

Abstract
Separation of proteins by SDS-PAGE followed by in-gel proteolytic digestion of resolved protein bands has produced high-resolution proteomic analysis of biological samples. Similar approaches, that would allow in-depth analysis of the glycans carried by glycoproteins resolved by SDS-PAGE, require special considerations in order to maximize recovery and sensitivity when using mass spectrometry (MS) as the detection method. A major hurdle to be overcome in achieving high-quality data is the removal of gel-derived contaminants that interfere with MS analysis. The sample workflow presented here is robust, efficient, and eliminates the need for in-line HPLC clean-up prior to MS. Gel pieces containing target proteins are washed in acetonitrile, water, and ethyl acetate to remove contaminants, including polymeric acrylamide fragments. O-linked glycans are released from target proteins by in-gel reductive β-elimination and recovered through robust, simple clean-up procedures. An advantage of this workflow is that it improves sensitivity for detecting and characterizing sulfated glycans. These procedures produce an efficient separation of sulfated permethylated glycans from non-sulfated (sialylated and neutral) permethylated glycans by a rapid phase-partition prior to MS analysis, and thereby enhance glycomic and sulfoglycomic analyses of glycoproteins resolved by SDS-PAGE.
Keywords: Chemistry, Issue 93, glycoprotein, glycosylation, in-gel reductive β-elimination, O-linked glycan, sulfated glycan, mass spectrometry, protein ID, SDS-PAGE, glycomics, sulfoglycomics
Introduction
Glycosylation is an essential protein post-translational modification, contributing to organismal physiology, tissue pathology, and cellular recognition 1-3. Despite major advances in analytical glycoscience, characterizing the complete diversity of glycans on a specific protein remains an extremely challenging task, especially on proteins isolated from primary biological sources. Nonetheless, the microheterogeneity of glycoprotein glycans frequently affects functional interactions with other proteins. Therefore, characterization of glycan diversity is essential for understanding the physiological significance of cellular and tissue glycosylation 4,5. In order to understand the contribution of glycoprotein glycosylation to tissue physiology and pathophysiology, robust, sensitive, and comprehensive glycomic analytical techniques have become increasingly important. In proteomic analysis, protein identifications are generally achieved by LC-MS/MS analysis of tryptic peptides 6. Protein digestion can be carried out using a purified protein or proteins resolved by SDS-PAGE following in-gel digestion with proteases such as trypsin 7-9. Pre-enrichment of the protein mixture by SDS-PAGE enhances the depth and accuracy of protein ID. The development of analogous strategies for glycomic analysis of glycoprotein glycosylation lies at the forefront of glycoscience.
The two major classes of glycans are attached to protein backbones through either N-linkage or O-linkage. N-linked glycans are attached to asparagine (Asn) residues found as part of a sequon defined as Asn-X-Ser/Thr/Cys (X is any amino acid except proline), and can be released by enzymatic digestion with peptide-N-glycanase (PNGaseF or A), either in solution, in-gel, or on-blot 10-12. O-linked glycans are mainly attached to serine (Ser) or threonine (Thr) residues. However, only one enzyme has been identified that is capable of releasing O-linked glycans from glycoprotein and it has an extremely limited glycan specificity, releasing only the simplest O-linked glycans. Chemical release strategies remain the method of choice for comprehensive release of O-linked glycans from glycoproteins. Reductive or non-reductive β-elimination, or hydrazinolysis are well-characterized chemical release techniques and are currently the most commonly used approaches for releasing O-linked glycans from glycoproteins 13,14. Although reductive β-elimination has been used to release O-linked glycans from glycoproteins separated by SDS-PAGE, previous approaches required HPLC separation for subsequent analysis 15-17.
Multidimensional MS (MSn) analysis currently provides the richest source of structural data for characterizing glycans released from glycoproteins isolated in the amounts expected from most biological sources. The depth of MS-based structural characterization is greatly facilitated by permethylating the released glycans prior to their analysis. Permethylation enhances ionization and tends to equalize molar signal responses across a broad range of glycan structures 18,19. In addition, permethylation unambiguously tags terminal and substituted monosaccharide moieties with distinctive masses, thereby enhancing structural elucidation 20-23. For example, acidic glycans are generally difficult to detect as non-permethylated species by MS. Although acidic glycans can be detected in negative ion mode by MS, it is impossible to detect both acidic and neutral glycans in the same ion mode. A major advantage of glycan permethylation is that all of the free hydroxyl groups (OH) on a glycan’s monosaccharide substituents will be capped with a methyl group (OCH3 or OMe), thus a sialylated glycan’s charges are neutralized, making them as detectable as permethylated neutral (asialo) glycans. However, the hydroxyls of sulfate moieties on sulfoglycans are resistant to permethylation, resulting in retention of anionic charge, which suppresses ionization and decreases sensitivity. This suppression currently prevents comprehensive glycomic analysis of very complex glycoproteins such as mucins, which carry a high abundance of sulfated glycans 24-26.
Recent reports on purifying sulfated glycans used charged, reverse-phase chromatography to purify and separate permethylated glycans prior to MALDI analysis. This method relies on complete separation of sulfated and non-sulfated glycans using different mobile phases for elution, which we have found to be less stringent than organic phase partitioning. Therefore, new techniques suitable for the detection and enrichment of sulfoglycans are presented here. These techniques allow for the quantitative recovery of sulfated glycans in the aqueous phase following water:DCM (dichloromethane) extraction, which is routinely performed at the end of glycan permethylation reactions 27. Importantly, this robust separation of permethylated sulfoglycans from a mixture of permethylated non-sulfated glycans concomitantly enriches for charged species while also simplifying MS2 fragmentation patterns. A comprehensive protocol for improved in-gel O-linked glycan analysis is also presented. The improved protocol enhances glycan recovery, increases the structural information obtainable through MSn analysis of permethylated glycans, and improves the sensitivity of sulfoglycomic analyses applied to essential glycoproteins isolated from biological sources.
This protocol is intended for O-linked glycan analysis of whole glycoprotein extracts or of a specific glycoprotein of interest resolved by SDS-PAGE and is composed of three experimental procedures; A) gel clean-up, B) in-gel reductive β-elimination, and C) glycan permethylation. The goal is to obtain comprehensive O-linked glycomic data for glycoproteins harvested from primary sources of biological interest (Figure 1). Glycoproteins separated by SDS-PAGE are visualized by staining and bands of interest are excised and the resulting gel band is sliced into small pieces. The gel pieces are destained and subjected to ethyl acetate washes to remove gel contaminants (Figure 2A). Glycan release is achieved by in-gel reductive β-elimination (Figure 2B) and the released glycans are permethylated. Aqueous-organic extraction following permethylation quantitatively partitions the anionic sulfated glycans away from non-sulfated neutral glycans (Figure 2C). In-gel reductive β-elimination coupled to aqueous-organic extraction enables the characterization of O-linked glycans and sulfoglycans released from small amounts of glycoprotein separated by SDS-PAGE. The strategic overview is summarized in Figure 1 and the details are shown in Figure 2. In addition, a portion of the destained and washed gel pieces can be used for protein ID by standard LC-MS/MS proteomic techniques.
Protocol
NOTE: Lab Safety Concerns
In keeping with standard laboratory best practices, observe the following. Store all organic solvents in appropriate locations. Keep all waste materials in chemical waste containers with clear labeling of chemical compositions. As several reagents used in these protocols are potential carcinogens or generate volatile combustible gases, handle all reagents in a fume hood with ventilation. Wear personal protective equipment such as gloves, lab coat, and eye protection when working with organic solvents.
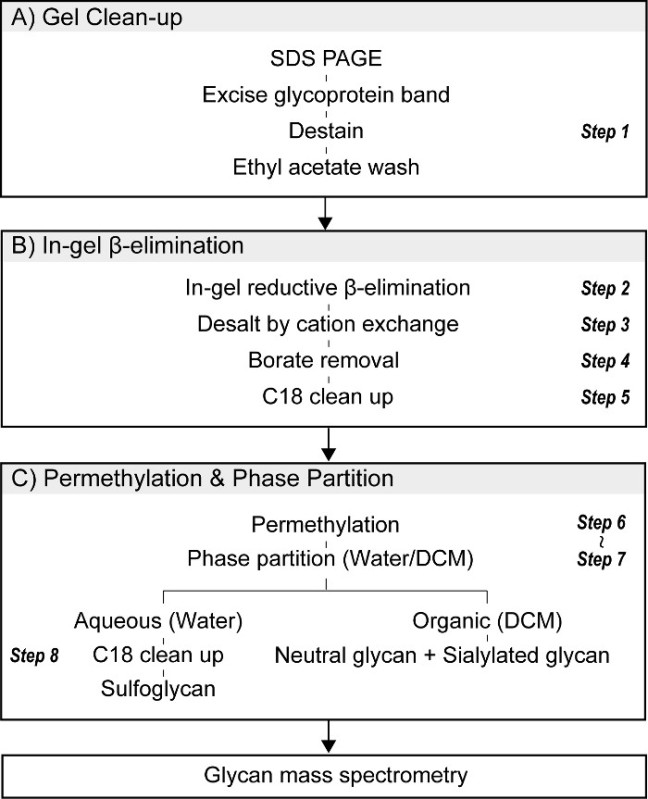 Figure 1. Procedure for in-gel O-glycomics. (A) Proteins of interest are resolved by SDS-PAGE, detected by appropriate staining procedures (Coomassie or silver), and excised. Excised gel pieces are sliced into small cubes, destained, and washed with ethyl acetate to reduce contaminants that interfere with subsequent MS analysis. A portion of the gel slice can be reserved for in-gel tryptic digestion and subsequent proteomic characterization by LC-MS/MS. (B) O-linked glycans are released from resolved glycoproteins by in-gel reductive β-elimination. Essential steps include desalting and borate removal by azeotrope with methanol. (C) O-linked glycans released by reductive β-elimination are permethylated and subsequently partitioned into aqueous and organic phases. Sulfoglycans are quantitatively recovered in the upper (aqueous) phase while neutral and sialylated glycans partition into the lower (DCM) layer.
Figure 1. Procedure for in-gel O-glycomics. (A) Proteins of interest are resolved by SDS-PAGE, detected by appropriate staining procedures (Coomassie or silver), and excised. Excised gel pieces are sliced into small cubes, destained, and washed with ethyl acetate to reduce contaminants that interfere with subsequent MS analysis. A portion of the gel slice can be reserved for in-gel tryptic digestion and subsequent proteomic characterization by LC-MS/MS. (B) O-linked glycans are released from resolved glycoproteins by in-gel reductive β-elimination. Essential steps include desalting and borate removal by azeotrope with methanol. (C) O-linked glycans released by reductive β-elimination are permethylated and subsequently partitioned into aqueous and organic phases. Sulfoglycans are quantitatively recovered in the upper (aqueous) phase while neutral and sialylated glycans partition into the lower (DCM) layer.
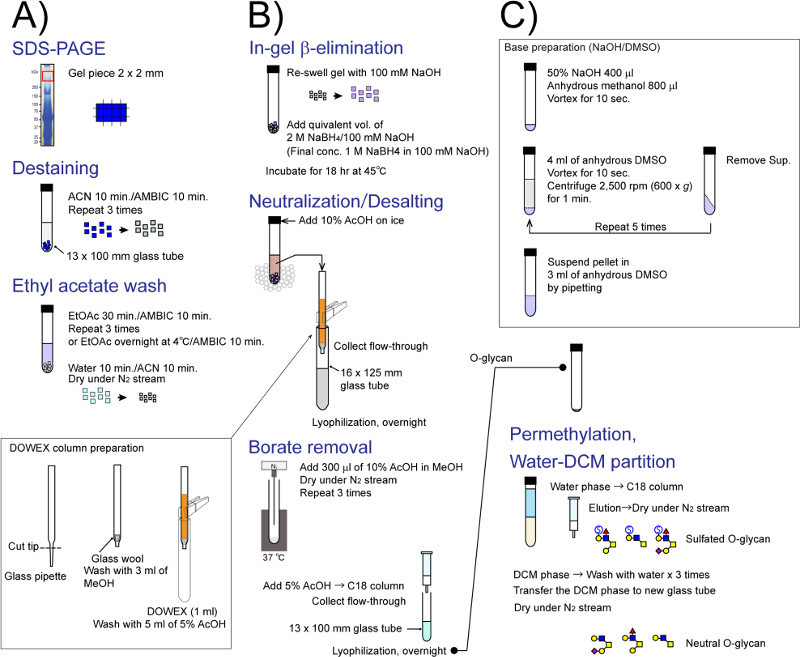 Figure 2. Detail flow chart for in-gel glycomics. Each experimental step shown in Figure 1 is individually illustrated. (A) gel destaining and EtOAc extraction, (B) direct in-gel reductive β-elimination for O-linked glycan release, and (C) permethylation and phase partition. Please click here to view a larger version of the figure.
Figure 2. Detail flow chart for in-gel glycomics. Each experimental step shown in Figure 1 is individually illustrated. (A) gel destaining and EtOAc extraction, (B) direct in-gel reductive β-elimination for O-linked glycan release, and (C) permethylation and phase partition. Please click here to view a larger version of the figure.
1. Gel Excision and Removal of Gel-derived Contaminants
Before starting the procedure, prepare chemicals and reagents listed in Materials List.
Resolve protein on SDS-PAGE gel using standard Tris-Glycine buffer systems. Stain resolved proteins with either Coomassie Brilliant Blue or Silver Stain. In general, recovery of O-linked glycans from silver stained gels is approximately 80–90% of the recovery achieved from Coomassie stained gels.
After electrophoresis, place the gel on a glass plate and excise the region of interest using a clean scalpel or razor blade.
To increase yield of O-glycans, transfer the gel band of interest onto another glass plate and cut the excised gel piece into approximately 2 mm cubes. Avoid making gel pieces smaller than 1 mm cubes because glycan recovery is reduced. Transfer the small gel pieces into a screw top glass tube (13 x 100 mm) with a stainless steel microspatula.
Add 1 ml of 25 mM ammonium bicarbonate (AMBIC, NH4HCO3) to the sample tube, cap the glass tube with a Teflon-lined screw top cap, and gently mix by flicking the tube before letting stand for 10 min. Do not employ vigorous agitation in order to avoid ripping the gel pieces.
Carefully remove AMBIC from the glass tube using either a Pasteur glass pipette or a pipette equipped with an extra-long plastic tip. Avoid smashing and transferring the small gel pieces while removing the AMBIC solution.
Add 1 ml of acetonitrile (ACN) to the glass tube, cap, and gently mix by flicking tube before letting stand for 10 min. Ensure that the gel pieces are completely covered with solvent. If necessary, push gel pieces off of tube wall and into solvent with pipette tip.
Pipette off ACN from the glass tube and repeat steps 1.5–1.7 until the bright blue color is eliminated. Repeat a minimum of five sequential AMBIC/ACN washes if necessary. For silver stained gels, use a destain kit. Proceed to the next step when the gel piece turns uniformly white while in ACN.
Pipette off any liquid from the glass tube, recap the tube, add 1 ml of AMBIC and let stand for 5 min.
Remove AMBIC and add 2 ml of ethyl acetate (EtOAc) to the glass tube. Recap tube with Teflon-lined cap and place at 4 °C overnight with end-over-end agitation or alternatively perform three EtOAc washes at room temperature for 30 min each. The longer the EtOAc wash, the more efficient the removal of contaminants.
Remove the final EtOAc wash and perform three washes each with 2 ml of H2O. Complete removal of EtOAc will ensure a robust reductive β-elimination reaction.
Pipette off H2O and dehydrate the gel by washing once with 1 ml of ACN. NOTE: After drying, the dehydrated gels are ready for in-gel β-elimination. Dehydrated gel pieces can be stored at -20 °C until use.
Stop the washing process at any step and store the gel pieces at 4 °C overnight, regardless of which buffer or solution is applied to the gel (AMBIC, ACN, H2O). Restart sample preparation anytime within the next 2 days.
Use a portion of the destained and dehydrated gel pieces for protein ID by standard proteomic approaches.
2. In-gel O-glycan Release by Reductive β-Elimination
- Prepare a stock solution of 1 M sodium hydroxide (NaOH) using 50% NaOH or solid NaOH and store at 4 °C until use.
- Take 5.2 ml of 50% NaOH (or 4 g of solid NaOH) and adjust volume to 100 ml to make 1 M NaOH solution. Dilute the 1 M NaOH stock solution to 100 mM NaOH, which can be stored at 4 °C up to 6 months without loss of efficiency in reductive β-elimination reactions.
Make 2 M sodium borohydride (NaBH4) in 100 mM NaOH. Dissolve 38 mg of NaBH4 in 0.5 ml of 100 mM NaOH. Generally, use 0.5–1.0 ml of the borohydride solution for each sample. Vary the volume of the reaction solution on the amount of dried gel pieces prepared in step 1. Expect to use 0.5 ml of borohydride solution for 1–2 excised gel pieces (step 1.3).
Add 500 µl of 100 mM NaOH to the dehydrated gel pieces and let stand for 3–5 min on ice in order to equilibrate the gel to the basic conditions which enhance glycan recovery.
- Add 500 µl of 2 M NaBH4 in 100 mM NaOH, resulting in final concentrations of 1 M NaBH4 in 100 mM NaOH. Gently mix the tube and incubate at 45 °C for 18 hr.
- During the first hour of incubation, gently mix the tube every 15 min. Avoid strong agitation, because it may fragment the gel pieces. Perform the reaction in a well-equilibrated incubator/oven or in a heating block. Ensure that the gel pieces are covered with reaction solution.
Stop the reaction by removing the sample tube from the incubator or heating block and placing it on ice. NOTE: After allowing the β-elimination reaction to proceed for 18 hr, the desalting process must be performed within the same day.
3. Desalting on Cation-exchange Chromatography
- While maintaining the sample tube on ice to prevent excessive heat formation, slowly add 10% acetic acid (AcOH) dropwise to neutralize the base. Gently vortex the sample tube between additions of AcOH. Be careful to add acid slowly and drop wise as the addition of acid will produce a “volcano” of bubbles. Centrifuge the tube to eliminate bubbles and prevent spillover, if necessary.
- In order to remove the large amount of sodium in the reaction mixture, pass the reaction mixture through a small cation-exchange column (Dowex or AG-50W-X8 resin, H+ form).
- Wash the cation exchange resin with 1 M NaOH and 1 M HCl prior to use in order to remove contaminants that interfere with MS analysis, even if the resin is of analytical grade.
- Soak the resin in 1 M NaOH and remove solution by decantation. Add de-ionized water, remove water, then add 1 M HCl.
Repeat these steps until the solution above the resin is colorless. Wash the cleaned resin with deionized water and store in 5% AcOH at 4 °C.
To make a small glass column, scratch and break the tip of a Pasteur glass pipette using a ceramic cutter so that the taper of the pipette tip is approximately 1 cm long. Tease apart a plug of glass wool and push towards the tip forming a support for the resin bed. Secure the pipette column with a clamp or clothespin and place over a glass test tube.
Wash the empty pipette with 1 ml of methanol (MeOH) and 3 ml of 5% AcOH. Swirl H+ Dowex or AG50 cation exchange resin slurry stored in 5% AcOH and transfer enough of the slurry to produce a 1 ml bed volume in the Pasteur pipette column.
Rinse the column using five volumes of 5% AcOH. Check flow through for the appearance of resin particles and do not use the column if resin is detected.
Place column over a new screw top glass tube (16 x 125 mm) and load sample. Collect the flow through and elute glycans with at least 3 volumes of 5% AcOH into the same tube.
Cover the tube with Parafilm and make tiny holes with a needle. Place the sample tube at -80 °C or on dry ice. Once frozen, dry the sample by lyophilization. Store the dried material at -20 °C until use.
4. Borate Removal
Prepare 10% AcOH in MeOH. Take 10 ml of glacial acetic acid and adjust volume to 100 ml with methanol. Store this reagent in a glass bottle with Teflon-lined cap for up to 6 months.
Add 300 µl of 10% AcOH in MeOH to the dried, lyophilized sample tube and vortex. Remove the resulting trimethyl borate by evaporation under a N2 stream. NOTE: Mixing borate salts with methanol under acidic conditions produces trimethyl borate which is a volatile compound.
- Dry under N2 stream at 37 °C. After drying, add another aliquot of AcOH in MeOH as described in step 4.2. Dry the liquid for 5 min under nitrogen (N2) stream at 37 °C.
- Repeat resuspension and drying at least 3 times. Store the dried material at -20 °C until use.
5. C18 Clean-up
Equilibrate a C18 cartridge column (size 1 ml, 100 mg resin) using three volumes of ACN and five volumes of 5% AcOH. Accelerate passage of the equilibrating solutions by application of mild positive air pressure.
Add 500 µl of 5% AcOH to the desalted and borate-free sample tube and dissolve by vortex.
Load resuspended sample onto the equilibrated C18 column and collect the flow through into a glass screw cap tube (13 x 100 mm). Elute O-glycans with 3 ml of 5% AcOH into the same tube.
Parafilm the tube, make small holes with a small needle or use loosely closed PTFE-lined cap to cover the tube, and freeze the sample at -80 °C or on dry ice. Remove solvent by lyophilization. Store the dried sample at -20 °C until use.
6. Base Preparation for Permethylation
NOTE: In order to achieve robust permethylation, prepare NaOH slurry fresh. All glassware used for permethylation reactions should be extensively cleaned.
To prepare the base reagent for permethylation, add 400 µl of 50% NaOH to a clean glass screw-top tube (13 x 100 mm). Add 800 µl of anhydrous MeOH and vortex.
Add 4 ml of anhydrous dimethyl sulfoxide (DMSO) and vortex to generate a white precipitate.
Centrifuge at 600 x g for 1 min to pellet the precipitate. Pipette off supernatant and discard. Add an additional 4 ml of anhydrous DMSO to the pellet. Vortex.
Repeat step 6.2 and 6.3 a minimum of three more times or until no white precipitate forms.
Dissolve the pelleted base in 3 ml anhydrous DMSO and gently mix by pipetting up-and-down with a clean Pasteur pipette.
Use the base immediately for the following permethylation reaction as it cannot be stored.
7. Permethylation of Released Glycans
Add 200 µl of anhydrous DMSO to the C18-purified sample and vortex or sonicate to resuspend.
Add 300 µl of the resuspended base slurry to the sample and immediately add 100 µl of iodomethane (MeI). Seal tube with Teflon-lined cap and vigorously mix for 5 min by vortex.
To stop the permethylation reaction, add 2 ml of 5% AcOH on ice and vortex. Pipette solution up and down 5 times to reduce the remaining volume of MeI by evaporation. NOTE: The AcOH neutralizes the solution and removal of MeI increases the extraction efficiency of sulfated glycans into the water phase during the phase-partition.
Add 2 ml of dichloromethane (DCM) and vortex. Centrifuge at 600 x g for 1 min at room temperature to separate the aqueous and organic phases.
Transfer the top layer (aqueous phase that contains the majority of permethylated sulfated glycans) into a new glass tube (13 x 100 mm).
Add 2 ml of H2O to the organic phase and vortex. Centrifuge at 600 x g for 1 min to separate the aqueous and organic phases and combine this second aqueous phase with the first aqueous phase (step 7.5).
Repeat step 7.6 and 7.5 three more times, but there is no need to save the aqueous phases resulting from the subsequent partitions.
Remove the final top layer (aqueous phase) as much as possible from the bottom layer (organic phase) and transfer the bottom layer (organic phase) into a separate new glass tube using clean Pasteur pipette.
Dry the organic phase under N2 stream at 42 °C.
Cover the tube with Parafilm and store at -20 °C until use.
8. C18 Clean-up of Permethylated Sulfated O-glycans from the Aqueous Phase
Equilibrate a C18 cartridge column with three volumes of ACN and five volumes of 5% AcOH.
Load the aqueous phases collected and combined from steps 7.5 and 7.6 onto the column.
Wash the column with 10 ml of H2O for desalting.
Elute the permethylated sulfated O-glycans into a new glass tube with 2 ml of 50% ACN. Use an additional elution with 85% ACN to enhance recovery of permethylated sulfated O-glycans on larger oligosaccharides.
Dry eluates under nitrogen stream at 42 °C.
Store the dried material at -20 °C up to 6 months prior to MS analysis.
9. Mass Spectrometry of Permethylated O-glycans
Analyze permethylated O-glycans by direct infusion into an appropriate mass spectrometer using a nanoelectrospray source at a syringe flow rate of 0.40–0.60 µl/min at 210 °C capillary temperature. For fragmentation by CID in MS/MS and MSn of an ion trap instrument, apply 30–40% collision energy.
For negative ion mode, reconstitute permethylated sulfated O-glycans in 50 μl of methanol/2-propanol/1-propanol/13 mM aqueous ammonium acetate (16:3:3:2 by volume). For positive ion mode, reconstitute permethylated neutral/sialylated O-glycans in 50 µl of 1 mM sodium hydroxide in methanol/water (1:1).
Use the total ion mapping (TIM) and neutral loss scan (NL scan) functionality of the Xcalibur software package version 2.0 for MSn analysis to characterize individual O-glycan structures 13,19.
Representative Results
Effect of Ethyl Acetate Treatment Prior to In-gel Reductive β-Elimination
A representative mass spectrum of permethylated O-linked glycan samples released from bovine mucin using in-gel reductive β-elimination is shown in Figure 3. The EtOAc wash of the gel pieces effectively removes SDS and polyacryl contaminants which interfere with subsequent MS analysis 27.
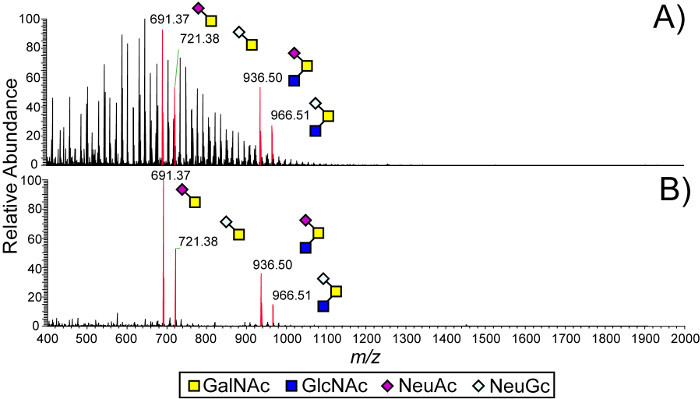 Figure 3. Ethyl acetate wash of polyacrylamide gel piece prior to in-gel reductive β-elimination enhances detection of released glycans. (A) Without wash with ethyl acetate, the mass spectrum of permethylated O-linked glycans released by in-gel β-elimination from bovine submaxillary mucin is dominated by an abundance of a polydisperse contaminant, which almost completely obscures the MS signals associated with O-linked glycans. (B) Washing the gel piece with ethyl acetate eliminates the contaminant peaks, allowing sensitive detection of glycans. Modified from Kumagai25.
Figure 3. Ethyl acetate wash of polyacrylamide gel piece prior to in-gel reductive β-elimination enhances detection of released glycans. (A) Without wash with ethyl acetate, the mass spectrum of permethylated O-linked glycans released by in-gel β-elimination from bovine submaxillary mucin is dominated by an abundance of a polydisperse contaminant, which almost completely obscures the MS signals associated with O-linked glycans. (B) Washing the gel piece with ethyl acetate eliminates the contaminant peaks, allowing sensitive detection of glycans. Modified from Kumagai25.
Recovery of O-linked Glycan is Greater from Small Gel Slices
O-linked glycans were released from bovine submaxillary mucin by in-gel reductive β-elimination using gel pieces that were either sliced small (~2 x 2 mm) or large (~5 x 5 mm). Gel pieces smaller than ~2 x 2 mm were not efficiently recovered through the washing steps. Following permethylation, a known amount of a permethylated external glycan standard (maltotri- and maltotetrasaccharide, Dp3 and Dp4) was added to each to facilitate quantification of glycan recovery 27. The recovery of O-linked glycans from small gel pieces was almost 10-fold greater than from large gel slices (Figure 4).
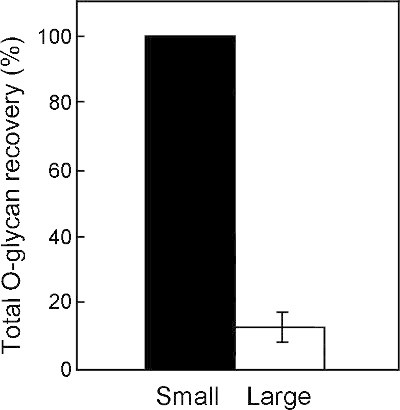 Figure 4. Glycan recovery from small gel pieces is more efficient than from larger pieces. Small pieces (~2 x 2 mm) yielded more glycan than larger cubes (~5 x 5 mm). Bars show relative signal intensities for the sum of all O-linked glycan structures normalized to the signal detected for an external standard (maltotrisaccharide Dp3, set to 100%), which was added to the released glycan before MS analysis. Values are mean standard deviation for n=3. Modified from Kumagai K25.
Figure 4. Glycan recovery from small gel pieces is more efficient than from larger pieces. Small pieces (~2 x 2 mm) yielded more glycan than larger cubes (~5 x 5 mm). Bars show relative signal intensities for the sum of all O-linked glycan structures normalized to the signal detected for an external standard (maltotrisaccharide Dp3, set to 100%), which was added to the released glycan before MS analysis. Values are mean standard deviation for n=3. Modified from Kumagai K25.
Enrichment of Sulfoglycans by Phase Partition
A permethylated sulfated glycan standard (sulfo-Lea) was completely recovered in the aqueous phase. Permethylated sialylated glycans released from the mucin glycoprotein partitioned into the DCM phase (Figure 5). The recovery of each permethylated glycans by aqueous-organic partition was comparable to that of the previously characterized C18 Sep-Pak clean-up method 27. The efficiency and simplicity of the phase partition method greatly facilitates sample throughput and subsequent analytic approaches.
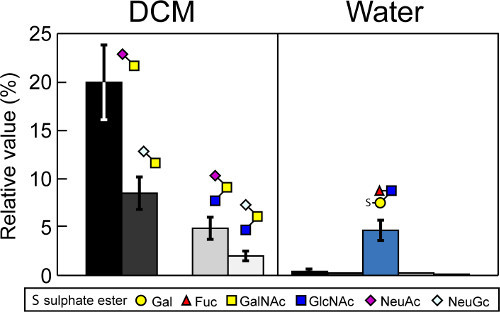 Figure 5. Differential recovery of sulfo- and sialo-glycans by phase partition. Bovine mucin glycoprotein was spiked with a known amount of a sulfo-glycan standard (sulfo-Lea) before being subjected to reductive β-elimination. Released glycans were permethylated and the permethylation reaction was adjusted to 1:1:water:DCM. The resulting organic and aqueous phases were separated and analyzed by MS. Glycan recovery was quantified relative to permethylated external glycan standards, which were spiked into the sample before MS analysis. Glycan recoveries into the organic (DCM) and aqueous (Water) phases are shown relative to the external standard, which was set to 100%. Sulfoglycans were undetectable in the organic phase but quantitatively recovered in the aqueous phase. Results represent the mean ± S.E. of three independent experiments. Modified from Kumagai25.
Figure 5. Differential recovery of sulfo- and sialo-glycans by phase partition. Bovine mucin glycoprotein was spiked with a known amount of a sulfo-glycan standard (sulfo-Lea) before being subjected to reductive β-elimination. Released glycans were permethylated and the permethylation reaction was adjusted to 1:1:water:DCM. The resulting organic and aqueous phases were separated and analyzed by MS. Glycan recovery was quantified relative to permethylated external glycan standards, which were spiked into the sample before MS analysis. Glycan recoveries into the organic (DCM) and aqueous (Water) phases are shown relative to the external standard, which was set to 100%. Sulfoglycans were undetectable in the organic phase but quantitatively recovered in the aqueous phase. Results represent the mean ± S.E. of three independent experiments. Modified from Kumagai25.
Application to In-depth Proteomic and Glycomic Analyses in Biological Samples
Human saliva proteins were separated by SDS-PAGE and stained with Coomassie Brilliant Blue (G-250). A high molecular weight protein at MW ~600 kDa was excised from the gel and a portion of the gel piece was subjected to in-gel tryptic digestion and LC-MS/MS-based proteomic analysis, which identified this band as MUC5B 16,24. The remainder of the gel piece was subjected to in-gel reductive β-elimination for O-glycan analysis 27. Following permethylation and aqueous-organic phase partition (Water:DCM, 1:1), permethylated glycans in the aqueous and organic phases were analyzed by NSI-MS. Non-sulfated permethylated O-glycans were recovered from the organic phase and all of the permethylated sulfoglycans were recovered from the aqueous phase, which facilitates the identification and characterization of nearly isobaric sulfated and non-sulfated glycans, e.g., Fuc1Hex3HexNAc2GalNAc-ol (m/z=1606.8, [M+Na]+) and (SO3)1NeuAc1Fuc1Hex2HexNAc1GalNAc-ol (m/z=1606.7, [M+2Na-H]+) differ by only 0.1 mass units and would be difficult to resolve without physically separating the two species by phase partition (Figure 6).
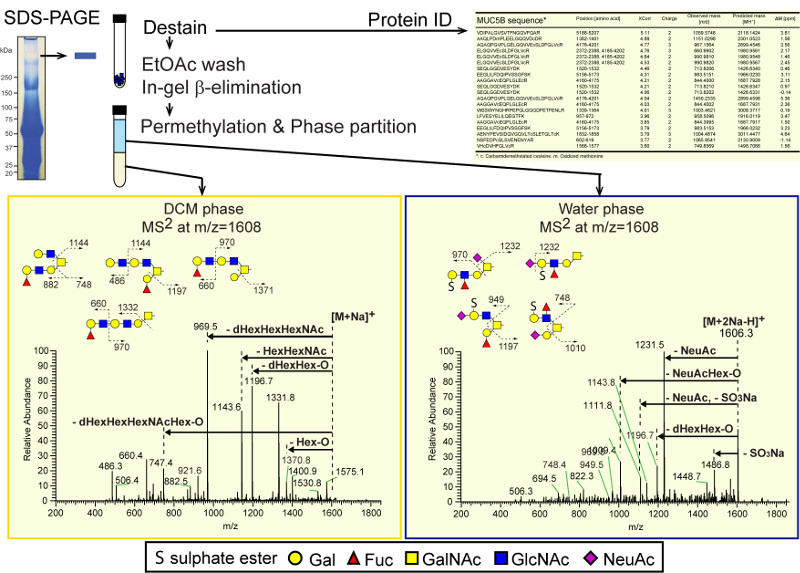 Figure 6. Detection of isobaric complexity in neutral and sulfo-glycans separated by aqueous-organic partition. MS2 fragmentation patterns obtained from parent ions detected by total ion mapping (TIM) analysis of permethylated O-glycans released from human salivary mucin by in-gel β-elimination and water:DCM partition following permethylation. (A) MS2 from TIM analysis of DCM phase for a 2.8 mass unit window around m/z = 1608. (B) The MS2 spectrum of the same mass window for TIM analysis of the water phase. In the DCM-phase, the major fragment ions correspond to loss of Hex1-O (m/z 1370.8), Fuc1Hex3HexNAc2+Na (1331.8), loss of Fuc1Hex1-O (1196.7), loss of Hex1HexNAc1 (1143.6), loss of Fuc1Hex1HexNAc1 (969.5), loss of Fuc1Hex2HexNAc1-O (747.4), Fuc1Hex1HexNAc1+Na (660.4), and Hex1HexNAc1+Na (486.3). Based on these fragment ions, a mixture of non-sulfated structures with a composition of Fuc1Hex3HexNAc2GalNAc-ol is proposed as shown to the right of the spectrum. In contrast, the major fragment ions for the water phase are loss of SO3Na (1486.8), loss of NeuAc (1231.5), loss of Fuc1Hex1-O (1196.7), loss of combination of NeuAc and SO3Na (1111.8), and loss of NeuAc1Hex1-O (1009.4). A mixture of sulfated structures with a composition of (SO3-)1NeuAc1Fuc1Hex2HexNAc1GalNAc-ol is proposed. Without physical separation of the neutral and sulfoglycans by phase partition, interpreting MS2 spectra of such mixtures is significantly more challenging. Modified from Kumagai25. Please click here to view a larger version of the figure.
Figure 6. Detection of isobaric complexity in neutral and sulfo-glycans separated by aqueous-organic partition. MS2 fragmentation patterns obtained from parent ions detected by total ion mapping (TIM) analysis of permethylated O-glycans released from human salivary mucin by in-gel β-elimination and water:DCM partition following permethylation. (A) MS2 from TIM analysis of DCM phase for a 2.8 mass unit window around m/z = 1608. (B) The MS2 spectrum of the same mass window for TIM analysis of the water phase. In the DCM-phase, the major fragment ions correspond to loss of Hex1-O (m/z 1370.8), Fuc1Hex3HexNAc2+Na (1331.8), loss of Fuc1Hex1-O (1196.7), loss of Hex1HexNAc1 (1143.6), loss of Fuc1Hex1HexNAc1 (969.5), loss of Fuc1Hex2HexNAc1-O (747.4), Fuc1Hex1HexNAc1+Na (660.4), and Hex1HexNAc1+Na (486.3). Based on these fragment ions, a mixture of non-sulfated structures with a composition of Fuc1Hex3HexNAc2GalNAc-ol is proposed as shown to the right of the spectrum. In contrast, the major fragment ions for the water phase are loss of SO3Na (1486.8), loss of NeuAc (1231.5), loss of Fuc1Hex1-O (1196.7), loss of combination of NeuAc and SO3Na (1111.8), and loss of NeuAc1Hex1-O (1009.4). A mixture of sulfated structures with a composition of (SO3-)1NeuAc1Fuc1Hex2HexNAc1GalNAc-ol is proposed. Without physical separation of the neutral and sulfoglycans by phase partition, interpreting MS2 spectra of such mixtures is significantly more challenging. Modified from Kumagai25. Please click here to view a larger version of the figure.
Discussion
Combining in-gel reductive β-elimination with aqueous-organic extraction enhances the sensitivity and depth of structural data that can be acquired for characterizing sulfated and non-sulfated O-linked glycans harvested from small amounts of mucin-type glycoproteins resolved from other proteins by SDS-PAGE. The essential advances of the technical approaches presented in this study are: (a) facile removal of gel derived contaminants by simple washing steps; (b) quantitative recovery of permethylated sulfoglycans in the aqueous phase following a rapid water-DCM partition. The workflow described here dramatically improves the sensitivity of detection for sulfated and non-sulfated glycans released from glycoproteins expressed at physiological levels in biological samples.
While the methods described here are robust and highly reproducible when applied with careful attention to standard analytic practices, a few considerations regarding possible sources of contamination are worth a brief mention. Ensuring that all glassware is exceptionally clean and free of dust and detergent is critical. Upon noticing contaminant peaks in MS spectra, all glassware should be washed thoroughly and rinsed with MeOH prior to final drying. Another source of contamination is the use of rubber-lined screw caps in place of PTFE-lined caps. The lack of organic chemical resistance characteristic of the rubber lining results in samples contaminated with an interfering polymeric compound. Although this protocol was specifically designed with the intention of utilizing MSn analysis by NSI-MS on an ion trap instrument, other types of mass spectrometers, such as MALDI-TOF/TOF can be also applied to dissecting sulfoglycoforms in biological materials using the preparative methods described here.
In comparison to other methodologies currently in use for in-gel glycan release and analysis, the techniques described here do not require HPLC separation prior to MS analysis and are applicable to permethylated glycans, producing greater structural characterization than can be extracted from non-derivatized samples. Moreover, the methods described here are capable of separating permethylated forms of sulfated O-glycans from non-sulfated neutral and sialylated O-glycans by a simple phase partition. The optimized sample clean-up and simple phase partition methodologies minimize sample manipulation and losses that can occur through chromatography steps. Methods requiring greater sample handling may lead to an underestimate of sulfoglycan diversity due to losses during work-up. Furthermore, detection of permethylated sulfated glycans in negative mode is significantly less sensitive than detection of permethylated glycans in positive mode. Desulfation of permethylated sulfoglycans by solvolysis specifically removes sulfate groups, leaving naked hydroxyls that can be subsequently permethylated with deuterated methyl iodide, increasing MS-signal intensities approximately 5-fold and providing a unique isotopic mass tag at the site of sulfation25.
Disclosures
The authors declare no competing financial interest.
Acknowledgments
This work was supported by the grant P01HL107151 from the NHLBI/NIH. The authors also gratefully acknowledge the support and access to instrumentation provided through grant P41GM103490 from the NIGMS/NIH.
References
- Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3(2):97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126(5):855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012;13(7):448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe JB, Marth JD. A genetic approach to Mammalian glycan function. Annu. Rev. Biochem. 2003;72:643–691. doi: 10.1146/annurev.biochem.72.121801.161809. [DOI] [PubMed] [Google Scholar]
- Yan A, Lennarz WJ. Unraveling the mechanism of protein N-glycosylation. J. Biol. Chem. 2005;280(5):3121–3124. doi: 10.1074/jbc.R400036200. [DOI] [PubMed] [Google Scholar]
- Washburn MP. Utilisation of proteomics datasets generated via multidimensional protein identification technology (MudPIT) Brief Funct. Genomic Proteomic. 2004;3(3):280–286. doi: 10.1093/bfgp/3.3.280. [DOI] [PubMed] [Google Scholar]
- Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal. Biochem. 1992;203(1):173–179. doi: 10.1016/0003-2697(92)90061-b. [DOI] [PubMed] [Google Scholar]
- Hellman U, Wernstedt C, Gonez J, Heldin CH. Improvement of an 'In-Gel' digestion procedure for the micropreparation of internal protein fragments for amino acid sequencing. Anal. Biochem. 1995;224(1):451–455. doi: 10.1006/abio.1995.1070. [DOI] [PubMed] [Google Scholar]
- Morelle W, Canis K, Chirat F, Faid V, Michalski JC. The use of mass spectrometry for the proteomic analysis of glycosylation. Proteomics. 2006;6(14):3993–4015. doi: 10.1002/pmic.200600129. [DOI] [PubMed] [Google Scholar]
- Mortz E, Sareneva T, Haebel S, Julkunen I, Roepstorff P. Mass spectrometric characterization of glycosylated interferon-gamma variants separated by gel electrophoresis. Electrophoresis. 1996;17(5):925–931. doi: 10.1002/elps.1150170514. [DOI] [PubMed] [Google Scholar]
- Kuster B, Wheeler SF, Hunter AP, Dwek RA, Harvey DJ. Sequencing of N-linked oligosaccharides directly from protein gels: in-gel deglycosylation followed by matrix-assisted laser desorption/ionization mass spectrometry and normal-phase high-performance liquid chromatography. Anal. Biochem. 1997;250(1):82–101. doi: 10.1006/abio.1997.2199. [DOI] [PubMed] [Google Scholar]
- Kuster B, Mann M. 18O-labeling of N-glycosylation sites to improve the identification of gel-separated glycoproteins using peptide mass mapping and database searching. Anal. Chem. 1999;71(7):1431–1440. doi: 10.1021/ac981012u. [DOI] [PubMed] [Google Scholar]
- Aoki K, et al. The diversity of O-linked glycans expressed during Drosophila melanogaster development reflects stage- and tissue-specific requirements for cell signaling. J. Biol. Chem. 2008;283(44):30385–30400. doi: 10.1074/jbc.M804925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak RP, Royle L, Gardner RA, Fernandes DL, Wuhrer M. Suppression of peeling during the release of O-glycans by hydrazinolysis. Anal. Biochem. 2012;423(1):119–128. doi: 10.1016/j.ab.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Schulz BL, Packer NH, Karlsson NG. Small-scale analysis of O-linked oligosaccharides from glycoproteins and mucins separated by gel electrophoresis. Anal. Chem. 2002;74(23):6088–6097. doi: 10.1021/ac025890a. [DOI] [PubMed] [Google Scholar]
- Thomsson KA, Schulz BL, Packer NH, Karlsson NG. MUC5B glycosylation in human saliva reflects blood group and secretor status. Glycobiology. 2005;15(8):791–804. doi: 10.1093/glycob/cwi059. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Holst O, Thomas-Oates J. Mass spectrometric profiling of O-linked glycans released directly from glycoproteins in gels using in-gel reductive β-elimination. Proteomics. 2006;6(10):2936–2946. doi: 10.1002/pmic.200500331. [DOI] [PubMed] [Google Scholar]
- Anumula KR, Taylor PB. A comprehensive procedure for preparation of partially methylated alditol acetates from glycoprotein carbohydrates. Anal. Biochem. 1992;203(1):101–108. doi: 10.1016/0003-2697(92)90048-c. [DOI] [PubMed] [Google Scholar]
- Aoki K, Perlman M, Lim JM, Cantu R, Wells L, Tiemeyer M. Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J. Biol. Chem. 2007;282(12):9127–9142. doi: 10.1074/jbc.M606711200. [DOI] [PubMed] [Google Scholar]
- Domon B, Costello CE. A Systematic Nomenclature for Carbohydrate Fragmentations in Fab-Ms Ms Spectra of Glycoconjugates. Glycoconj. J. 1988;5(4):397–409. [Google Scholar]
- Ashline D, Singh S, Hanneman A, Reinhold V. Congruent strategies for carbohydrate sequencing. 1. Mining structural details by MSn. Anal. Chem. 2005;77(19):6250–6262. doi: 10.1021/ac050724z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapadula AJ, et al. Congruent strategies for carbohydrate sequencing. 3. OSCAR: an algorithm for assigning oligosaccharide topology from MSn data. Anal. Chem. 2005;77(19):6271–6279. doi: 10.1021/ac050726j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashline DJ, et al. Carbohydrate structural isomers analyzed by sequential mass spectrometry. Anal. Chem. 2007;79(10):3830–3842. doi: 10.1021/ac062383a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsson KA, et al. The salivary mucin MG1 (MUC5B) carries a repertoire of unique oligosaccharides that is large and diverse. Glycobiology. 2002;12(1):1–14. doi: 10.1093/glycob/12.1.1. [DOI] [PubMed] [Google Scholar]
- Yu SY, Wu SW, Hsiao HH, Khoo KH. Enabling techniques and strategic workflow for sulfoglycomics based on mass spectrometry mapping and sequencing of permethylated sulfated glycans. Glycobiology. 2009;19(10):1136–1149. doi: 10.1093/glycob/cwp113. [DOI] [PubMed] [Google Scholar]
- Yu SY, et al. Priming mass spectrometry-based sulfoglycomic mapping for identification of terminal sulfated lacdiNAc glycotope. Glycoconj J. 2013;30(2):183–194. doi: 10.1007/s10719-012-9396-z. [DOI] [PubMed] [Google Scholar]
- Kumagai T, Katoh T, Nix DB, Tiemeyer M, Aoki K. In-gel β-elimination and aqueous-organic partition for improved O- and sulfoglycomics. Anal. Chem. 2013;85(18):8692–8699. doi: 10.1021/ac4015935. [DOI] [PMC free article] [PubMed] [Google Scholar]