

Abstract
S-Adenosyl-l-methionine (AdoMet or SAM)-dependent methyltransferases (MTase) catalyze the transfer of the activated methyl group from AdoMet to specific positions in DNA, RNA, proteins and small biomolecules. This natural methylation reaction can be expanded to a wide variety of alkylation reactions using synthetic cofactor analogues. Replacement of the reactive sulfonium center of AdoMet with an aziridine ring leads to cofactors which can be coupled with DNA by various DNA MTases. These aziridine cofactors can be equipped with reporter groups at different positions of the adenine moiety and used for Sequence-specific Methyltransferase-Induced Labeling of DNA (SMILing DNA). As a typical example we give a protocol for biotinylation of pBR322 plasmid DNA at the 5’-ATCGAT-3’ sequence with the DNA MTase M.BseCI and the aziridine cofactor 6BAz in one step. Extension of the activated methyl group with unsaturated alkyl groups results in another class of AdoMet analogues which are used for methyltransferase-directed Transfer of Activated Groups (mTAG). Since the extended side chains are activated by the sulfonium center and the unsaturated bond, these cofactors are called double-activated AdoMet analogues. These analogues not only function as cofactors for DNA MTases, like the aziridine cofactors, but also for RNA, protein and small molecule MTases. They are typically used for enzymatic modification of MTase substrates with unique functional groups which are labeled with reporter groups in a second chemical step. This is exemplified in a protocol for fluorescence labeling of histone H3 protein. A small propargyl group is transferred from the cofactor analogue SeAdoYn to the protein by the histone H3 lysine 4 (H3K4) MTase Set7/9 followed by click labeling of the alkynylated histone H3 with TAMRA azide. MTase-mediated labeling with cofactor analogues is an enabling technology for many exciting applications including identification and functional study of MTase substrates as well as DNA genotyping and methylation detection.
Keywords: Biochemistry, Issue 93, S-adenosyl-l-methionine, AdoMet, SAM, aziridine cofactor, double activated cofactor, methyltransferase, DNA methylation, protein methylation, biotin labeling, fluorescence labeling, SMILing, mTAG
Introduction
Specific labeling of nucleic acids1,2 and proteins3,4 is of major interest for functional characterizations, medical diagnosis and (nano)biotechnology. Here we present an enzymatic labeling method for these biopolymers which is based on S-adenosyl-l-methionine (AdoMet or SAM)-dependent methyltransferases (MTases). This class of enzymes (EC 2.1.1.) targets individual nucleophilic positions (nitrogen, oxygen, sulfur and carbon atoms) within specific residues of nucleic acids and proteins and naturally transfers the activated methyl group of the cofactor AdoMet (Figure 1A)5. In addition, MTases can utilize synthetic cofactor analogues for specific labeling with affinity tags, fluorophores or other labels (Figure 1B)6. Two classes of AdoMet analogues have been developed: Aziridine cofactors for Sequence-specific Methyltransferase-Induced Labeling (SMILing)7 and double activated AdoMet analogues for methyltransferase-directed Transfer of Activated Groups (mTAG)8.
 Figure 1: Reactions catalyzed by methyltransferases (MTases). A. Methyl group transfer from the natural cofactor AdoMet (SAM) to various substrates including DNA, RNA, proteins and small biomolecules. B. Labeling/functionalization of nucleic acids and proteins (NNNNN = base pairs for DNA, nucleotides for RNA and amino acids for proteins; XXXXX = recognition sequence of the MTase with target residue in green) with synthetic cofactor analogues. Aziridine cofactors containing a reporter group (blue sphere) attached to the adenine ring are sequence specifically coupled with the target residue (left) and double-activated AdoMet analogues lead to transfer of extended alkyl chains carrying a chemical reporter Y (right) which can be labeled by bioorthogonal click reaction in a second step. Please click here to view a larger version of this figure.
Figure 1: Reactions catalyzed by methyltransferases (MTases). A. Methyl group transfer from the natural cofactor AdoMet (SAM) to various substrates including DNA, RNA, proteins and small biomolecules. B. Labeling/functionalization of nucleic acids and proteins (NNNNN = base pairs for DNA, nucleotides for RNA and amino acids for proteins; XXXXX = recognition sequence of the MTase with target residue in green) with synthetic cofactor analogues. Aziridine cofactors containing a reporter group (blue sphere) attached to the adenine ring are sequence specifically coupled with the target residue (left) and double-activated AdoMet analogues lead to transfer of extended alkyl chains carrying a chemical reporter Y (right) which can be labeled by bioorthogonal click reaction in a second step. Please click here to view a larger version of this figure.
Aziridine cofactors work best with DNA MTases. They contain a three membered ring with a nitrogen atom9 (or an N-mustard10,11) instead of the sulfonium center as reactive group. Protonation of this nitrogen atom activates the aziridine ring for nucleophilic attack by the target nucleotide which leads to covalent coupling of the whole cofactor with DNA. By attaching reporter groups to the adenine ring the aziridine cofactors can be used in combination with DNA MTases to label DNA in one step (Figure 1B, left)7,12. This is demonstrated in detail for the biotinylation of DNA with 6BAz13–15 (aziridine cofactor with biotin attached to the 6 position of the adenine ring) and the adenine-specific DNA MTase from Bacillus stearothermophilus (M.BseCI)16 (Figure 2, see protocol section 2: One-step labeling of DNA via aziridine cofactors). In addition to M.BseCI (5’-ATCGAT-3’ recognition sequence), the DNA MTases from Thermus aquaticus (M.TaqI, 5’-TCGA-3’), from Haemophilus heamolyticus (M.HhaI, 5’-GCGC-3’) and from Spiroplasma (M.SssI, 5’-CG-3’) have been successfully used to biotinylate DNA with 6BAz17. Furthermore, aziridine cofactors can be employed for one-step fluorescence DNA labeling18,19.
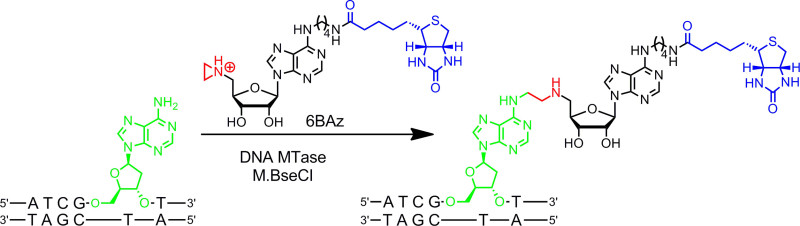 Figure 2: Sequence specific one-step biotinylation of DNA with M.BseCI and 6BAz. The DNA MTase M.BseCI recognizes the double-stranded DNA sequence 5’-ATCGAT-3’ and naturally methylates the amino group of the second adenine residue (green) using AdoMet. With the aziridine cofactor 6BAz the course of the reaction is changed and M.BseCI leads to sequence specific DNA biotinylation by coupling the whole cofactor including biotin (blue) with the target adenine. Please click here to view a larger version of this figure.
Figure 2: Sequence specific one-step biotinylation of DNA with M.BseCI and 6BAz. The DNA MTase M.BseCI recognizes the double-stranded DNA sequence 5’-ATCGAT-3’ and naturally methylates the amino group of the second adenine residue (green) using AdoMet. With the aziridine cofactor 6BAz the course of the reaction is changed and M.BseCI leads to sequence specific DNA biotinylation by coupling the whole cofactor including biotin (blue) with the target adenine. Please click here to view a larger version of this figure.
Double activated AdoMet analogues contain extended unsaturated side chains instead of a methyl group at the sulfonium center (Figure 1B, right)20. The unsaturated double or triple bond in β-position to the sulfonium center electronically compensates unfavorable steric effects within the transition state by conjugative stabilization. Since both the sulfonium center and the unsaturated bond activate the side chain for enzymatic transfer, these cofactors were named double-activated AdoMet analogues. Typically, they are used to transfer side chains with unique chemical groups (chemical reporters), like amino, alkyne and azide groups, for chemo-selective labeling in a second step8,21. In general, double-activated AdoMet analogues can not only function as cofactors for DNA MTases8,20,21 but also for RNA MTases22,23 and protein MTases24–28 allowing additional labeling of RNA and proteins. However, the extended side chains are sterically more demanding than a methyl group and enlarging the MTase active sites by protein engineering is often required to obtain efficient transfer rates. Another solution to this problem is to use an AdoMet analogue with a small propargyl group (three carbons) where the terminal alkyne serves two functions: 1. Stabilization of the transition state during enzymatic transfer and 2. reactive handle for following chemical modifications by copper-catalyzed azide-alkyne cycloaddition (CuAAC) click chemistry. It turned out that the resulting propargylic AdoMet analogue29 is quite unstable under neutral or slightly basic conditions and only of limited use. This drawback can be fixed by replacing the sulfur atom with selenium. The resulting cofactor 5‘-[(Se)[(3S)-3-amino-3-carboxypropyl]prop-2-ynylselenonio]-5‘-deoxyadenosine (SeAdoYn, Figure 3) is accepted by wild-type DNA, RNA and protein MTases30–32 which abrogate the need for protein engineering in many cases. This is exemplified by fluorescence protein labeling with the histone H3 lysine 4 (H3K4) MTase Set7/933 (Figure 3, see protocol section 3: Two-step protein labeling via double activated cofactors).
 Figure 3: Sequence-specific two-step fluorescence labeling of histone H3 with Set7/9, SeAdoYn and TAMRA azide. The protein MTase Set7/9 naturally methylates the amino group of lysine 4 in histone H3 (H3K4, green) using AdoMet. With the double-activated cofactor SeAdoYn the MTase transfers a small propargyl group (red) to the lysine residue. The attached terminal triple bond is then selectively modified in a bioorthogonal click reaction (copper-catalyzed azide-alkyne cycloaddition, CuAAC) with azide-derivatized TAMRA (tetramethylrhodamine, blue) fluorophore. Please click here to view a larger version of this figure.
Figure 3: Sequence-specific two-step fluorescence labeling of histone H3 with Set7/9, SeAdoYn and TAMRA azide. The protein MTase Set7/9 naturally methylates the amino group of lysine 4 in histone H3 (H3K4, green) using AdoMet. With the double-activated cofactor SeAdoYn the MTase transfers a small propargyl group (red) to the lysine residue. The attached terminal triple bond is then selectively modified in a bioorthogonal click reaction (copper-catalyzed azide-alkyne cycloaddition, CuAAC) with azide-derivatized TAMRA (tetramethylrhodamine, blue) fluorophore. Please click here to view a larger version of this figure.
Protocol
1. General Instructions
Store aziridine cofactor 6BAz (in DMSO) and protein MTase Set7/9 at -80 °C and all other reagents including double-activated cofactor SeAdoYn and DNA MTase M.BseCI (in 50% glycerol) at -20 °C.
Determine the concentration of 6BAz and SeAdoYn via UV/Vis spectroscopy using the extinction coefficients ε269nm (6BAz) = 16,000 cm-1 M-1 and ε260nm (SeAdoYn) = 15,400 cm-1 M-1 in deionized water. Determine the concentration of MTases by the Bradford assay or, if the extinction coefficient is available, via direct absorption at 280 nm.
Try to avoid creating bubbles by intensive pipetting or vortexing to prevent loss of enzyme activity. Instead, mix by gently pipetting up and down.
When adding aziridine cofactors from stock solutions in DMSO make sure that final DMSO concentration in the assay is less than 5%. Always include 10 mM magnesium ions in the assay buffer to prevent non-specific reactions with DNA.
When adding double activated cofactors from acidic stock solutions use small volumes (highly concentrated stock solutions) to avoid pH changes and make sure that the pH of the assay solution does not change significantly. Avoid thiols, e.g., β-mercaptoethanol or dithiothreitol (DTT), in the assay buffer because they can interfere with the click reaction by complexation of the required copper ions.
2. One-step Labeling of DNA via Aziridine Cofactors
- Sequence-specific Methyltransferase-Induced Label ing (SMILing) of plasmid DNA with M.BseCI DNA MTase and aziridine cofactor 6BAz.
- Thaw the cofactor solution at 20 °C and prepare the reaction mixtures on ice.
- In addition to the assay perform a “cofactor” control, to visualize any non specific modifications, and an “ enzyme” control, to make sure that the MTase preparation is free of the natural cofactor AdoMet.
- For the assay mix 2 µl of 10x modification buffer (containing 100 mM Tris-HCl, 100 mM MgCl2, 20 mM β-mercaptoethanol, pH 7.4), 2 µl of pBR322 (0.5 µg/µl), 10 eq. M.BseCI per recognition sequence on the DNA (1 recognition sequence in pBR322) and the aziridine cofactor 6BAz to a final concentration of 60 µM within a total volume of 20 µl. Add cofactor and DNA MTase last. NOTE: β-Mercaptoethanol is toxic, corrosive and environmentally damaging.
- For the “cofactor” control add deionized water instead of M.BseCI and for the “enzyme” control add deionized water instead of 6BAz.
- Mix the solutions by gently pipetting up and down.
- Incubate the tubes at 55 °C for 1 hr.
- Centrifuge briefly to collect all liquid at the bottom of the tubes.
- Restriction-modification assay to verify DNA modification.
- Prepare a solution by mixing 10 µl 10x R.TaqI buffer (containing 100 mM Tris-HCl, 50 mM MgCl2, 1 M NaCl, 1 mg/ml bovine serum albumin, pH 8.0), 80 µl deionized water and 3.3 µl of the restriction endonuclease (REase) from Thermus aquaticus (R.TaqI, 10 U/µl). Make sure to add the REase in the last step.
- To each tube from 2.1.7 add 2 µl of 10x R. TaqI buffer and 28 µl of the solution from above (2.2.1).
- Mix the solutions by gently pipetting up and down.
- Incubate the tubes at 65 °C for 30 min.
- Centrifuge briefly to collect all liquid at the bottom of the tubes.
- Electromobility shift assay (EMSA) with streptavidin to verify functional modification.
- Remove 25 µl from each tube (2.2.5) and add 2.4 µl of a streptavidin solution (1 mM with respect to streptavidin monomer in streptavidin buffer containing 100 mM Na2HPO4, 100 mM NaCl, pH 7.5; 4 equivalents of total biotin). Add 2.4 µl of streptavidin buffer to the remaining tubes.
- Incubate all tubes at 37 °C for 1 hr.
- Analysis via agarose gel electrophoresis.
- Add 5 µl of 6x loading buffer (0.25% bromophenol blue, 30% glycerol) to each tube.
- Mix the solutions gently.
- Load 10 µl of each sample into the wells of an agarose gel (1% agarose in 0.5x TBE buffer containing 1x GelRed from a 10,000x stock solution).
- Run the gel in 0.5x TBE buffer with 80 V for approx. 1 hr.
- Visualize DNA bands on a UV table (312 nm) with a CCD camera equipped with a filter (540 ± 50 nm). NOTE: UV light is damaging to eyes and skin.
3. Two-step Protein Labeling via Double Activated Cofactors
- Methyltransferase-Directed Transfer of Activated Groups (mTAG) with Set7/9 and double-activated cofactor SeAdoYn for histone H3 lysine 4 labeling (modification step).
- Thaw the components and prepare the reaction mixtures on ice. NOTE: Always keep SeAdoYn cooled to avoid degradation.
- In addition to the assay perform a “cofactor” control, to visualize any non specific modifications, and an “enzyme” control, to exclude non-specific reactions of the fluorescent probe.
- Prepare an assay solution (20 µl) containing modification buffer (50 mM Tris-HCl, 5% glycerol, pH 8.5), 10 µM histone H3, 10 µM Set7/9 and 600 µM SeAdoYn (mixture of both epimers at selenium). In the last steps add cofactor and then MTase.
- For the “cofactor” control prepare an assay solution as in 3.1.3 and add 60 mM AdoMet to compete with the synthetic cofactor. For the “enzyme” control add deionized water instead of SeAdoYn.
- Mix the solutions by slowly pipetting up and down. Check the pH by adding 1 µl of each solution to the upper field of a pH strip (pH range 5 - 10).
- Incubate at 37 °C for 2 hr.
- In the meanwhile prepare a 12% SDS polyacrylamide gel (running gel: 357 mM Bis-Tris pH 6.5-6.8, 0.1% (w/v) APS, 0.04% (v/v) TEMED and 12% acrylamide/bisacrylamide 37.5:1; loading gel: 357 mM Bis-Tris pH 6.5-6.8, 0.1% (w/v) APS, 0.04% (v/v) TEMED and 5% acrylamide/bisacrylamide 37.5:1). NOTE: Acrylamide/bisacrylamide is toxic and health hazardous. Wear gloves during this procedure.
- Chemical labeling of alkinylated lysine 4 in histone H3 via copper-catalyzed azide-alkyne cycloaddition (CuAAC) (labeling step).
- Just before the end of the modification reaction prepare a 5x click mix containing 3 mM CuSO4, 3 mM tris(3-hydroxypropyl-triazolylmethyl)amine (THPTA), 250 mM sodium ascorbate and 6 mM TAMRA azide with a total volume of 20 µl.
- Add 5 µl of the freshly prepared 5x click mix to each tube to start the CuAAC and quench the modification reaction.
- Mix gently by pipetting up and down.
- Protect all tubes with aluminum foil from light to avoid photo-bleaching of the fluorophore.
- Incubate at 20 °C for 1 hr.
- Protein precipitation to remove excess of free TAMRA fluorophore.
- To avoid outshining of the fluorescent labeled histone H3 by intensive in-gel fluorescence of free TAMRA fluorophore, remove excess fluorophore by precipitation of proteins (3.3.2 – 3.3.4)34.
- Add 75 µl methanol, 18.8 µl chloroform and 50 µl deionized water to each tube and vortex briefly after each addition. Centrifuge at 16,000 x g for 5 min. Remove the upper phase without disturbing the interface layer, which contains the protein.
- Add 56.3 µl methanol to the remaining phase in each tube, vortex and centrifuge at 16,000 x g for 5 min to pellet the protein. Remove the supernatant. Repeat this step to wash the pellet.
- Cover the open tubes with a lint free tissue and let them dry for 15 – 30 min.
- Analysis via SDS PAGE.
- Dissolve the precipitated proteins from 3.3.4 in 20 µl SDS loading buffer (50 mM Tris-HCl, 2.5% (w/v) SDS, 10% (v/v) glycerol, 320 mM β-mercaptoethanol and 0.05% (w/v) bromophenol blue, pH 6.8). Make sure to completely dissolve the pellet by rinsing the walls of the tubes with a pipette.
- Incubate the samples at 95 °C for 10 min and let them cool down to 20 °C.
- Centrifuge briefly to collect all liquid at the bottom of the tubes.
- Load the whole amount of each sample into the wells of an SDS polyacrylamide gel (3.1.7). Use 50 mM MOPS, 50 mM Tris-X (Tris-base), 5 mM EDTA, 0.1% (w/v) SDS as running buffer for electrophoresis.
- Run the gel with 120 V for approx. 90 min.
- Visualize the in-gel fluorescence on a UV table (312 nm) with a CCD camera equipped with a filter (540 nm ± 50 nm). NOTE: UV light is damaging to eyes and skin.
Representative Results
One-step Labeling of DNA via Aziridine Cofactors
This example reaction is carried out with the DNA MTase M.BseCI, which modifies the second adenine residue within the double-stranded 5’-ATCGAT-3’ sequence and has one recognition site on the pBR322 plasmid (Figure 4A). To test plasmid labeling, pBR322 is challenged with the restriction endonuclease (REase) R.TaqI (5‘-TCGA-3‘). R.TaqI has seven sites on pBR322, one of which is included in the M.BseCI site. If labeling occurs, the M.BseCI site will be protected against cleavage and a new fragment with 683 base pairs (bp) will form while two other fragments will disappear (315 and 368 bp). Of course, when analyzing DNA labeling with other DNA MTases different REases with corresponding recognition sequences should be employed.
The agarose gel in Figure 4B clearly shows the appearance of a new fragment with 683 bp (lane 3). This demonstrates that labeling of the M.BseCI site occurred. Only the assay (lane 3) shows this fragment. The “cofactor” control (lane 5) as well as the “enzyme” control (lane 7) are missing this band. Especially the “enzyme” control is important because it demonstrates that the natural cofactor AdoMet is not present in the enzyme preparation. Specificity of the labeling reaction is demonstrated by electromobility shift assay (EMSA) upon addition of streptavidin (Figure 4B and detail in 4C). Electromobility of the 683 bp is retarded while the mobility of all other fragments without M.BseCI site is unchanged compared to the controls (compare lane 4 with lanes 6 and 8).
 Figure 4: Sequence specific biotinylation of pBR322 plasmid DNA with M.BseCI and 6BAz. A. Plasmid map of pBR322 showing recognition sites for M.BseCI (red) and R.TaqI (green) as well as the length of expected DNA fragments in base pairs (bp) after restriction with R.TaqI. B. Analysis of DNA fragmentation and EMSA by agarose gel electrophoresis. M = Marker, lanes 1 and 2: plasmid DNA only, lanes 3 and 4: assay, lanes 5 and 6: 'cofactor' control, lanes 7 and 8: 'enzyme' control. Lanes 2, 4, 6 and 8 additionally contain streptavidin. C. Enlarged inset from B. Please click here to view a larger version of this figure.
Figure 4: Sequence specific biotinylation of pBR322 plasmid DNA with M.BseCI and 6BAz. A. Plasmid map of pBR322 showing recognition sites for M.BseCI (red) and R.TaqI (green) as well as the length of expected DNA fragments in base pairs (bp) after restriction with R.TaqI. B. Analysis of DNA fragmentation and EMSA by agarose gel electrophoresis. M = Marker, lanes 1 and 2: plasmid DNA only, lanes 3 and 4: assay, lanes 5 and 6: 'cofactor' control, lanes 7 and 8: 'enzyme' control. Lanes 2, 4, 6 and 8 additionally contain streptavidin. C. Enlarged inset from B. Please click here to view a larger version of this figure.
Two-step Labeling of Proteins via Double Activated Cofactors
As an example for two-step labeling of MTase substrates with double-activated AdoMet analogues we chose the histone H3 lysine 4 (H3K4) MTase Set7/9 and the small SeAdoYn cofactor. After enzymatic transfer of the activated propargyl group to the histone H3 substrate (first step), the terminal alkyne is chemically labeled with an azide-modified TAMRA fluorophore by CuAAC click chemistry (second step) (compare Figure 3). Analysis of the labeling reaction is done by SDS-PAGE and in-gel fluorescence detection (Figure 5A). The assay (lane 1) shows a fluorescent band corresponding to histone H3 indicating that the side chain of the cofactor was successfully transferred and the modified protein labeled with the TAMRA fluorophore. No bands are visible in the “cofactor” and “enzyme” controls (lanes 2 and 3) demonstrating that labeling of histone H3 is mediated by the MTase. Therefore, non-specific chemical labeling either by the cofactor alone (first step) or during CuAAC (second step) can be ruled out. The “cofactor” control is performed differently than in the protocol for one-step DNA labeling. This is because precipitation of histone H3 is more efficient in the presence of Set7/9 and omitting the protein MTase can lead to reduced amounts of histone H3 in the loading control. Thus, we added a high concentration of the natural cofactor AdoMet to compete with SeAdoYn. Loading of histone H3 can be easily controlled by staining the gel with Coomassie Blue after fluorescence detection.
MTase substrates can also be labeled with biotin instead of fluorophores by using azide-derivatized biotin in the second chemical step30. Biotinylation of proteins is conveniently analyzed by Western blotting with horseradish peroxidase-conjugated avidin. This is shown in Figure 5B for the biotinylation of histone H3 with Set7/9, SeAdoYn and azide-modified biotin. The assay in lane 1 shows a clear band for the labeled histone H3. The absence of bands in the “enzyme” (lane 2) and “cofactor” control (lane 3) demonstrates again that labeling is specific for the MTase. The “cofactor” control is simply performed in the absence of protein MTase because the Western blot analysis does not require removal of excess biotin by protein precipitation.
 Figure 5: Labeling of histone H3 with the protein MTase Set7/9 and SeAdoYn followed by click reactions with azide-derivatized labels. A. In-gel fluorescence of TAMRA-labeled histone H3 and controls as well as staining with Coomassie Blue for loading control. B. Western blot analysis of biotinylated histone H3 and controls using avidin-horseradish peroxidase (HRP) conjugate. Experimental conditions were very similar to those for fluorescence labeling in A (enzymatic modification: 6.5 µM histone H3, 10 µM Set7/9, 600 µM SeAdoYn, in 50 mM Tris-HCl, 5 mM MgCl2, 5% glycerol, pH 9.0, 30 °C, 3 hr; chemical labeling: 0.6 mM CuSO4, 0.6 mM THPTA, 50 mM sodium ascorbate, 1.2 mM biotin-azide, 37 °C, 1 hr). Please click here to view a larger version of this figure.
Figure 5: Labeling of histone H3 with the protein MTase Set7/9 and SeAdoYn followed by click reactions with azide-derivatized labels. A. In-gel fluorescence of TAMRA-labeled histone H3 and controls as well as staining with Coomassie Blue for loading control. B. Western blot analysis of biotinylated histone H3 and controls using avidin-horseradish peroxidase (HRP) conjugate. Experimental conditions were very similar to those for fluorescence labeling in A (enzymatic modification: 6.5 µM histone H3, 10 µM Set7/9, 600 µM SeAdoYn, in 50 mM Tris-HCl, 5 mM MgCl2, 5% glycerol, pH 9.0, 30 °C, 3 hr; chemical labeling: 0.6 mM CuSO4, 0.6 mM THPTA, 50 mM sodium ascorbate, 1.2 mM biotin-azide, 37 °C, 1 hr). Please click here to view a larger version of this figure.
Discussion
One-step labeling of DNA with DNA MTases and aziridine cofactors (SMILing DNA) is a robust method but some aspects should be considered when planning the experiment.
Aziridine cofactor: The 6BAz concentration for DNA labeling with M.BseCI was 60 µM. When using other DNA MTases the cofactor concentration should be optimized, e.g. concentrations as low as 20 µM have been employed with the DNA MTase M.TaqI19. Low 6BAz concentrations have the advantage that a fourfold excess of streptavidin (binding sites with respect to the whole amount of biotin in the assay) can be directly added after incubation with REase without interfering with analysis by EMSA. Otherwise the biotinylated DNA should be purified to remove excess cofactor and avoid smearing of the DNA during agarose gel electrophoresis. When adding aziridine cofactors from stock solutions in DMSO make sure that the final DMSO concentration in the assay is less than 5%. Too much DMSO can inactivate enzymes. Always store aziridine cofactors at -80 °C to avoid degradation.
DNA MTase: Enzymatic coupling of aziridine cofactors with DNA can lead to strong product inhibitors and the DNA MTases have to be used in stoichiometric amounts. Therefore always use more than 1 equivalent of DNA MTase. Most DNA MTases copurify with bound AdoMet which needs to be removed by extensive dialysis or washing the enzymes on a column with large volumes of buffer. The “enzyme” control will tell whether AdoMet is still present in the enzyme preparation. Labeling with M.BseCI can also be done by incubation over night at 37 °C.
Modification buffer: Include 10 mM magnesium ions in the buffer to prevent non-specific reactions of aziridine cofactors with DNA. If magnesium ions lead to activation of contaminating nucleases, barium ions might be added instead.
For the sequence specific labeling of DNA not only SMILing DNA but also mTAG can be employed. Here double activated cofactors are used for enzymatic transfer of side chains with unique functional groups to DNA. These functional groups, e.g. primary amines, can be modified with a wide variety of reporter groups, including biotin or fluorophores, by NHS ester chemistry in a second step8,35,36. Alternatively we have started to synthesize double activated AdoMet analogues with large side chains already containing the reporter group and used them for DNA labeling in one step.
The two-step mTAG labeling procedure for proteins with the double activated cofactor SeAdoYn has been successfully used with a number of protein MTases30,31. However, the following comments should be considered before performing the experiment.
Double activated cofactor: These cofactors, including SeAdoYn, are stored under acidic conditions and care must be taken that the pH of the modification solution does not significantly change upon cofactor addition. We always check the pH by adding 1 µl of the modification solution to a pH strip and, if the pH is too low, add small amounts of sodium hydroxide solution (50 mM) to adjust the pH. In addition cofactor analogues should be added in a low volume to avoid pH changes. Thus, minimal cofactor concentrations needed for full modification should be determined for other MTases and highly concentrated cofactor stock solutions are preferred. Cofactor concentrations are typically varied between 10 µM and 600 µM. The chemical synthesis of double-activated cofactors typically yields an approximately 1:1 mixture of epimers at sulfur or selenium and separation of the biologically active epimer, corresponding to the S-epimer of AdoMet, from the inactive epimer is often difficult to achieve. Thus, cofactor concentrations are typically given for a mixture of isomers. Double activated cofactor analogues are quite unstable at room temperature and should be stored at -20 °C. Also make sure to thaw the stock solutions on ice and keep them cold during pipetting. They can be frozen again but to ensure reproducible activity we recommend to make aliquots.
Protein MTase: For transformations with double activated cofactors the MTases can be used in catalytic amounts. However, MTases are often slow enzymes having turnover numbers in the min-1 range with the natural cofactor AdoMet. For practical reasons we often use the MTases in stoichiometric amounts and minimize incubation time and temperature (typically 10 min to 2 hr and 20 °C to 37 °C). The “cofactor” control varies, depending on the type of reporter group and detection. For biotin labeling the MTase is simply omitted and analysis can be done by Western blotting (Figure 5B). For fluorescence labeling with in-gel-fluorescence detection the free fluorophore should be removed from the labeled protein before electrophoresis. In the current protocol this is achieved by precipitation of histone H3 in the presence of Set7/9 MTase. However, without Set7/9 precipitation is incomplete. Therefore, the “cofactor” control is performed in the presence of protein MTase but a high concentration of the natural cofactor AdoMet is added to displace SeAdoYn from the active site and prevent enzymatic transfer of the propargyl group (Figure 5A). However, if precipitation of histone H3 leads to loss of protein, SDS-PAGE can be extended and excess of free TAMRA fluorophore will run out of the gel with the bromphenol blue front.
Modification buffer: The optimal enzyme buffer can be used, but thiols, like dithiothreitol (DTT) and β-mercaptoethanol, should be avoided. They bind to copper ions and block the following CuAAC click reaction.
CuAAC click reaction: The final concentration of TAMRA azide should be twice as high as the alkyne concentration. When using biological extracts the copper and ligand concentration should be increased up to 5 mM each. TAMRA azide is better soluble in DMSO than in water. Make sure that the final concentration of DMSO is less than 12%. Extended incubation times can lead to protein damage and smearing on the SDS polyacrylamide gel. Thus, the labeling reaction should be either stopped by protein precipitation or addition of thiols.
This two-step labeling procedure can also be used to label protein MTase substrates with biotin either for Western blot detection (Figure 5B) or isolation with streptavidin-coated magnetic beads. In addition, double activated AdoMet analogues can be used to label DNA, RNA and small natural products with corresponding MTases.
Compared to most other labeling methods for nucleic acids and proteins, MTase-mediated labeling has the advantage that native substrates can be directly used and no further modification is required. Of course, care must be taken that the natural substrates are not blocked by methylation through a corresponding MTase in vivo. In addition, MTase-mediated labeling is very flexible both in terms of reporter groups, which include biotin, fluorophores or other molecular tags, as well as targets for MTases. REBASE, a data base for restriction endonucleases and DNA MTases, lists more than 700 experimentally characterized DNA MTases with hundreds of different recognition sequences ranging from two to eight bp37 and it is expected that more than 200 different MTases of all types are produced in the human cell38. An important determinant for MTase activity is whether the AdoMet analogue fits into the enzyme active site. Although the presented cofactors 6BAz and SeAdoYn are designed to have small reactive groups, some MTases may not readily accept them. In these cases the active sites could be enlarged by protein engineering as has been demonstrated for DNA35, RNA23 and protein MTases25–28 with sterically more demanding double activated AdoMet analogues.
Sequence specific labeling of DNA and RNA with AdoMet analogues is of major interest for genotyping (DNA mapping)36 and methylation detection39, functional studies of DNA/RNA and DNA/RNA-modifying enzymes15 as well as for (nano)biotechnology13,14 and gene delivery19. Other methods for sequence specific covalent DNA labeling rely on synthetic triple helix-forming oligodeoxynucleotides, peptide nucleic acids or hairpin polyamides as targeting devices or on sequence specific nicking endonucleases (NEases) followed by nick translation labeling.1,2 However, the number of target sequences for triple helix formation or NEases (about 30 NEases are listed in REBASE37) are limited which makes DNA MTase-mediated labeling more general.
Specific labeling of proteins with double activated AdoMet analogues has been mainly directed towards applications in proteomic research, e.g. identification of new substrates for protein MTases in complex biological mixtures27,28, but other applications, like functional studies, should also be feasible. Although MTase-mediated labeling has been mainly performed with purified MTases in vitro, labeling can also be achieved in living cells as has been reported recently40. Of course, many other methods for specific protein labeling are available3,4. Besides incorporation of unnatural amino acids using engineered cells they typically require genetic fusions of the protein of interest with self-labeling tags/proteins or tags for enzyme-mediated labeling. In this respect short peptide sequences serving as substrates for protein MTases, e.g. N-terminal histone tails, could be fused to a protein of interest and specifically labeled with AdoMet analogues and corresponding protein MTases.
Finally, the biocatalytic repertoire of small molecule MTases can be expanded with synthetic AdoMet analogues41–44. This represents a new approach to introduce structural diversity into natural products, e.g. antibiotics or polyketides, which should find interesting applications in screening for novel biological activities.
Disclosures
The authors disclose the following competing financial interest: E.W. is inventor on related patents.
Acknowledgments
The authors thank Kerstin Glensk for preparing the MTases M.BseCI and Set7/9 and gratefully acknowledge funding by the Excellence Initiative of the German Federal and State Governments and RWTH Aachen University. The authors are happy to provide 6BAz and SeAdoYn or other cofactor analogues for collaborative research.
References
- Gottfried A, Weinhold E. Sequence-specific covalent labelling of DNA. Biochem. Soc. Trans. 2011;39:623–628. doi: 10.1042/BST0390623. [DOI] [PubMed] [Google Scholar]
- Zohar H, Muller SJ. Labeling DNA for single-molecule experiments: methods of labeling internal specific sequences on double-stranded DNA. Nanoscale. 2011;3:3027–3039. doi: 10.1039/c1nr10280j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinner MJ, Johnsson K. How to obtain labeled proteins and what to do with them. Curr. Opin. Biotechnol. 2010;21:766–776. doi: 10.1016/j.copbio.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Wua Y-W, Goody RS. Probing protein function by chemical modification. J. Pept. Sci. 2010;16:514–523. doi: 10.1002/psc.1287. [DOI] [PubMed] [Google Scholar]
- Struck A-W, Thompson ML, Wong LS, Micklefield J. S-Adenosyl-methionine-dependent methyltransferases: Highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem. 2012;13:2642–2655. doi: 10.1002/cbic.201200556. [DOI] [PubMed] [Google Scholar]
- Klimasauskas S, Weinhold E. A new tool for biotechnology: AdoMet-dependent methyltransferases. Trends Biotechnol. 2007;25:99–104. doi: 10.1016/j.tibtech.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Pljevaljcic G, Schmidt F, Weinhold E. Sequence-specific Methyltransferase-Induced Labeling of DNA (SMILing DNA) ChemBioChem. 2004;5:265–269. doi: 10.1002/cbic.200300739. [DOI] [PubMed] [Google Scholar]
- Lukinavicius G, Lapiene V, Stasevskij Z, Dalhoff C, Weinhold E, Klimasauskas S. Targeted labeling of DNA by methyltransferase-directed Transfer of Activated Groups (mTAG) J. Am. Chem. Soc. 1021;129:2758–2759. doi: 10.1021/ja0691876. [DOI] [PubMed] [Google Scholar]
- Pignot M, Siethoff C, Linscheid M, Weinhold E. Coupling of a nucleoside with DNA by a methyltransferase. Angew. Chem. Int. Ed. 1998;37:2888–2891. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2888::AID-ANIE2888>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Weller RL, Rajski SR. Design, synthesis, and preliminary biological evaluation of a DNA methyltransferase-directed alkylating agent. ChemBioChem. 2006;7:243–245. doi: 10.1002/cbic.200500362. [DOI] [PubMed] [Google Scholar]
- Du Y, Hendrick CE, Frye KS, Comstock LR. Fluorescent DNA Labeling by N-Mustard Analogues of S-adenosyl-l-methionine. ChemBioChem. 2012;13:2225–2233. doi: 10.1002/cbic.201200438. [DOI] [PubMed] [Google Scholar]
- Pljevaljcic G, Schmidt F, Scheidig AJ, Lurz R, Weinhold E. Quantitative labeling of long plasmid DNA with nanometer precision. ChemBioChem. 1002;8:1516–1519. doi: 10.1002/cbic.200700294. [DOI] [PubMed] [Google Scholar]
- Wilkinson S, et al. Molecular scale architecture: engineered three- and four-way junctions. Bioconjugate Chem. 2008;19:470–475. doi: 10.1021/bc700270k. [DOI] [PubMed] [Google Scholar]
- Braun G, et al. Enzyme-directed positioning of nanoparticles on large DNA templates. Bioconjugate Chem. 2008;19:476–479. doi: 10.1021/bc700275h. [DOI] [PubMed] [Google Scholar]
- Kim S, et al. Enzymatically incorporated genomic tags for optical mapping of DNA binding proteins. Chem. Int. Ed. 2012;51:3578–3581. doi: 10.1002/anie.201107714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rina M, Bouriotis V. Cloning purification and characterization of the BseCI DNA methyltransferase from Bacillus stearothermophilus. Gene. 1993;133:91–94. doi: 10.1016/0378-1119(93)90229-v. [DOI] [PubMed] [Google Scholar]
- Weinhold E, Meier T, Düfel H, Markert-Hahn C, Schmuck R, inventors. Sequence-specific detection of methylation in biomolecules. 8,129,106. US Patent. 2012
- Pljevaljcic G, Pignot M, Weinhold E. Design of a new fluorescent cofactor for DNA methyltransferases and sequence-specific labeling of DNA. J. Am. Chem. Soc. 2003;125:3492–3410. doi: 10.1021/ja021106s. [DOI] [PubMed] [Google Scholar]
- Schmidt FH-G, Hüben M, Gider B, Renault F, Teulade-Fichou M-P, Weinhold E. Sequence-specific Methyltransferase-Induced Labelling (SMILing) of plasmid DNA for studying cell transfection. Bioorg. Med. Chem. 2008;16:40–48. doi: 10.1016/j.bmc.2007.04.054. [DOI] [PubMed] [Google Scholar]
- Dalhoff C, Lukinavicius G, Klimasauskas S, Weinhold E. Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nat. Chem. Biol. 2006;2:31–32. doi: 10.1038/nchembio754. [DOI] [PubMed] [Google Scholar]
- Lukinavicius G, Tomkuviene M, Masevicius V, Klimasauskas S. Enhanced chemical stability of AdoMet analogues for improved methyltransferase-directed labeling of DNA. ACS Chem. Biol. 2013;8:1134–1139. doi: 10.1021/cb300669x. [DOI] [PubMed] [Google Scholar]
- Motorin Y, et al. Expanding the chemical scope of RNA:methyltransferases to site-specific alkynylation of RNA for click labeling. Nucleic Acids Res. 1943;39:1943–1952. doi: 10.1093/nar/gkq825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Holstein JM, Rentmeister A. A chemo-enzymatic approach for site-specific modification of the RNA cap. Angew. Chem. Int. Ed. 2013;52:7874–7878. doi: 10.1002/anie.201302874. [DOI] [PubMed] [Google Scholar]
- Peters W, et al. Enzymatic site-specific functionalization of protein methyltransferase substrates with alkynes for click labeling. Angew. Chem. Int. Ed. 2010;49:5170–5173. doi: 10.1002/anie.201001240. [DOI] [PubMed] [Google Scholar]
- Islam K, Zheng W, Yu H, Deng H, Luo M. Expanding cofactor repertoire of protein lysine methyltransferase for substrate labeling. ACS Chem. Biol. 2011;6:679–684. doi: 10.1021/cb2000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng W, Yu H, Deng H, Luo M. Labeling substrates of protein arginine methyltransferase with engineered enzymes and matched S-adenosyl-l-methionine analogues. J. Am. Chem. Soc. 2011;133:7648–7651. doi: 10.1021/ja2006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K, et al. Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-l-methionine. J. Am. Chem. Soc. 2012;134:5909–5915. doi: 10.1021/ja2118333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K, et al. Defining efficient enzyme-cofactor pairs for bioorthogonal profiling of protein methylation. Proc. Natl. Acad. Sci. U.S.A. 2013;110:16778–16783. doi: 10.1073/pnas.1216365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda O, Boyce M, Rush JS, Palaniappan KK, Bertozzi CR, Gozani O. A chemical method for labeling lysine methyltransferase substrates. ChemBioChem. 2011;12:330–334. doi: 10.1002/cbic.201000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willnow S, Martin M, Lüscher B, Weinhold E. A selenium-based click AdoMet analogue for versatile substrate labeling with wild-type protein methyltransferases. ChemBioChem. 2012;13:1167–1173. doi: 10.1002/cbic.201100781. [DOI] [PubMed] [Google Scholar]
- Bothwell IR, et al. Se-Adenosyl-l-selenomethionine cofactor analogue as a reporter of protein methylation. J. Am. Chem. Soc. 2012;134:14905–14912. doi: 10.1021/ja304782r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkuviene M, Clouet-d’Orval B, Cerniauskas I, Weinhold E, Klimasauskas S. Programmable sequence-specific click-labeling of RNA using archaeal box C/D RNP methyltransferases. Nucleic Acids Res. 2012;40:6765–6773. doi: 10.1093/nar/gks381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, et al. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PM, et al. Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins. J. Am. Chem. Soc. 2008;130 doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G, Lapinaite A, Urbanaviciute G, Gerasimaite R, Klimasauskas S. Engineering the DNA cytosine-5 methyltransferase reaction for sequence-specific labeling of DNA. Nucleic Acids Res. 2012;40:11594–11602. doi: 10.1093/nar/gks914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely RK, Dedecker P, Hotta J, Urbanaviciute G, Klimasauskas S, Hofkens J. DNA fluorocode: A single molecule, optical map of DNA with nanometre resolution. Chem. Sci. 2010;1:453–460. [Google Scholar]
- Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:234–236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrossian TC, Clarke SG. Uncovering the human methyltransferasome. Mol. Cell. Proteomics. 2011;10:1–12. doi: 10.1074/mcp.M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriukiene E, et al. DNA unmethylome profiling by covalent capture of CpG sites. Nat. Commun. 2013;4:2190. doi: 10.1038/ncomms3190. [DOI] [PubMed] [Google Scholar]
- Wang R, et al. Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. J. Am. Chem. Soc. 2013;135:1048–1056. doi: 10.1021/ja309412s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Weller RL, Thorson JS, Rajski SR. Natural product diversification using a non-natural cofactor analogue of S-adenosyl-l-methionine. J. Am. Chem. Soc. 2006;128:2760–2761. doi: 10.1021/ja056231t. [DOI] [PubMed] [Google Scholar]
- Stecher H, et al. Biocatalytic Fiedel-Crafts alkylation using non-natural cofactors. Angew. Chem. Int. Ed. 2009;48:9546–9548. doi: 10.1002/anie.200905095. [DOI] [PubMed] [Google Scholar]
- Lee BWK, Sun HG, Zang T, Kim BJ, Alfaro JF, Zhou ZS. Enzyme-catalyzed transfer of a ketone group from an S-adenosylmethionine analogue: A tool for the functional analysis of methyltransferases. J. Am. Chem. Soc. 2010;132:3642–3643. doi: 10.1021/ja908995p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter JM, et al. Expanding the structural diversity of polyketides by exploring the cofactor tolerance of an inline methyltransferase domain. Org. Lett. 2013;15:3774–3777. doi: 10.1021/ol401723h. [DOI] [PMC free article] [PubMed] [Google Scholar]