

Abstract
To investigate the influence of the mitochondrial calcium uniporter on the mitochondrial permeability transition pore, the present study observed mitochondrial morphology in cortical neurons isolated from adult rats using transmission electron microscopy, and confirmed the morphology and activity of isolated mitochondria by detecting succinic dehydrogenase and monoamine oxidase, two mitochondrial enzymes. Isolated mitochondria were treated with either ruthenium red, an inhibitor of the uniporter, spermine, an activator of the uniporter, or in combination with cyclosporin A, an inhibitor of the mitochondrial permeability transition pore. Results showed that ruthenium red inhibited CaCl2-induced mitochondrial permeability transition pore opening, spermine enhanced opening, and cyclosporin A attenuated the effects of spermine. Results demonstrated that the mitochondrial calcium uniporter plays a role in regulating the mitochondrial permeability transition pore in mitochondria isolated from the rat brain cortex.
Keywords: mitochondria, calcium uniporter, permeability transition pore, neuron, cortex, cell death, cerebroprotection
INTRODUCTION
Mitochondria play an important role in energy metabolism, Ca2+ signaling, aging and apoptotic cell death by releasing several apoptotic factors such as cytochrome c, apoptosis-inducing factor and procaspases[1]. Mitochondria are spatiotemporal modulators of cytosolic Ca2+ as they have the ability to both sequester and release Ca2+[2,3,4,5]. Moreover, calcium signals have been identified as one of the major signals that converge on mitochondria to trigger the mitochondrial-dependent pathway of apoptotic cell death[1,6,7,8,9,10,11,12].
Physiological mitochondrial Ca2+ efflux is attributed to either activation of the permeability transition pore (PTP) or reversal of the mitochondrial calcium uniporter (MCU)[4,6,10]. The MCU, located on the inner mitochondrial membrane, is the primary influx pathway for Ca2+ in mitochondria, and hence is a key regulator of mitochondrial Ca2+[5,13,14,15,16]. The MCU controls the rate of energy production, shapes the amplitude and spatiotemporal patterns of intracellular Ca2+ signals, and is instrumental to cell death[5]. The mitochondrial PTP (mPTP) is defined as a voltage-dependent, cyclosporine A-sensitive, high-conductance inner membrane channel[17,18,19,20,21]. If a Ca2+ concentration gradient exists between the matrix and the external medium, onset of permeability transition will lead to Ca2+ release[22,23]. However, ischemia/reperfusion injury of neurons was closely related to opening of the mPTP[24,25], which has been recognized as a critical area in mitochondrial-mediated apoptosis[19,26,27,28,29,30].
Recent evidence has shown that the perfusion of Ru360, which is a selective and potent MCU blocker, prevented opening of the mPTP in mitochondria isolated from reperfused hearts[31]. In addition, it has been demonstrated that the MCU is involved in the cardioprotection conferred by ischemic preconditioning, and that activity of the mPTP may be related to MCU activity[32]. Accordingly, we conclude that the MCU may play a role in regulating the mPTP, and this regulatory relationship could be of great significance to cerebral protection. The present study attempted to verify the conclusion using electron microscopic observation, enzyme activity detection and spectrophotometry.
RESULTS
Ultrastructure of isolated mitochondria from cortical neurons
Following transmission electron microscopy, normal mitochondria showed intact membranes and a dense matrix space (Figure 1A). However, incubation with CaCl2 induced mitochondrial swelling (Figure 1B), separated the inner and outer mitochondrial membrane, ruptured cristae, and a non-dense matrix was evident[32,33,34,35]. At the end of the experiment, we examined the treated mitochondria by transmission electron microscopy. The degree of swelling in the ruthenium red group (Figure 1C) was attenuated compared with the control group. Mitochondrial electron photomicrographs from the spermine group (Figure 1D) and cyclosporine A + spermine group (Figure 1E) were similar to the control group, and in some cases they were difficult to distinguish.
Figure 1.
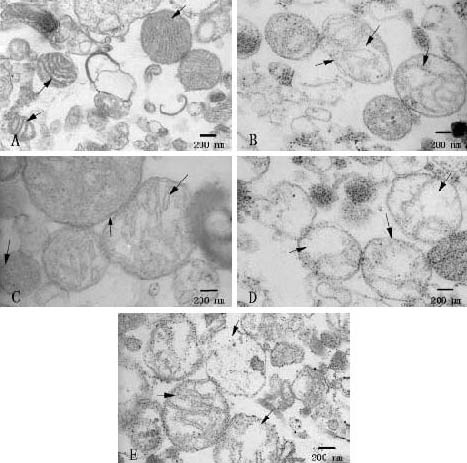
Electron photomicrographs of mitochondria (scale bars: 200 nm).
(A) Normal mitochondria: following fixation, intact membranes and dense matrix space were observed (arrows).
(B) Control group: incubation with CaCl2 induced mitochondrial swelling, separated inner and outer mitochondrial membrane, ruptured cristae, and a non-dense matrix was observed (arrows).
(C) Ruthenium red group: some comparatively intact membranes and dense matrix space were also observed (arrows).
(D) Spermine group: separated inner and outer mitochondrial membrane, ruptured cristae, and a non-dense matrix space were clearly observed (arrows).
(E) Cyclosporine A plus spermine group: the degree of swelling (arrows) was similar to that of the control group.
Succinic dehydrogenase (SDH) and monoamine oxidase (MAO) activities in isolated mitochondria from cortical neurons
The activities of the mitochondrial marker enzymes were assessed as soon as possible to prevent enzyme inactivation. The activities of SDH and MAO were higher in isolated mitochondria than in the cytosol (P < 0.01), especially SDH activity (Table 1), suggesting that isolated mitochondria have normal structure and function.
Table 1.
Activities of succinic dehydrogenase (U/min/mg) and monoamine oxidase (U/h/mg) in mitochondria and the cytosol (specific activity)

Regulatory effect of MCU activity on mPTP in isolated mitochondria (Figure 2)
Figure 2.
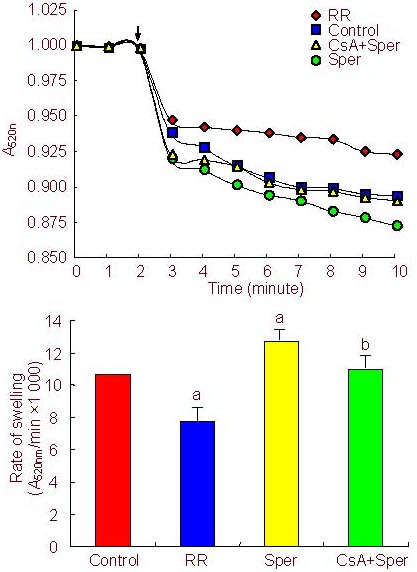
Effects of ruthenium red (RR), spermine (Sper), and cyclosporine A (CsA)+Sper treatment on mitochondrial permeability transition pore opening.
(A) Pore opening was determined by an absorbance decrease at 520 nm (A520nm) after the addition of CaCl2(200 μM). RR (5 μM) or CsA (2 μM) was added 2 minutes before Ca2+ addition, Sper (100 μM) and Ca2+ were added together[32]. The amplitude of swelling was observed for 10 minutes. All data are expressed relative to an initial absorbance of 1[36].
(B) Maximal swelling rates of mitochondria. Values represent mean ± SD of 8 independent experiments. aP < 0.01, vs. control group; bP < 0.01, vs. Sper group (one-way analysis of variance).
Induction of mitochondrial swelling in isolated mitochondria was determined by absorbance decreases at 520 nm. Pore opening was initiated by the addition of CaCl2(200 µM). The absorbance decreased rapidly after the addition of Ca2+. To explore the possibility that mPTP opening may be regulated by the activity of the MCU, a group of isolated mitochondria were preincubated (2 minutes) with the MCU inhibitor ruthenium red (5 µM). This treatment inhibited pore opening induced by Ca2+. Meanwhile, spermine (100 µM), an activator of the MCU, added together with Ca2+ accelerated the opening of the pore, and this was attenuated by cyclosporine A (2 µM), a specific pore inhibitor (Figure 2).
DISCUSSION
In the present study, we detected that inhibition of the MCU with ruthenium red diminished the opening of the mPTP in isolated brain mitochondria, and activation of the MCU with spermine increased pore opening. Furthermore, cyclosporin A, a specific pore inhibitor, attenuated the effect of spermine. In addition, the effect of Ru360, a highly specific MCU inhibitor, was similar and more obvious than ruthenium red (data not shown). Accordingly, the MCU may be involved in the regulation of the mPTP in mitochondria isolated from the rat brain. Regulation of the mPTP by the MCU in liver and heart isolated mitochondria has been mentioned. For example, the MCU and rapid mode of calcium uptake, a mechanism of calcium uptake, mediated the two physiological roles-control of the rate of cellular adenosine triphosphate (ATP) production and induction of permeability transition and apoptosis. In addition, a previous study suggested that the MCU, but not the rapid mode, played the primary role in permeability transition induction[3]. Inhibition of the MCU by Ru360 could prevent opening of the mPTP in post-ischemic rat hearts[31], demonstrating that the MCU was likely to be involved in the regulation of the mPTP. This hypothesis has been preliminarily proven by another study that showed activity of the mPTP may be related to MCU activity in mitochondria isolated from the heart[32].
MCU is a key regulator of mitochondrial Ca2+, and is instrumental to cell death[5,13,14,15,16]. In addition, the mPTP has been shown to signal necrotic and apoptotic cell death[37,38]. Previous studies of rat heart mitochondria indicated that preventing or reducing mitochondrial accumulation by inhibiting the MCU may benefit the heart during ischemia and reperfusion, i.e., blockade of the MCU during reperfusion provided protection against ischemia/reperfusion injury. In contrast, activating the MCU prohibited myocardial protection of ischemic preconditioning, and cyclosporin A abolished this effect[32]. In addition, regulation of the mPTP by the MCU was involved in myocardial protection of remote preconditioning.
In the present study, two properties of brain mPTP were detected, which were different to those observed in mitochondria from other tissues, as described previously[33,39,40,41]. Rat brain mitochondrial swelling was slower and milder; moreover, rat brain mitochondria were less sensitive to cyclosporin A, compared with the rat liver and heart mPTP[33,39,40,41].
However, the exact relationship between the MCU and the mPTP in isolated brain mitochondria remains unclear. The mPTP open-closed states are modulated by the transmembrane electrical potential, by matrix pH, by redox potential, by adenine nucleotides, and by divalent cations (e.g. Me2+, Mg2+, Sr2+ and Mn2+)[21,28,42,43,44,45]. Zhang et al[32] speculated that the activity of the MCU was related to the mitochondrial membrane via mitochondrial ATP-sensitive potassium channel activation, i.e., mitochondrial membrane depolarization inhibited MCU activity, and subsequently mPTP opening was inhibited. Similarly, in one of our previous studies, we found that inhibiting the MCU alleviated ischemia/reperfusion injury, which may be related to the decline in reactive oxygen species generation and caspase-3 activation[46]. Accordingly, we assume that the mechanism may include oxidation-reduction status, calcium content of the mitochondrial matrix, and transmembrane electrical potential. Further studies should verify the hypothesis at the tissue and cellular levels, and clarify the significance of the regulatory effect on cerebral protection.
MATERIALS AND METHODS
Design
An in vitro controlled animal experiment.
Time and setting
The study was performed at Cerebrovascular Disease Institute of Qingdao University Medical College and Central Laboratory of the Affiliated Hospital of Qingdao University Medical College, China, from August 2010 to April 2011.
Materials
Eight male Wistar rats of clean grade, aged 3 months and weighing 250–300 g, were purchased from the Laboratory Animal Center of Qingdao (License No. SCXK (Lu) 20090007). Animals were housed in a temperature-controlled (22–24 °C) room under a 12-hour light/12-hour dark cycle and allowed free access to water and food. All procedures used in this study were performed in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology of China[47].
Methods
Extraction and quantification of mitochondria
Mitochondria were isolated from rats according to a standard differential centrifugation procedure using a mitochondria isolation kit (Genmed Scientifics Inc., Shanghai, China). Briefly, rats were anesthetized by intraperitoneal injection of 10% (v/v) chloral hydrate (35 mg/100 g) and sacrificed by decapitation, then the cerebral cortex was rapidly removed. The following experimental operation was performed according to previously described methods with some modifications[33,35,36]. All procedures were conducted at 0–4 °C using a 3K30 refrigerated centrifuge (Sigma, St. Louis, MO, USA). Mitochondrial protein was measured by the Bradford method using the total protein quantification kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), with bovine serum albumin as a standard.
Transmission electron microscopy for monitoring the ultrastructure of isolated mitochondria
The ultrastructure of isolated mitochondrial preparations was observed by transmission electron microscopy using a JEM-1200EX transmission electron microscope (JEOL, Tokyo, Japan)[33]. Mitochondrial pellets were fixed in a 3.0% (v/v) solution of glutaraldehyde and the sample was fixed in a 1% (w/v) solution of osmium tetroxide. The fixed mitochondria were embedded in Epon812-Araldite and ultrathin slices (50 nm thick) were obtained.
Spectrophotometry for measuring mitochondrial enzyme activities
The marker enzyme activities of mitochondria were assessed spectrophotometrically using the SDH detection kit and MAO detection kit (Nanjing Jiancheng Bioengineering Institute). The activities of SDH were measured as an inner membrane marker enzyme, and MAO as an outer membrane marker enzyme. Enzyme activity was expressed as the specific activity units per hour or minute per milligram of protein[32,34,48]. Enzyme activities were assessed after mitochondrial lysis without delay.
Mitochondrial swelling for the detection of mPTP opening
To measure mPTP opening, isolated brain mitochondria were suspended in reaction buffer (150 mM KCl, 10 mM Tris, 20 mM 3-(N-Morpholino) propanesulfonic acid, 2 µM rotenone (Sigma), and 2 µM A23187 (Sigma), pH 7.4) to a final concentration of 0.5 mg/mL[34,44,45]. mPTP opening was determined by an absorbance decrease in light scattering at 520 nm using a Safire2 multimode microplate reader (Tecan, Zurich, Switzerland). The temperature was maintained at room temperature. Ruthenium red (5 µM; Sigma) or cyclosporine A (2 µM; Sigma) was added 2 minutes before Ca2+ addition, spermine (100 µM; Sigma) and Ca2+ were added together. Mitochondrial swelling was initiated by addition of CaCl2(200 µM), and the extent of mPTP opening was expressed in terms of the maximal rate of mitochondrial swelling[34,35,49,50,51]. In the present study, the swelling rate was calculated from the initial linear change in light scattering following the addition of CaCl2[35].
Statistical analysis
All data were expressed as mean ± SD. Statistical significance was determined by one-way analysis of variance, using SPSS 13.0 software (SPSS, Chicago, IL, USA). A value of P < 0.01 was considered statistically significant.
Acknowledgments:
We thank Dr. Ruyong Yao, Central Laboratory of the Affiliated Hospital of Qingdao University Medical College, Prof. Yunliang Guo, Cerebrovascular Disease Institute, Associate professors Yingyi Hou and Jinshan Tan, Electron Microscopy Research Office of Qingdao University Medical College, China for providing technical support.
Footnotes
Conflict of interest: None declared.
Funding: This work was supported by the National Natural Science Foundation of China, No. 30972855/C160203 and Natural Science Foundation of Shandong Province No. ZR2009CM062.
Ethical approval: This study was approved by the Animal Ethics Committee of the Affiliated Hospital of Qingdao University Medical College, China.
(Edited by Zhang H, Zhou YF/Su LL/Wang L)
REFERENCES
- [1].Pacher P, Csordás G, Hajnóczky G. Mitochondrial Ca2+ signaling and cardiac apoptosis. Biol Signals Recept. 2001;10(3-4):200–223. doi: 10.1159/000046888. [DOI] [PubMed] [Google Scholar]
- [2].Csordás G, Thomas AP, Hajnóczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999;18(1):96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gunter TE, Gunter KK. Uptake of calcium by mitochondria: transport and possible function. IUBMB Life. 2001;52(3-5):197–204. doi: 10.1080/15216540152846000. [DOI] [PubMed] [Google Scholar]
- [4].Chinopoulos C, Starkov AA, Fiskum G. Cyclosporin A-insensitive permeability transition in brain mitochondria: inhibition by 2-aminoethoxydiphenyl borate. J Biol Chem. 2003;278(30):27382–27389. doi: 10.1074/jbc.M303808200. [DOI] [PubMed] [Google Scholar]
- [5].Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- [6].Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89(7):1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- [7].Pörn-Ares MI, Ares MP, Orrenius S. Calcium signalling and the regulation of apoptosis. Toxicol In Vitro. 1998;12(5):539–543. doi: 10.1016/s0887-2333(98)00032-0. [DOI] [PubMed] [Google Scholar]
- [8].Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhu LP, Yu XD, Ling S, et al. Mitochondrial Ca2+ homeostasis in the regulation of apoptotic and necrotic cell deaths. Cell Calcium. 2000;28(2):107–117. doi: 10.1054/ceca.2000.0138. [DOI] [PubMed] [Google Scholar]
- [10].Montero M, Alonso MT, Albillos A, et al. Mitochondrial Ca2+ -induced Ca2+ release mediated by the Ca2+ uniporter. Mol Biol Cell. 2001;12(1):63–71. doi: 10.1091/mbc.12.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zecchini E, Siviero R, Giorgi C, et al. Mitochondrial calcium signalling: message of life and death. Ital J Biochem. 2007;56(4):235–242. [PubMed] [Google Scholar]
- [12].Murgia M, Giorgi C, Pinton P, et al. Controlling metabolism and cell death: at the heart of mitochondrial calcium signaling. J Mol Cell Cardiol. 2009;46(6):781–788. doi: 10.1016/j.yjmcc.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Deryabina YI, Isakova EP, Zvyagilskaya RA. Mitochondrial calcium transport systems: properties, regulation, and taxonomic features. Biochemistry (Mosc) 2004;69(1):91–102. doi: 10.1023/b:biry.0000016357.17251.7b. [DOI] [PubMed] [Google Scholar]
- [14].Vay L, Hernández-Sanmiguel E, Santo-Domingo J, et al. Modulation of Ca2+ release and Ca2+ oscillations in HeLa cells and fibroblasts by mitochondrial Ca2+ uniporter stimulation. J Physiol. 2007;580(Pt 1):39–49. doi: 10.1113/jphysiol.2006.126391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dash RK, Qi F, Beard DA. A biophysically based mathematical model for the kinetics of mitochondrial calcium uniporter. Biophys J. 2009;96(4):1318–1332. doi: 10.1016/j.bpj.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pradhan RK, Qi F, Beard DA, et al. Characterization of membrane potential dependency of mitochondrial Ca2+ uptake by an improved biophysical model of mitochondrial Ca2+ uniporter. PLoS One. 2010;5(10):e13278. doi: 10.1371/journal.pone.0013278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84(2-3):153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- [18].Crompton M. On the involvement of mitochondrial intermembrane junctional complexes in apoptosis. Curr Med Chem. 2003;10(16):1473–1484. doi: 10.2174/0929867033457197. [DOI] [PubMed] [Google Scholar]
- [19].Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- [20].Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46(6):821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- [21].Zorov DB, Juhaszova M, Yaniv Y, et al. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res. 2009;83(2):213–225. doi: 10.1093/cvr/cvp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability Transition. Physiol Rev. 1999;79(4):1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- [23].Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J. 2001;358(Pt 1):147–155. doi: 10.1042/0264-6021:3580147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kristal BS, Stavrovskaya IG, Narayanan MV, et al. The mitochondrial permeability transition as a target for neuroprotection. J Bioenerg Biomembr. 2004;36(4):309–312. doi: 10.1023/B:JOBB.0000041759.35731.70. [DOI] [PubMed] [Google Scholar]
- [25].Martin LJ, Adams NA, Pan Y, et al. The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. J Neurosci. 2011;31(1):359–370. doi: 10.1523/JNEUROSCI.2225-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- [27].Zamzami N, Larochette N, Kroemer G. Mitochondrial permeability transition in apoptosis and necrosis. Cell Death Differ. 2005;12(Suppl 2):1478–1480. doi: 10.1038/sj.cdd.4401682. [DOI] [PubMed] [Google Scholar]
- [28].Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34(Pt 2):232–237. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- [29].Shoshan-Barmatz V, Israelson A, Brdiczka D, et al. The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr Pharm Des. 2006;12(18):2249–2270. doi: 10.2174/138161206777585111. [DOI] [PubMed] [Google Scholar]
- [30].Honda HM, Ping P. Mitochondrial permeability transition in cardiac cell injury and death. Cardiovasc Drugs Ther. 2006;20(6):425–432. doi: 10.1007/s10557-006-0642-0. [DOI] [PubMed] [Google Scholar]
- [31].de Jesús García-Rivas G, Guerrero-Hernández A, Guerrero-Serna G, et al. Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart. FEBS J. 2005;272(13):3477–3488. doi: 10.1111/j.1742-4658.2005.04771.x. [DOI] [PubMed] [Google Scholar]
- [32].Zhang SZ, Gao Q, Cao CM, et al. Involvement of the mitochondrial calcium uniporter in cardioprotection by ischemic preconditioning. Life Sci. 2006;78(7):738–745. doi: 10.1016/j.lfs.2005.05.076. [DOI] [PubMed] [Google Scholar]
- [33].Kobayashi T, Kuroda S, Tada M, et al. Calcium-induced mitochondrial swelling and cytochrome c release in the brain: its biochemical characteristics and implication in ischemic neuronal injury. Brain Res. 2003;960(1-2):62–70. doi: 10.1016/s0006-8993(02)03767-8. [DOI] [PubMed] [Google Scholar]
- [34].Paul MK, Kumar R, Mukhopadhyay AK. Characterization of rat liver mitochondrial permeability transition pore by using mitochondrial swelling assay. Afr J Pharm Pharmacol. 2008;2(2):14–21. [Google Scholar]
- [35].Friberg H, Connern C, Halestrap AP, et al. Differences in the activation of the mitochondrial permeability transition among brain regions in the rat correlate with selective vulnerability. J Neurochem. 1999;72(6):2488–2497. doi: 10.1046/j.1471-4159.1999.0722488.x. [DOI] [PubMed] [Google Scholar]
- [36].Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160(1):226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- [37].De Giorgi F, Lartigue L, Bauer MK, et al. The permeability transition pore signals apoptosis by directing Bax translocation and multimerization. FASEB J. 2002;16(6):607–509. doi: 10.1096/fj.01-0269fje. [DOI] [PubMed] [Google Scholar]
- [38].Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol. 2009;104(2):181–188. doi: 10.1007/s00395-009-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schild L, Huppelsberg J, Kahlert S, et al. Mitochondria are primed by moderate Ca2+ rise upon hypoxia/reoxygenation for functional breakdown and morphological disintegration. J Biol Chem. 2003;278(28):25454–25460. doi: 10.1074/jbc.M302743200. [DOI] [PubMed] [Google Scholar]
- [40].Panov AV, Andreeva L, Greenamyre JT. Quantitative evaluation of the effects of mitochondrial permeability transition pore modifiers on accumulation of calcium phosphate: comparison of rat liver and brain mitochondria. Arch Biochem Biophys. 2004;424(1):44–52. doi: 10.1016/j.abb.2004.01.013. [DOI] [PubMed] [Google Scholar]
- [41].Votyakova TV, Reynolds IJ. Ca2+-induced permeabilization promotes free radical release from rat brain mitochondria with partially inhibited complex I. J Neurochem. 2005;93(3):526–537. doi: 10.1111/j.1471-4159.2005.03042.x. [DOI] [PubMed] [Google Scholar]
- [42].Fontaine E, Eriksson O, Ichas F, et al. Regulation of the permeability transition pore skeletal muscle mitochondria. J Biol Chem. 1998;273(20):12662–12668. doi: 10.1074/jbc.273.20.12662. [DOI] [PubMed] [Google Scholar]
- [43].Bernardi P, Krauskopf A, Basso E, et al. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273(10):2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- [44].Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc Res. 2006;70(2):191–199. doi: 10.1016/j.cardiores.2006.01.016. [DOI] [PubMed] [Google Scholar]
- [45].Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12(5):815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- [46].Liu ZL, Wang SL, Wang P, et al. The role of mitochondrial calcium uniporter on cerebral ischemia reperfusion injury in rats. Zhongfeng yu Shenjing Jibing Zazhi. 2011;18(1):9–12. [Google Scholar]
- [47].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006-09-30 [Google Scholar]
- [48].Kosenko E, Venediktova N, Kaminsky Y, et al. Preparation and handling of brain mitochondria useful to study uptake and release of calcium. Brain Res Brain Res Protoc. 2001;7(3):248–254. doi: 10.1016/s1385-299x(01)00071-x. [DOI] [PubMed] [Google Scholar]
- [49].Dedov VN, Demin OV, Chernyak VY, et al. Induction of nonselective permeability of the inner membrane in deenergized mitochondria. Biochemistry (Mosc) 1999;64(7):809–816. [PubMed] [Google Scholar]
- [50].Wu L, Shen F, Lin L, et al. The neuroprotection conferred by activating the mitochondrial ATP-sensitive K + channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci Lett. 2006;402(1-2):184–189. doi: 10.1016/j.neulet.2006.04.001. [DOI] [PubMed] [Google Scholar]
- [51].Kristal BS, Dubinsky JM. Mitochondrial permeability transition in the central nervous system: induction by calcium cycling-dependent and -independent pathways. J Neurochem. 1997;69(2):524–538. doi: 10.1046/j.1471-4159.1997.69020524.x. [DOI] [PubMed] [Google Scholar]