

Abstract
This study sought to analyze the genotype and gene mutations of human seizure-related gene 6 in 98 patients with idiopathic generalized epilepsy (non-febrile seizures), who were selected from three generations of the Chinese Han population living in Shanghai, Zhejiang Province, Wuxi of Jiangsu Province, and Jiangxi Province of Southern China. Twenty-six patients’ parents were available as a first-degree relatives group and 100 biologically unrelated healthy controls were collected as the control group. Based on the age of onset and seizure type, the patients were divided into six subgroups. Polymerase chain reaction and DNA direct sequencing analysis showed that the most frequent mutations c.1249dupC (p.Gly418Argfx31) and c.1636A > G (p.Thr546Ala) were detected in some idiopathic generalized epilepsy patients and their asymptomatic first-degree relatives (30.6% vs. 19.2% and 11.2% vs. 26.9%). A novel mutation c.1807G > A (p.Val603Met) was found in a patient with late-onset idiopathic generalized epilepsy. There was no significant difference in the incidence of these three mutations among the different subgroups of idiopathic generalized epilepsy and controls. Thus, further analysis of a larger population is needed to confirm the assumption that human seizure-related gene 6 is a susceptibility gene for idiopathic generalized epilepsy with various sub-syndromes.
Keywords: human seizure-related gene 6, non-febrile seizure, generalized epilepsy, mutation, polymorphism, genetic, neural regeneration
INTRODUCTION
Epilepsy is a frequently-occurring nervous disease with an incidence of 0.5–2.0% in the general population[1,2]. The etiology of epilepsy and epileptic seizures is multifactorial. Genetic factors are believed to contribute to some epilepsy syndromes, especially idiopathic epilepsy[2]. Idiopathic generalized epilepsy (IGE; OMIM: 600669), a common form, usually has no evidence of an underlying cause and is believed to be largely genetic in origin[3]. IGE is clinically characterized by absence, myoclonic seizures, and tonic-clonic seizures, typically beginning in childhood or adolescence with the characteristic electroencephalographic pattern of generalized spike-wave discharge. Based on the age of onset and seizure type, IGE can be divided into four syndromes according to the still valid classification of the International League Against Epilepsy: childhood absence epilepsy, occurring after the neonatal period; juvenile absence epilepsy; juvenile myoclonic epilepsy; and IGE with tonic–clonic seizures alone[4]. In addition, adult-onset IGE has also been described[5,6]. Although mutations in >25 genes have been linked to epilepsy, the relationship between the molecular defect and the clinical phenotype varies greatly[2,3,7,8].
Seizure-related gene 6 (SEZ-6) was originally identified as a gene with an mRNA that is upregulated in the cortex after administration of the convulsant drug pentylenetetrazole in mice[9]. Restriction of the expression pattern of SEZ-6 protein to cortical neurons implies that it plays an important role in the development of the forebrain and cognitive function in the adult mouse brain[10]. SEZ-6 protein contains one threonine-rich domain, five short consensus repeats, two CUB-like domains (CUB: Clr/Cls, urinary epidermal growth factor, and bone morphogenic protein), one transmembrane domain, and one short cytoplasmic segment in the C-terminal region, which suggests that it is involved in membrane receptor signaling[11,12].
An association between human –SEZ-6 mutations and febrile seizure has been found[13,14]. Another recent genome-wide linkage study of a Finnish family with various epilepsy phenotypes has suggested the presence of an epileptic underlying gene on chromosome 17q12–q24, around the region also associated with the SEZ-6 gene[15]. This raises the question: is SEZ-6 a susceptibility gene for IGE, with complex inheritance? The aim of the present study was to investigate the distribution of specific mutations or polymorphisms of SEZ-6 in Chinese IGE patients.
RESULTS
Quantitative analysis of subjects
A total of 224 subjects was included in this study and divided into three groups: an IGE group (n = 98), a first-degree relatives group (n = 26) and a control group (n = 100). All of the individuals were included in the final analysis without drop-outs.
Frequency of SEZ-6 gene variants
Genomic DNA was amplified by PCR and direct DNA sequencing to identify SEZ-6 gene variants or polymorphisms. Of the 98 IGE patients examined, 34 (34.7%) showed SEZ-6 mutations (Table 1). Of these 34 subjects, genomic DNA was available from 26 sets of parents and 21 of them also carried the mutations. Results showed the genotypes in IGD patients and controls were in accordance with the Hardy-Weinberg equilibrium.
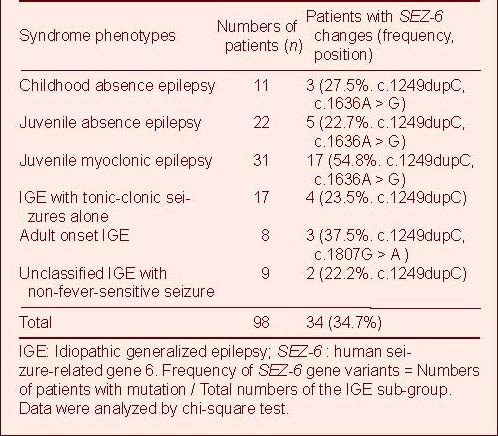
Table 1.
Epileptic phenotypes of 98 IGE patients screened for SEZ-6 variations
Distribution of the most common variant of SEZ-6 in the subjects
c.1249dupC
The most common variant was a single nucleotide duplication at position 1 249 within exon 5, c.1249dupC, resulting in a translational reading frameshift and a premature stop codon: p.Gly418Argfx31. Thirty of the IGE patients (30.6%) had this variant, among whom those with juvenile myoclonic epilepsy made up a high percentage (16/31, 51.6%), while the frequency in first-degree relatives was 19.2% (5/26) (Table 2). Most of c.1249dupC mutation was heterozygous, compared with the wild-type DNA sequence (Figures 1A and B). The homozygous mutation form was rarely identified in some patients with juvenile myoclonic epilepsy and IGE with tonic-clonic seizures alone, as well as their parents (Figure 1C). In addition, three of 100 unrelated healthy controls were found to carry the heterozygous form of this variant (3%).
Table 2.
IGE patients and their available relatives with the commonest variant, c.1249dupC
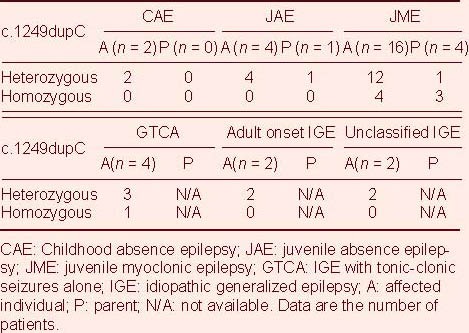
Figure 1.
c:1249dupC sequencing results of human seizure-related gene 6.
(A) Wild-type DNA sequence.
(B) Heterozygous form (the double peak; arrow) of this mutation; the inserted base C (red box) is indicated.
(C) Homozygous form of this mutation (arrow).
p.Thr546Ala
The second most frequent mutation was a missense mutation within exon 7, c.1636A > G (p.Thr546Ala). In the majority of patients, this mutation is concomitantly present in the same patients with the c.1249dupC variant, considering as compound mutations (Figure 2). Eleven patients (including two cases of childhood absence epilepsy, two of juvenile absence epilepsy, six of juvenile myoclonic epilepsy, and one of IGE with tonic-clonic seizures alone) carried the heterozygous form of this variant. There was only one exception in which the homozygotic form occurred in the parental DNA of a juvenile myoclonic epilepsy patient (Figure 2C). The frequencies in the patients and asymptomatic relatives were 11.2% (11/98) and 26.9% (7/26), respectively. In addition, the heterozygous form of c.1636A > G was identified in five controls (5%).
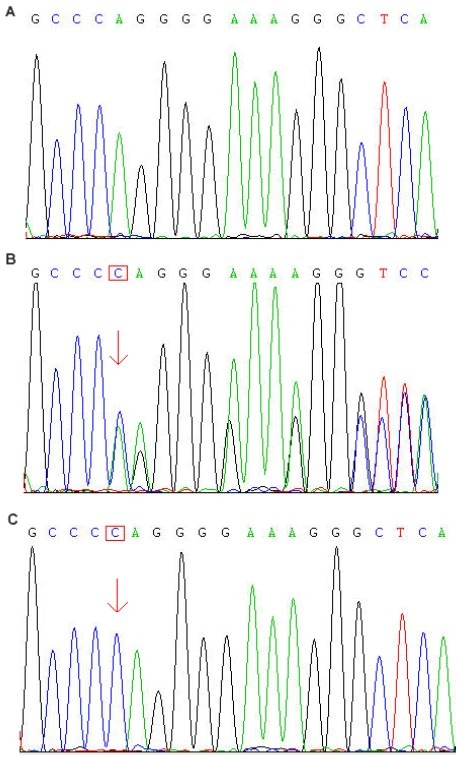
Figure 2.
DNA sequencing results for mutation c.1636A > G of human seizure-related gene 6.
(A) Control. ACA (red box) indicates the normal codon.
(B) Heterozygous form (the double peak. arrow) of this mutation from a male juvenile myoclonic epilepsy patient.
(C) Homozygous form of this mutation from the juvenile myoclonic epilepsy patient's first-degree relative. Codon ACA was substituted by GCA (red box), which resulted in an amino acid change from threonine to alanine.
c.1807G > A
A novel missense mutation c.1807G > A within exon 8, resulting in the substitution of methionine for valine at codon 603 (p.Val603Met), was found in an adult-onset IGE. Amino acid alignment of this mutation showed that it affected a highly conserved domain of SEZ-6 protein in other organisms (cow, mouse and Xenopus) and the SEZ-6 gene family (SEZ-L1 and SEZ-L2; Figure 3). DNA samples of 100 controls were screened for this mutation, successfully excluding the hypothesis of a genetic polymorphism.
Figure 3.
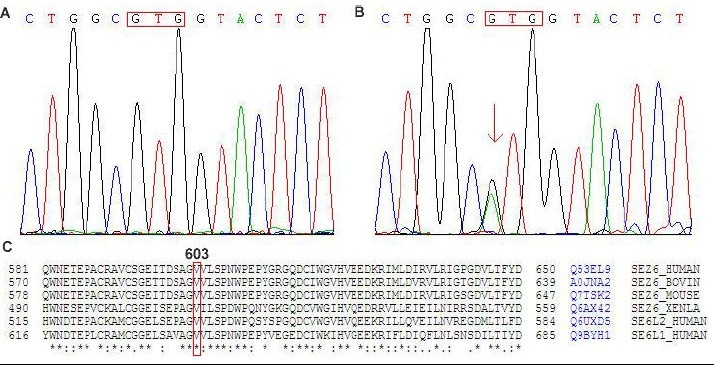
DNA sequencing results for mutation c.1807G > A within exon 8 of human seizure-related gene 6.
(A) Control. GTG (red box) indicates the normal codon.
(B) A heterozygous missense mutation G→A (the double peak, arrow) was identified at position 1807 in a late-onset IGE patient, which resulted in an amino acid change from valine to methionine.
(C) Valine 603 is an evolutionarily invariant in the paralogous human seizure-related gene 6 family and in homologous from other species (cow, mouse and Xenopus).
There was no significant difference in frequency of the above three mutations among different IGE subgroups and controls.
DISCUSSION
Since the homologous SEZ-6 gene was cloned by homologous screening, the possible molecular mechanisms of its function and association with epilepsy are gradually being investigated. Human SEZ-6 gene is located on chromosome 17q12, encoding at least three alternative splice variants, SEZ-6 types I, II and III[16]. SEZ-6 types I and II appear to be transmembrane proteins with long extracellular domains and short C-terminal cytoplasmic domains, whereas the type III isoform appears to be a secreted protein. Expression analysis shows highly restricted and stage-specific expression of SEZ-6 mRNA in the developing forebrain. SEZ-6 mRNA is expressed strongly in the cortical plate of the fetal brain, and is moderately expressed in the adult mouse brain, except for strong expression in the hippocampus, piriform cortex, and olfactory tubercle[10,12,17,18]. Recently, a new anti-SEZ-6 antibody has been used to show spatiotemporal changes in SEZ-6 distribution during the postnatal period, which suggests developmental regulation of the expression of SEZ-6 mRNA and implies thatS EZ-6 plays crucial roles in the development of the forebrain and in cognitive function in the adult mouse brain[10]. Although SEZ-6-deficient mice only show moderate signs of impaired spatial memory, motor deficits, and decreased anxiety, this is not discouraging, because complex conditions like epilepsy are probably caused by abnormal expression of multiple genes, not just by mutation or deletion of a single gene[17]. The heritability of epilepsy on a population basis is logistically complex and driven by polygenic susceptibility loci[19,20]. In addition, reduced numbers of excitatory synapses and unaltered inhibitory inputs, along with greater seizure resistance in SEZ-6 null mice suggest to some extent that levels of SEZ-6 play a role in determining excitability and therefore may be indirectly involved in seizure disorders[21,22].
IGE is considered to be caused by a genetic predisposition with no other evident etiology[2,3,23]. Patients with one of the four IGE sub-syndromes usually have seizure onset during childhood or adolescence, which used to be defined as the classic age at onset[24]. However, IGE cases with a later-than-usual onset have been regularly described, with an incidence less rare than the previously reported[25,26,27,28,29,30,31,32]. It has been suggested that IGE with late onset shares biological determinants common with IGE, while etiological factors and relations between each IGE syndrome are still under investigation.
In this study, we performed genetic screening focusing on SEZ-6 mutations in a small cohort of various Chinese IGE patients. To the best of our knowledge, this is the first report on the correlation between SEZ-6 and IGE. Thirty-eight sequence variants were identified in the 34 mutation-positive patients, while four affected children each had two changes. We described two frequent variants, c.1249dupC and c.1636A > G, in almost all types of IGE patients with no significant difference in occurrence among them. In addition, a small proportion of first-degree relatives of patients were found to be carriers of these mutations. Many reasonable causes could account for this phenomenon; for example, these two variants could be non-pathogenic, or certain individuals in the population could be tolerant of these mutations. What is more, a novel SEZ-6 mutation, c.1807G > A, was found in a late-onset IGE patient, which seemed to be pathogenic, because it was located in a highly conserved domain of SEZ-6. The patient carrying this unique mutation was a 48-year old man, who was referred to the clinic after his first tonic-clonic seizure at the age of 26 years. No evidence of an underlying cause was found upon clinical and electroencephalographic examination. Only eight patients were defined as adult-onset IGE in our study, with a broad age of seizure onset ranging from 21 to 58 years. Further analysis of a larger population might reveal interactions between SEZ-6 mutations and adult-onset IGE. Here, we cannot yet confirm the assumption that SEZ-6 is a susceptibility gene for IGE with various sub-syndromes.
SUBJECTS AND METHODS
Design
A case-control study focused on gene mutations or polymorphisms.
Time and setting
The study was performed at the Morbid Genomics Laboratory, School of Medicine, Zhejiang University, China, between April 2010 and May 2011.
Subjects
IGE group
A total of 98 Southern Chinese Han patients (56 males and 42 females) from Shanghai, Zhejiang Province, Wuxi of Jiangsu Province and Jiangxi Province in China with different types of IGE, were enrolled. The mean age of the patients was 26.5 years (range: 7–78 years). All affected individuals were inpatients at the Department of Neurology, Changhai Hospital, Shanghai, China. The forms of epilepsy were subdivided into clinical syndromes on the basis of age of onset, seizure pattern and other clinical, electroencephalography and imaging features according to the International League Against Epilepsy classification of epileptic syndromes[4]. Patients were evaluated by three neurologists independently. The epilepsy phenotypes of the 98 patients are shown in Table 1.
Inclusion criteria: (1) electroencephalography with generalized spike-wave and normal background; (2) compatible clinical history; (3) no pre-existing known neurological deficit; or (4) normal brain neuroimaging unless abnormality due to an acquired unrelated condition.
Exclusion criteria: (1) evidence for structural lesions, metabolic or degenerative diseases of the brain; (2) atonic/astatic or tonic seizures; (3) complex partial seizures; (4) epilepsy with myoclonic absences; (5) exclusively stimulus induced seizures.
First-degree relatives group
Twenty-six IGE patients’ parents were available as a first-degree relatives group. Exclusion criteria comprised the presence of any abnormal neurological finding, mental retardation, and morphological or metabolic brain disorders.
Control group
One hundred biologically unrelated healthy controls, including 55 men and 45 women, aged 20–79 years, with a similar ethnic background, were enrolled as the control group.
This study was conducted according to the 33rd Rule of Hospital Administration Regulation of China[33], and was approved by the Second Military Medical University of Chinese PLA Review Board, Shanghai, China. Informed consent was given by all participants.
Methods
Genotyping of SEZ-6 gene and mutation analysis
Genomic DNA was extracted from the peripheral blood sample by standard extraction protocols. PCR was carried out to amplify the target DNA frames of the SEZ-6 gene as previously described[13]. PCR products were sequenced directionally with an automated sequencer (ABI 3770; Applied Biosystems, Foster City, CA, USA) and analyzed by the DNA Assist 2.0 analysis software package (http://www.dnassist.com)[34]. Mutated sites were identified through comparison with the normal sequences of the exons of the SEZ-6 gene. All mutation sites were expressed with reference to SEZ-6 cDNA (GenBank accession number GI: 20143984)[13].
Statistical analysis
All data were entered into a database and analyzed by standard statistical methodology with SPSS 10.0 software (SPSS, Chicago, IL, USA). Comparison of mutation incidences among the IGE, first-degree relatives and control groups were carried out using a chi-square test. Statistical significance was accepted at P < 0.05.
Footnotes
Conflict of interest: None declared.
Funding: The study was supported by Shanghai Natural Science Foundation, China, No. ZR1404500.
Ethical approval: The experiment complied with the Ethics Committee of the Second Military Medical University of Chinese PLA, Shanghai, China.
(Edited by Zhao R, Shi J/Yang Y/Wang L)
REFERENCES
- [1].Lakhan R, Misra UK, Kalita J, et al. No association of ABCB1 polymorphisms with drug-refractory epilepsy in a north Indian population. Epilepsy Behav. 2009;14(1):78–82. doi: 10.1016/j.yebeh.2008.08.019. [DOI] [PubMed] [Google Scholar]
- [2].Mefford HC, Muhle H, Ostertag P, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6(5):e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Greenberg DA, Subaran R. Blinders, phenotype, and fashionable genetic analysis: a critical examination of the current state of epilepsy genetic studies. Epilepsia. 2011;52(1):1–9. doi: 10.1111/j.1528-1167.2010.02904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Engel J, Jr International League Against Epilepsy (ILAE) A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42(6):796–803. doi: 10.1046/j.1528-1157.2001.10401.x. [DOI] [PubMed] [Google Scholar]
- [5].Marini C, King MA, Archer JS, et al. Idiopathic generalized epilepsy of adult onset: clinical syndromes and genetics. J Neurol Neurosurg Psychiatry. 2003;74(2):192–196. doi: 10.1136/jnnp.74.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reichsoellner J, Larch J, Unterberger I, et al. Idiopathic generalized epilepsy of late onset: a separate nosological entity? J Neurol Neurosurg Psychiatry. 2010;81(11):1218–1222. doi: 10.1136/jnnp.2009.176651. [DOI] [PubMed] [Google Scholar]
- [7].Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87(1):41–57. doi: 10.1016/j.pneurobio.2008.09.016. [DOI] [PubMed] [Google Scholar]
- [8].Kleefuss-Lie A, Friedl W, Cichon S, et al. CLCN2 variants in idiopathic generalized epilepsy. Nat Genet. 2009;41(9):954–955. doi: 10.1038/ng0909-954. [DOI] [PubMed] [Google Scholar]
- [9].Shimizu-Nishikawa K, Kajiwara K, Sugaya E. Cloning and characterization of seizure-related gene, SEZ-6. Biochem Biophys Res Commun. 1995;216(1):382–389. doi: 10.1006/bbrc.1995.2635. [DOI] [PubMed] [Google Scholar]
- [10].Osaki G, Mitsui S, Yuri K. The distribution of the seizure-related gene 6 (SEZ-6) protein during postnatal development of the mouse forebrain suggests multiple functions for this protein: An analysis using a new antibody. Brain Res. 2011;1386:58–69. doi: 10.1016/j.brainres.2011.02.025. [DOI] [PubMed] [Google Scholar]
- [11].Bork P, Beckmann G. The CUB domain a widespread module in developmentally regulated proteins. J Mol Biol. 1993;231(2):539–545. doi: 10.1006/jmbi.1993.1305. [DOI] [PubMed] [Google Scholar]
- [12].Herbst R, Nicklin MJ. SEZ-6: promoter selectivity, genomic structure and localized expression in the brain. Mol Brain Res. 1997;44(2):309–322. doi: 10.1016/s0169-328x(96)00274-4. [DOI] [PubMed] [Google Scholar]
- [13].Yu ZL, Jiang JM, Wu DH, et al. Febrile seizures are associated with mutation of seizure-related (SEZ) 6, a brain-specific gene. J Neurosci Res. 2007;85(1):166–172. doi: 10.1002/jnr.21103. [DOI] [PubMed] [Google Scholar]
- [14].Mulley JC, Iona X, Hodgson B, et al. The role of seizure-related SEZ6 as a susceptibility gene in febrile seizures. Neurol Res Int 2011. 2011 doi: 10.1155/2011/917565. 917565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Siren A, Polvi A, Chahine L, et al. Suggestive evidence for a new locus for epilepsy with heterogeneous phenotypes on chromosome 17q. Epilepsy Res. 2010;88(1):65–75. doi: 10.1016/j.eplepsyres.2009.09.022. [DOI] [PubMed] [Google Scholar]
- [16].Kim MH, Gunnersen JM, Tan SS. Localized expression of the seizure-related gene SEZ-6 in developing and adult forebrains. Mech Dev. 2002;118(1-2):171–174. doi: 10.1016/s0925-4773(02)00238-1. [DOI] [PubMed] [Google Scholar]
- [17].Gunnersen JM, Kuek A, Phipps JA, et al. Seizure-related gene 6 (SEZ-6) in amacrine cells of the rodent retina and the consequence of gene deletion. PLoS One. 2009;4(8):e6546. doi: 10.1371/journal.pone.0006546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gunnersen JM, Kim MH, Fuller SJ, et al. SEZ-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007;56(4):621–639. doi: 10.1016/j.neuron.2007.09.018. [DOI] [PubMed] [Google Scholar]
- [19].Lu Y, Wang X. Genes associated with idiopathic epilepsies: a current overview. Neurol Res. 2009;31(2):135–143. doi: 10.1179/174313209X393942. [DOI] [PubMed] [Google Scholar]
- [20].Rees MI. The genetics of epilepsy--the past, the present and future. Seizure. 2010;19(10):680–683. doi: 10.1016/j.seizure.2010.10.029. [DOI] [PubMed] [Google Scholar]
- [21].Wong RO, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci. 2002;3(10):803–812. doi: 10.1038/nrn941. [DOI] [PubMed] [Google Scholar]
- [22].Swann JW, Smith KL, Lee CL. Neuronal activity and the establishment of normal and epileptic circuits during brain development. Int Rev Neurobiol. 2001;45:89–118. doi: 10.1016/s0074-7742(01)45007-0. [DOI] [PubMed] [Google Scholar]
- [23].Jallon P, Latour P. Epidemiology of idiopathic generalized epilepsies. Epilepsia. 2005;46(Suppl 9):10–14. doi: 10.1111/j.1528-1167.2005.00309.x. [DOI] [PubMed] [Google Scholar]
- [24].Janz D. The idiopathic generalized epilepsies of adolescence with childhood and juvenile age of onset. Epilepsia. 1997;38(1):4–11. doi: 10.1111/j.1528-1157.1997.tb01073.x. [DOI] [PubMed] [Google Scholar]
- [25].Cutting S, Lauchheimer A, Barr W, et al. Adult-onset idiopathic generalized epilepsy: clinical and behavioral features. Epilepsia. 2001;42(11):1395–1398. doi: 10.1046/j.1528-1157.2001.14901.x. [DOI] [PubMed] [Google Scholar]
- [26].Gilliam F, Steinhoff BJ, Bittermann HJ, et al. Adult myoclonic epilepsy: a distinct syndrome of idiopathic generalized epilepsy. Neurology. 2000;55(7):1030–1033. doi: 10.1212/wnl.55.7.1030. [DOI] [PubMed] [Google Scholar]
- [27].Loiseau J, Crespel A, Picot MC, et al. Idiopathic generalized epilepsy of late onset. Seizure. 1998;7(6):485–487. doi: 10.1016/s1059-1311(98)80007-1. [DOI] [PubMed] [Google Scholar]
- [28].Luef G, Schauer R, Bauer G. Idiopathic generalized epilepsy of late onset: a new epileptic syndrome. Epilepsia. 1996;37(Suppl 4):4. [Google Scholar]
- [29].Marini C, King MA, Archer JS, et al. Idiopathic generalized epilepsy of adult onset: clinical syndromes and genetics. J Neurol Neurosurg Psychiatry. 2003;74(2):192–196. doi: 10.1136/jnnp.74.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nicolson A, Chadwick DW, Smith DF. A comparison of adult onset and classical idiopathic generalized epilepsy. J Neurol Neurosurg Psychiatry. 2004;75(1):72–74. [PMC free article] [PubMed] [Google Scholar]
- [31].Panayiotopoulos CP, Koutroumanidis M, Giannakodimos S, et al. Idiopathic generalized epilepsy in adults manifested by phantom absences, generalized toniceclonic seizures, and frequent absence status. J Neurol Neurosurg Psychiatry. 1997;63(5):622–627. doi: 10.1136/jnnp.63.5.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yenjun S, Harvey AS, Marini C, et al. EEG in adult-onset idiopathic generalized epilepsy. Epilepsia. 2003;44(2):252–256. doi: 10.1046/j.1528-1157.2003.26402.x. [DOI] [PubMed] [Google Scholar]
- [33].State Council of the People's Republic of China. Administrative Regulations on Medical Institution. 1994 [Google Scholar]
- [34].Patterton HG, Graves S. DNAssist, a C++ program for editing and analysis of nucleic acid and protein sequences on PC-compatible computers running Windows 95, 98, NT4.0 or 2000. Biotechniques. 2000;28(6):1192–1197. doi: 10.2144/00286bc02. [DOI] [PubMed] [Google Scholar]