8) (Garcia-Alvarez et al., 2003) and the N-ter lobe of Ca2+-CaM (PDB, 3CLN) (Babu et al., 1988) as search models.
Crystals of P1aABDΔ22 were grown by the sitting-drop vapor diffusion method at 22°C from a solution containing 50 mM monobasic potassium phosphate and 20% PEG 8000. The structure was solved by molecular replacement using human plectin ABD (PDB, 1MB8) (Garcia-Alvarez et al., 2003).
An equimolar mixture of P1aABDΔ22 and β4Fn12 was concentrated to 12 mg/ml. Crystals of the protein complex were grown at 22°C using hanging-drop vapor diffusion, from a solution containing 20 mM HEPES-NaOH (pH 6.5), 150 mM sodium formate, 7.5% PEG 550 MME, and 3% sucrose. Crystals were dehydrated by transferring coverslips to reservoirs containing the crystallization solution with increasing concentrations of PEG 550 MME. The structure was solved by molecular replacement using plectin ABD (PDB, 1MB8) and integrin β4 fragment (PDB, 3F7Q) as search models (de Pereda et al., 2009).
Diffraction data were collected at the beamlines ID14-1, ID14-4, and ID23-2 at ESRF (European Synchrotron Radiation Facility, Grenoble, France). Details on data collection, processing, structure determination, and refinement are described in the Supplemental Materials and Methods and in Table 1.
ITC
All protein samples were dialyzed against the ITC buffer (20 mM HEPES-NaOH [pH 7.5], 150 mM NaCl, and 5 mM CaCl2) overnight at 4°C. ITC was performed at 25°C or 30°C using an iTC200 Microcalorimeter (MicroCal, GE Healthcare). Thermodynamic parameters were obtained by fitting the one-site binding model or competitive binding model using the program Origin 7. The heat of dilution into buffer was subtracted from each reaction or the final titration point was used to estimate the reference baseline.
SAXS, Crosslinking, and Mass Spectrometry Analyses
SAXS data were collected on beamline X3 at EMBL (European Molecular Biology Laboratory) c/o DESY (Deutsches Elektronen-Synchrotron) (Hamburg, Germany) for WT, NEECK, and PIP2 mutants at three different concentrations and analyzed using the ATSAS package program (Petoukhov et al., 2012) following standard procedures.
Crosslinking experiments were performed using both a one-step and a two-step protocol. For the one-step method, with EDC and sulfo-NHS, bands from SDS-PAGE were excised, trypsinized, and analyzed by high-resolution LC-MS/MS. Details are described in the Supplemental Materials and Methods.
Pull-Down Assay
All plectin constructs were prepared at a concentration of 5 μM in buffer P (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% Tween 20). Plectin samples (1 ml) were mixed with 50 μl of CaM-Sepharose 4B beads (GE Healthcare) supplemented with either 5 mM CaCl2 or 1 mM EDTA. The samples were incubated for 2 hr at room temperature, followed by centrifugation at 3,000 x g for 2 min. Beads were washed with 1.5 ml of buffer P three times and incubated with an SDS-PAGE sample buffer at 95°C for 10 min to elute bound samples.
FRET Experiments
Rat bladder carcinoma 804G cells were grown in DMEM (Sigma Aldrich). FRET was measured by the acceptor photobleaching method as previously described (Stanek and Neugebauer, 2004) using the Leica SP5 confocal microscope. Details of the procedures are described in the Supplemental Materials and Methods.
Author Contributions
K.D.C. and G.W. conceived the project. J.-G.S. performed all structural and biochemical experiments and analyzed the data. F.D. and B.K. performed the XL-MS experiments and analyzed the data. E.d.A.R., P.K., and D.S. helped with analysis of SAXS data. M.G. performed FRET experiments and analyzed the data. I.G. helped with X-ray diffraction data collection and structure refinement. J.-G.S., J.K., and K.D.C. prepared the figures and wrote the manuscript. All authors commented on the manuscript.
Acknowledgments
K.D.C.’s research was supported by the Austrian Science Fund (FWF) through Grant Number W1221 DK, by the Federal Ministry of Economy, Family and Youth through the initiative “Laura Bassi Centres of Expertise” Funding Center of Optimized Structural Studies, Grant Number 253275, and by the Austrian Science Fund Grant (FWF) Number P22276. M.G. received support from Ministry of Education, Youth and Sports Grant Number 7AMB13AT012 and Operational Programme Research and Development for Innovation Grant Number CZ.1.05/1.1.00/02.0109 BIOCEV. The work of B.W. was supported by the Deutsche Forschungsgemeinschaft, Grant Number FOR1352 and by the Excellence Initiative of the German Federal & State Governments, Grant Number EXC 294 BIOSS Centre for Biological Signaling Studies). The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under BioStruct-X (Grant Agreement N°283570). We thank staff of X-ray beamlines at ESRF in Grenoble, SAXS beamline X33 (DESY, Hamburg), and SWING (Soleil, Saint-Aubin) for their excellent support. We are grateful to Oliviero Carugo (University of Vienna and University of Pavia) and Brooke Morriswood (University of Vienna, Max F. Perutz Laboratories) for critical reading of the manuscript.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Accession Numbers
The atomic coordinates of the P1aABDΔ22, P1aABDΔ22/CaMNL, and P1aABDΔ22/β4Fn12 complexes are deposited in the PDB under codes: 4Q59, 4Q57, and 4Q58, respectively.
Supplemental Information
References
- Andra K., Lassmann H., Bittner R., Shorny S., Fassler R., Propst F., Wiche G. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andra K., Kornacker I., Jorgl A., Zorer M., Spazierer D., Fuchs P., Fischer I., Wiche G. Plectin-isoform-specific rescue of hemidesmosomal defects in plectin (-/-) keratinocytes. J. Invest. Dermatol. 2003;120:189–197. doi: 10.1046/j.1523-1747.2003.12027.x. [DOI] [PubMed] [Google Scholar]
- Babu Y.S., Bugg C.E., Cook W.J. Structure of calmodulin refined at 2.2 A resolution. J. Mol. Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- Bennett V., Healy J. Organizing the fluid membrane bilayer: diseases linked to spectrin and ankyrin. Trends Mol. Med. 2008;14:28–36. doi: 10.1016/j.molmed.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Bernado P., Mylonas E., Petoukhov M.V., Blackledge M., Svergun D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- Borradori L., Sonnenberg A. Structure and function of hemidesmosomes: more than simple adhesion complexes. J. Invest. Dermatol. 1999;112:411–418. doi: 10.1046/j.1523-1747.1999.00546.x. [DOI] [PubMed] [Google Scholar]
- Bresnick A.R., Warren V., Condeelis J. Identification of a short sequence essential for actin binding by Dictyostelium ABP-120. J. Biol. Chem. 1990;265:9236–9240. [PubMed] [Google Scholar]
- Castanon M.J., Walko G., Winter L., Wiche G. Plectin-intermediate filament partnership in skin, skeletal muscle, and peripheral nerve. Histochem. Cell Biol. 2013;140:33–53. doi: 10.1007/s00418-013-1102-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya R., Meador W.E., Means A.R., Quiocho F.A. Calmodulin structure refined at 1.7 A resolution. J. Mol. Biol. 1992;228:1177–1192. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- Crivici A., Ikura M. Molecular and structural basis of target recognition by calmodulin. Annu. Rev. Biophys. Biomol. Struct. 1995;24:85–116. doi: 10.1146/annurev.bb.24.060195.000505. [DOI] [PubMed] [Google Scholar]
- de Pereda J.M., Lillo M.P., Sonnenberg A. Structural basis of the interaction between integrin alpha6beta4 and plectin at the hemidesmosomes. EMBO J. 2009;28:1180–1190. doi: 10.1038/emboj.2009.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunitz J.D. The entropic cost of bound water in crystals and biomolecules. Science. 1994;264:670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Frijns E., Sachs N., Kreft M., Wilhelmsen K., Sonnenberg A. EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin {beta}4. J. Biol. Chem. 2010;285:37650–37662. doi: 10.1074/jbc.M110.138818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijns E., Kuikman I., Litjens S., Raspe M., Jalink K., Ports M., Wilhelmsen K., Sonnenberg A. Phosphorylation of threonine 1736 in the C-terminal tail of integrin beta4 contributes to hemidesmosome disassembly. Mol. Biol. Cell. 2012;23:1475–1485. doi: 10.1091/mbc.E11-11-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs P., Zorer M., Rezniczek G.A., Spazierer D., Oehler S., Castanon M.J., Hauptmann R., Wiche G. Unusual 5′ transcript complexity of plectin isoforms: novel tissue-specific exons modulate actin binding activity. Hum. Mol. Genet. 1999;8:2461–2472. doi: 10.1093/hmg/8.13.2461. [DOI] [PubMed] [Google Scholar]
- Galkin V.E., Orlova A., Salmazo A., Djinovic-Carugo K., Egelman E.H. Opening of tandem calponin homology domains regulates their affinity for F-actin. Nat. Struct. Mol. Biol. 2010;17:614–616. doi: 10.1038/nsmb.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alvarez B., Bobkov A., Sonnenberg A., de Pereda J.M. Structural and functional analysis of the actin binding domain of plectin suggests alternative mechanisms for binding to F-actin and integrin beta4. Structure. 2003;11:615–625. doi: 10.1016/s0969-2126(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Geerts D., Fontao L., Nievers M.G., Schaapveld R.Q., Purkis P.E., Wheeler G.N., Lane E.B., Leigh I.M., Sonnenberg A. Binding of integrin alpha6beta4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J. Cell Biol. 1999;147:417–434. doi: 10.1083/jcb.147.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges-Labouesse E., Messaddeq N., Yehia G., Cadalbert L., Dierich A., Le Meur M. Absence of integrin alpha 6 leads to epidermolysis bullosa and neonatal death in mice. Nat. Genet. 1996;13:370–373. doi: 10.1038/ng0796-370. [DOI] [PubMed] [Google Scholar]
- Green K.J., Jones J.C. Desmosomes and hemidesmosomes: structure and function of molecular components. FASEB J. 1996;10:871–881. doi: 10.1096/fasebj.10.8.8666164. [DOI] [PubMed] [Google Scholar]
- Heidorn D.B., Trewhella J. Comparison of the crystal and solution structures of calmodulin and troponin C. Biochemistry. 1988;27:909–915. doi: 10.1021/bi00403a011. [DOI] [PubMed] [Google Scholar]
- Ishida T., Kinoshita K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007;35:W460–W464. doi: 10.1093/nar/gkm363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Korsgren C., Lux S.E. The carboxyterminal EF domain of erythroid alpha-spectrin is necessary for optimal spectrin-actin binding. Blood. 2010;116:2600–2607. doi: 10.1182/blood-2009-12-260612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsgren C., Peters L.L., Lux S.E. Protein 4.2 binds to the carboxyl-terminal EF-hands of erythroid alpha-spectrin in a calcium- and calmodulin-dependent manner. J. Biol. Chem. 2010;285:4757–4770. doi: 10.1074/jbc.M109.056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostan J., Gregor M., Walko G., Wiche G. Plectin isoform-dependent regulation of keratin-integrin alpha6beta4 anchorage via Ca2+/calmodulin. J. Biol. Chem. 2009;284:18525–18536. doi: 10.1074/jbc.M109.008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Levine B.A., Moir A.J., Patchell V.B., Perry S.V. The interaction of actin with dystrophin. FEBS Lett. 1990;263:159–162. doi: 10.1016/0014-5793(90)80728-2. [DOI] [PubMed] [Google Scholar]
- Litjens S.H., de Pereda J.M., Sonnenberg A. Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol. 2006;16:376–383. doi: 10.1016/j.tcb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Nakamura F., Hartwig J.H., Stossel T.P., Szymanski P.T. Ca2+ and calmodulin regulate the binding of filamin A to actin filaments. J. Biol. Chem. 2005;280:32426–32433. doi: 10.1074/jbc.M502203200. [DOI] [PubMed] [Google Scholar]
- Niessen C.M., Hulsman E.H., Oomen L.C., Kuikman I., Sonnenberg A. A minimal region on the integrin beta4 subunit that is critical to its localization in hemidesmosomes regulates the distribution of HD1/plectin in COS-7 cells. J. Cell Sci. 1997;110:1705–1716. doi: 10.1242/jcs.110.15.1705. [DOI] [PubMed] [Google Scholar]
- Novotny I., Blazikova M., Stanek D., Herman P., Malinsky J. In vivo kinetics of U4/U6.U5 tri-snRNP formation in Cajal bodies. Mol. Biol. Cell. 2011;22:513–523. doi: 10.1091/mbc.E10-07-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitz I., Tsomo L., Mercurio A.M. Protein kinase C-alpha phosphorylation of specific serines in the connecting segment of the beta 4 integrin regulates the dynamics of type II hemidesmosomes. Mol. Cell. Biol. 2004;24:4351–4360. doi: 10.1128/MCB.24.10.4351-4360.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads A.R., Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- Sigurskjold B.W. Exact analysis of competition ligand binding by displacement isothermal titration calorimetry. Anal. Biochem. 2000;277:260–266. doi: 10.1006/abio.1999.4402. [DOI] [PubMed] [Google Scholar]
- Stanek D., Neugebauer K.M. Detection of snRNP assembly intermediates in Cajal bodies by fluorescence resonance energy transfer. J. Cell Biol. 2004;166:1015–1025. doi: 10.1083/jcb.200405160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D., Barberato C., Koch M.H.J. CRYSOL–a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995;28:768–773. [Google Scholar]
- Tamura R.N., Rozzo C., Starr L., Chambers J., Reichardt L.F., Cooper H.M., Quaranta V. Epithelial integrin alpha 6 beta 4: complete primary structure of alpha 6 and variant forms of beta 4. J. Cell Biol. 1990;111:1593–1604. doi: 10.1083/jcb.111.4.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennenbaum T., Li L., Belanger A.J., De Luca L.M., Yuspa S.H. Selective changes in laminin adhesion and alpha 6 beta 4 integrin regulation are associated with the initial steps in keratinocyte maturation. Cell Growth Differ. 1996;7:615–628. [PubMed] [Google Scholar]
- Tompa P., Csermely P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004;18:1169–1175. doi: 10.1096/fj.04-1584rev. [DOI] [PubMed] [Google Scholar]
- van der Neut R., Krimpenfort P., Calafat J., Niessen C.M., Sonnenberg A. Epithelial detachment due to absence of hemidesmosomes in integrin beta 4 null mice. Nat. Genet. 1996;13:366–369. doi: 10.1038/ng0796-366. [DOI] [PubMed] [Google Scholar]
- Walko G., Vukasinovic N., Gross K., Fischer I., Sibitz S., Fuchs P., Reipert S., Jungwirth U., Berger W., Salzer U. Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet. 2011;7:e1002396. doi: 10.1371/journal.pgen.1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walko G., Castanon M.J., Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2014 doi: 10.1007/s00441-015-2216-6. Published online December 9, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K., Litjens S.H., Kuikman I., Margadant C., van Rheenen J., Sonnenberg A. Serine phosphorylation of the integrin beta4 subunit is necessary for epidermal growth factor receptor induced hemidesmosome disruption. Mol. Biol. Cell. 2007;18:3512–3522. doi: 10.1091/mbc.E07-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder S.J., Kendrick-Jones J. Calcium/calmodulin-dependent regulation of the NH2-terminal F-actin binding domain of utrophin. FEBS Lett. 1995;357:125–128. doi: 10.1016/0014-5793(94)01347-4. [DOI] [PubMed] [Google Scholar]
- Young P., Gautel M. The interaction of titin and alpha-actinin is controlled by a phospholipid-regulated intramolecular pseudoligand mechanism. EMBO J. 2000;19:6331–6340. doi: 10.1093/emboj/19.23.6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.