

Summary
Objective
Screening for specific coding mutations in the EFHC1 gene has been proposed as a means of assessing susceptibility to juvenile myoclonic epilepsy (JME). To clarify the role of these mutations, especially those reported to be highly penetrant, we sought to measure the frequency of exonic EFHC1 mutations across multiple population samples.
Methods
To find and test variants of large effect, we sequenced all EFHC1 exons in 23 JME and 23 non-JME idiopathic generalized epilepsy (IGE) Hispanic patients, and 60 matched controls. We also genotyped specific EFHC1 variants in IGE cases and controls from multiple ethnic backgrounds, including 17 African American IGE patients, with 24 matched controls, and 92 Caucasian JME patients with 103 matched controls. These variants are reported to be pathogenic, but are also found among unphenotyped individuals in public databases. All subjects were from the New York City metro area and all controls were required to have no family history of seizures.
Results
We found the reportedly pathogenic EFHC1 P77T-R221H (rs149055334-rs79761183) JME haplotype in one Hispanic control and in two African American controls. Public databases also show that the EFHC1 P77T-R221H JME haplotype is present in unphenotyped West African ancestry populations, and we show that it can be found at appreciable frequency in healthy individuals with no family history of epilepsy. We also found a novel splice-site mutation in a single Hispanic JME patient, the effect of which is unknown.
Significance
Our findings raise questions about the effect of reportedly pathogenic EFHC1 mutations on JME. One intriguing possibility is that some EFHC1 mutations may be pathogenic only when introduced into specific genetic backgrounds. By focusing on data from multiple populations, including the understudied Hispanic and Black/African American populations, our study highlights that for complex traits like JME, the body of evidence necessary to infer causality is high.
Keywords: Idiopathic epilepsy, Complex genetic disorders, Causative genetic variants, Disease prediction, Exonic variants, Ancestry-specific effects
It has been reported that coding mutations in the EFHC1 (for EF-hand containing) gene are the cause of perhaps up to 9% of all cases of juvenile myoclonic epilepsy (JME).1 The major evidence supporting this conclusion includes the following: (1) Hispanic and Dutch families with epilepsy show linkage to a locus that includes EFHC1,2–6 (2) highly penetrant EFHC1 coding mutations segregate with JME in families reporting Hispanic and Central American ancestry,7 (3) rare EFHC1 mutations have been reported to be both inherited and de novo in sporadic JME cases,8 (4) Efhc1-deficent mice exhibit spontaneous seizures,9 (5) EFHC1 regulates important neurodevelopmental events in cell culture.10,11 However, despite this evidence, notable contradictions still remain. For instance, in the study that initially proposed mutations in EFHC1 as a cause of JME,7 four separate coding mutations were found to segregate in 6 of the 30 JME families that showed linkage to the EFHC1-containing 6p12-p11 locus. Although these four mutations were not found in 382 Hispanic controls, neither they nor any other EFCH1 coding mutations were found segregating in the other 24 families in the study. Therefore, the six families were the only families in which these four EFHC1 mutations were found to segregate with JME. It is notable that when these six families were excluded from the analysis, evidence in favor of linkage remained.7 Although these observations support the hypothesis of a JME gene existing in the region, they cast some doubt on the importance of EFHC1 coding mutations for disease and/or on the role of EFHC1 in JME susceptibility. A subsequent study of Dutch JME patients also found linkage to the same region5 but failed to find any exonic mutations in EFHC1 associated with JME.12 Thus, in the majority of 6p12-p11-linked JME families from both Hispanic and Dutch populations, EFHC1 coding mutations do not segregate with disease. This means that other elements in the region, either within EFHC1 or in some other gene, must be responsible for the genetic signal. These elements might include regulatory factors affecting EFHC1. However, an additional possibility that cannot be ruled out is that EFHC1 is not the actual causative gene and that mutations in some unexamined nearby element functionally unrelated to EHFC1 are the cause of 6p12-p11-linked JME. One example that illustrates how causal mutations can be in a gene nearby a gene initially thought to be causal is in benign familial neonatal seizures (BFNS). Initially, given the observation of a nonsense mutation in CHRNA4, the gene encoding the neuronal nicotinic cholinergic receptor α4 subunit, segregating13 with BFNS in a linked region of chromosome 20q,14 this mutation was thought to be causal. At the time of its discovery, the evidence seemed convincing. However, it was ultimately shown that mutations in a nearby gene KCNQ2 were in fact the cause of 20q-linked BFNS.15,16 Thus, whether one considers CHRNA4 and BFNS or EFHC1 and JME, the existence of segregating mutations is not proof of causality, especially in the case of EFHC1, which also happens to have several coding mutations that are unrelated to epilepsy.7,17,18 (A fact often ignored in a cosegregation argument is that, in a linked region, all variants within the region, including those not in the disease gene itself, are more likely to cosegregate with the disease, by the definition of linkage analysis. This means that cosegregation alone cannot prove that a variant is the cause of the disease.) Importantly, if it is the case that another gene at 6p11-12 is causal for JME, the mutations are unlikely to be coding mutations, as mutation analysis of several genes in the region seems to rule out this possibility.18
More recently, exome-wide sequencing efforts have raised another issue regarding the role of EFHC1 in JME. Specifically, two of the four mutations initially reported to cause JME with high penetrance, and reported to co-segregate with JME,7 are variants found in individuals in the National Heart, Blood and Lung Institute Exome Variant Server (EVS: http://evs.gs.washington.edu/EVS) database.19 The EFHC1 F229L variant (rs137852776) is relatively rare (0.4%) and carriers in the EVS database could possibly be JME patients, since the sample is unphenotyped. However, the frequency of the other variant, the EFHC1 P77T-R221H haplotype (rs149055334-rs79761183), is found in >2% in the EVS data base among African Americans, a surprisingly high frequency for a haplotype reported to cause JME with roughly 80% penetrance. Without phenotypic data on these individuals, we cannot rule out the possibility that the EVS individuals with this haplotype are also JME patients, although a frequency of >2% significantly exceeds the prevalence of all forms of JME in any population. Therefore, an important question becomes, for example, “What does it mean to be an African American carrying the EFHC1 P77T-R221H allele and how does this impact individual susceptibility to epilepsy?”
The reported role of pathogenic EFHC1 variants in JME7,8 has been mentioned in reports weighing the benefits and harm of genetic testing in the epilepsies (e.g., Ottman et al.20), with the causative role of EFHC1 in JME listed as one of the most well-accepted findings. Ottman et al. note that being told that one carries a risk allele for epilepsy can lead to anxiety, stigmatization, and discrimination. Therefore, it is important to distinguish between “susceptibility” alleles and “causal” alleles. “Susceptibility” alleles interact with other genetic or environmental factors to increase the chances of developing disease, but they need not have any direct effect on the underlying disease mechanism and are not necessary for the disease to manifest. In contrast, the defining characteristic of “causal” alleles is their clear direct involvement in the disease mechanism with interactions between other genetic or environmental factors being of secondary importance in disease expression. If the putative EFHC1 disease alleles are in fact causative, as is the case for genes involved in several severe, monogenic forms of epilepsy (e.g., SCN1A in Dravet syndrome21), their interpretation in the clinic will be substantially different than if they are merely susceptibility alleles. Therefore, it is essential that the risk attributed to genetic variants not be overstated. Furthermore, understanding which category EFHC1 fits into is of importance in understanding the mechanisms underlying epilepsy susceptibility. This is especially important because, unlike the ion channel–encoding candidate genes, which have been studied extensively in epileptogenesis, EFHC1 appears to function in neural development and differentiation,22,23 a mechanism for epileptogenesis only recently being widely acknowledged.1,24
Because of the need to properly assess the effect of EFHC1 disease alleles in JME, we investigated the reportedly strong effect of EFHC1 coding mutations in Hispanic patients with epilepsy, where the effect is presumed to be particularly large. Our controls were carefully selected so as to have no family history of epilepsy (as opposed to the unphenotyped individuals in the EVS [http://evs.gs.washington.edu/EVS] and 1,000 Genomes databases [http://www.1000genomes.org]. We measured the frequency of the reportedly pathogenic EFHC1 P77T-R221H and F229L alleles in specific populations and used 1,000 Genomes data to supplement our findings. Our study does not find an excess of EFHC1 mutations in JME patients, but may suggest a possible ancestry-specific component governing susceptibility to EFHC1-based JME. Such possible interactions must be considered when assigning risk values to susceptibility alleles in JME and in all complex disorders, especially when considering such variants in applications such as designing genetic testing panels for assessing risk for a particular disease.
Materials and Methods
Study recruitment criteria
All of our JME patients were recruited by referral from epileptologists in the New York area. Diagnoses and the ethnicity of parents and grandparents were confirmed by direct interview or telephone interview. Ethnicity was determined by self-report, parental, and grand-parental information when available, and by the analysis of ancestry-informative markers.25 JME inclusion criteria required the presence of myoclonic jerks of the arms and shoulders occurring shortly after awakening, experienced with full consciousness and interictal bursts of a polyspike wave on electroencephalography (EEG) and no indication of focality. The onset of seizures ranged from 8 to 20 years of age; a family history of epilepsy was not required for recruitment. Furthermore, patients with myoclonic absence seizures, focal seizures, substance abuse, mental retardation, or suggestion of structural, metabolic, or degenerative disease were excluded from this study.26 Of the 46 Hispanic IGE patients (23 of whom were JME) in our genetic analyses, four (three of whom were JME) had a parent also diagnosed with IGE (two of which were familial JME). We recruited ethnically matched controls from the New York area, provided that they self-reported as having neither seizures nor a family history of seizure disorders. We also included 17 Black/African American IGE patients with 24 matched controls and 92 Caucasian IGE cases with 103 matched controls we recruited for measuring the frequency of EFHC1 P77T-R221H and F229L, as these match the populations where these variants are observed in EVS.
EFHC1 mutation analysis
Genomic DNA was extracted from saliva and sequenced with the primer pairs reported in previous studies.8,17 From the 46 Hispanic IGE cases, we amplified the 11 exons of EFHC1, their flanking exon/intron junctions, the promoter region, and the 5′ and 3′ untranslated regions (UTRs). Three of the EFHC1 exons and the EFHC1 promoter proved to have polymorphisms in those cases, so we sequenced those three polymorphism-containing exons and the promoter in 60 Hispanic controls. (Note that this approach of sequencing all the exons in cases and then genotyping the controls for only the specific variants found in cases is known to bias analyses toward finding association.27) In African American cases and controls, we screened for one particular variant that is a haplotype of two single-nucleotide polymorphisms (SNPs): rs149055334-rs79761183 (EFHC1 P77T-R221H). (Note that we refer to this two-allele haplotype as a single variant, as the minor alleles almost always occur together.) We also examined sequence data from the 1,000 Genomes Project for all populations, taken from http://www.1000genomes.org. The regions of interest included all EFHC1 exons, regulatory regions, and 39 ancestry-informative markers (AIMs) along chromosome 6. Validated variants, frequencies, and scores for predicted effect of amino acid change (if any) were obtained using ANNOVAR (annotation of variants, a software tool for annotation of genetic variants [http://wannovar.usc.edu]).28
Ancestry-informative markers (AIMs)
We used 39 AIMs along chromosome 6 to assess the proportion of European, African, and Native American ancestry in our Hispanic subjects. These AIMs cover EFHC1 and were chosen from a genome-wide list of 2,120 AIMs that showed the highest standard deviation of allele frequencies between Caucasians, Mesoamericans, South Americans, and Yorubans in Nigeria.25 Study participants were genotyped at these AIMs by Roswell Park Cancer Institute (Buffalo, NY, U.S.A.) using the MassARRAY platform from Sequenom (San Diego, CA, U.S.A.). We applied multidimensional scaling (MDS) (as implemented in PLINK29 (a software tool for genetic association analysis; [http://pngu.mgh.harvard.edu/˜purcell/plink/]) to the AIMs-related genotypes of 1,264 individuals: 116 Hispanics, 23 African Americans, 33 Caucasians (all ascertained in the New York area) and 1,092 individuals from the 1,000 Genomes Project data.
Results
We did not find any of the reportedly pathogenic EFHC1 mutations7,8 in our 23 JME or our 23 other-IGE Hispanic cases. The cases in our data set were found to have six different EFHC1 exonic variants; five of these six variants—rs116586919, rs142489544, rs3804506, rs3804505, and rs1266787 (Table1)—were found at equal frequencies in our 60 Hispanic controls, despite the sampling bias, which tends to increase the number of false positives (see Materials and Methods). The sixth variant is a novel mutation we found in a single Hispanic JME case, but was not observed in any other individual in our entire study and does not appear in any database we examined, including EVS, 1,000 Genomes, and the SNP database dbSNP (http://www.ncbi.nlm.nih.gov/SNP/). This is a splice-site mutation immediately 3′ to the exon/intron junction of exon 9 and transitions the completely conserved G to an A, suggesting it may alter function of the EFHC1 transcript. Nonetheless, without genotypes from family members, we can neither determine whether this variant is de novo or inherited, nor ascribe it any pathogenicity based on the available information.
Table 1.
All the mutations identified by sequencing the exons of EFHC1 in New York City Hispanic IGE patients
| Region | rsID | Nucleotide | AA change |
|---|---|---|---|
| 5′ Regulatory | rs116586919 | −520C>T | N/A |
| 5′ Regulatory | rs142489544 | −146_147delGC | N/A |
| Exon 3 | rs3804506 | 475C>T/G | R159W/G |
| Exon 3 | rs3804505 | 545G>A | R182H |
| Exon 8 | rs1266787 | 1343T>C | M448T |
| Splice-site | Novel | IVS9+1G>A | N/A |
All except the novel splice-site IVS9+1G>A mutation were found at equal frequencies in matched controls.
Given the known admixed composition of Hispanic genomes, we wanted to ensure that the genetic makeup of our New York City (NYC) Hispanic cases is indeed comparable with previous reports in the literature,7 which sampled mostly Mexicans from Los Angeles, CA. We used MDS to demonstrate the relatively short genetic distance that separates our Hispanics from the Los Angeles Mexican (MXL) individuals of 1,000 Genomes. To address the issue of power to detect significant differences in ancestry, we applied the same MDS technique to all Asian, African, and European individuals in 1,000 Genomes and demonstrated that the 39 AIMs do indeed provide sufficient information to distinguish different populations (see Fig. 2).
Figure 2.
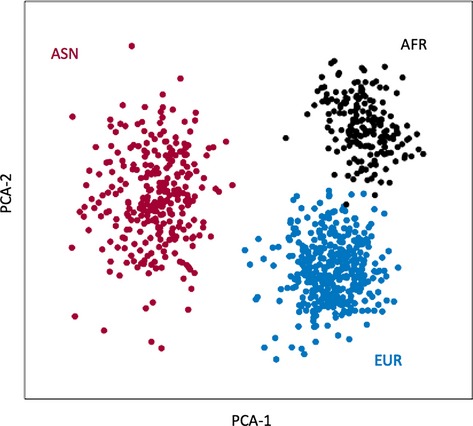
Multidimensional scaling (MDS) of three super groups: African, Asian, and European. The genotypes at 39 ancestry-informative markers (AIMs) along chromosome 6 were extracted from the 1,000 Genomes Project data. Based on the first two principal components, there is a clear separation between Africans (AFR: black), Asians (ASN: red), and Europeans (EUR: blue).
The EFHC1 F229L and P77T-R221H alleles have each been reported to be highly penetrant disease alleles with a relatively high prevalence among JME patients. However, these alleles can also be found in publicly available databases (e.g., EVS). In the 1,000 Genomes data, we found two individuals with the F229L allele, both from the Tuscan Italian (TSI; n = 92) population. Therefore, although the allele seems rare when examining databases collectively, the frequency increases significantly when considered from a population-specific perspective. We found similar and even more striking ethnicity-based enrichment of the P77T-R221H allele. In particular, this haplotype is found at a genotypic frequency of 8% in the 1,000 Genomes Nigerian Yoruba (YRI) population. It is also found in the African American (ASW) and Puerto Rico (PUR) populations, each known to have considerable West African ancestry. Because the occurrence of this allele greatly exceeds any estimate of JME prevalence, it is extremely unlikely that these unphenotyped carriers of reportedly pathogenic EFHC1 alleles are affected with JME. Furthermore, given the reported high penetrance of these alleles, it is also unlikely that these individuals are nonpenetrant carriers of a causal variant. This casts some doubt on the P77T-R221H allele being a highly penetrant cause of JME.
In contrast to 1,000 Genomes and EVS data, all of our subjects are phenotyped. So, we genotyped our samples of patients and controls for the EFHC1 P77T-R221H and F229L alleles as well. For EFCH1 P77T-R221, we genotyped our Hispanic controls (mentioned earlier), 17 Black/African American IGE patients from the New York City metro area, and 24 matched African American healthy controls. Even in this small sample, we found P77T-R221H in two African American controls, which is consistent with 1,000 Genomes frequency of 8% in YRI. This shows that this allele occurs at appreciable frequencies in unaffected individuals with West African ancestry. This allele was also found in one of our Hispanic controls who reported Caribbean ancestry, again suggesting an increased chance of West African ancestry. In addition to being healthy, our controls were required to have no family history of epilepsy. Furthermore, this allele was not found in our 17 African American epilepsy patients, which, given its frequency in African American controls, suggests that it does not influence JME susceptibility in individuals of West African ancestry. For the EFCH1 F229L, we genotyped our Hispanic controls (mentioned earlier), 92 Caucasian IGE patients from the NYC metro area, and 103 matched Caucasian controls. Although we found F229L in one of 92 Caucasian JME patients, this is hardly sufficient evidence to infer causality. But since F229L was found exclusively in the 1,000 Genomes TSI sample, it is interesting that this Caucasian patient happens to be of Mediterranean descent with a genetic background that most closely resembles the 1,000 Genomes TSI sample (data not shown).
Of note, neither the EFHC1 F229L nor the EFHC1 P77T-R221H allele is present in any 1,000 genomes individual in the MXL population. These two alleles were each found in two Hispanic JME families, accounting for four of the six families in which EFHC1 coding mutations reported to segregate with JME.7 That study did not observe these two mutations in 382 healthy Mexican controls from Los Angeles.7 That they are also absent in the 1,000 Genomes MXL population is consistent with these alleles not being common in individuals reporting Mexican ancestry.
Discussion
Coding mutations in the EFHC1 gene have been proposed as a major cause of JME, especially among individuals reporting Hispanic ancestry. Their penetrance is reported as high (˜80%)7 and their prevalence among JME patients has been estimated to be as high as 9%.1 Although our sample sizes were small, we have ample power to detect such large effects. Instead, we found that both our JME cases (and IGE cases) and our controls have about the same frequency of EFHC1 coding mutations. We did observe a single EFHC1 splice-site variant in a single JME case that may affect gene function, but simply identifying mutations predicted to affect function is not sufficient to imply causality, since many such mutations can be found in healthy individuals.30 Importantly, we show that the EFHC1 P77T-R221H allele, reported to cause JME with ˜80% penetrance does not cause JME in individuals of Black/African American ancestry as, in addition to being found, for example, 8% in the 1,000 genome YRI population, we find it in healthy controls with no family history of epilepsy. Therefore the effect of this allele in Mexicans in Los Angeles Hispanics does not apply to all ethnic groups.
The term “Hispanic” describes a heterogeneous group of ethnicities. One possible explanation for our inability to find pathogenic EFHC1 coding mutations with major effects in our sample might involve differences in ancestry between our New York Hispanic sample and Mexicans from Los Angeles, in which the originally reported mutations were found. Mexicans in Los Angeles are known to contain a substantial contribution of Native American ancestry,31,32 whereas the Hispanics from New York typically have a far greater contribution from Afro-Caribbean ancestry. However, an MDS analysis based on the ancestry informative markers we genotyped in our sample shows that our Hispanic cases and controls cocluster with the 1,000 Genomes Project MXL individuals (Fig.1). This same analysis also shows that the 39 AIMs of chromosome 6 are sufficient to identify clusters of major continental ancestry (Fig.2). Therefore, any differences in ancestry between our sample and Mexicans in Los Angeles are unlikely to explain the absence of pathogenic EFHC1 coding mutations in our Hispanic cases.
Figure 1.
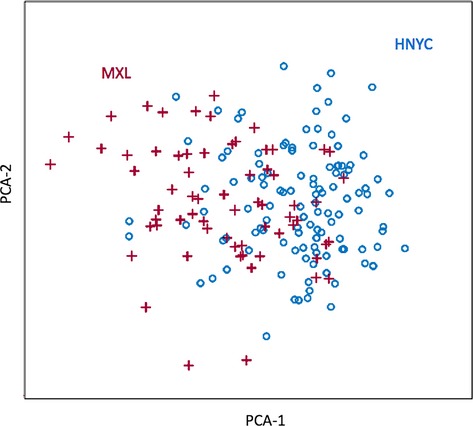
Multidimensional scaling (MDS) of our Hispanics from the New York Metro area (HNYC) and of the Mexicans (MXL) of the 1,000 Genomes Project data. The MDS is based on genotypes at 39 ancestry-informative markers (AIMs) of chromosome 6, and is equivalent to the first two principal components of variation. Clearly, there is considerable overlap between HNYC (blue) and MXL (red).
The importance of ethnicity in studying the genetics of epilepsy has received insufficient attention, perhaps because most of the epidemiology of epilepsy has been done in the Caucasian population.33 However, evidence suggests that ethnicity should be expected to play a critical role in determining which genes cause epilepsy in which populations. For example, the BRD2 locus, linked and associated with JME in Caucasians, showed evidence against linkage in non-Caucasians.26 Other JME loci have been identified from specific regions, such as southern India.34 We should expect different genetic backgrounds to harbor different genes that influence susceptibility to epilepsy. One possible and intriguing explanation for our observation that EFHC1 alleles common in non-Hispanic populations appear to be a cause of JME in the Mexican/Hispanic population could be that EFHC1 interacts with other elements of the genetic background in Hispanics to become pathogenic. This could explain why these mutations appear to be benign in several 1,000 Genomes Project populations but deleterious in a subpopulation of Hispanics and why they are absent in healthy Mexican controls. Evidence from human studies and animal models point to a prominent role for genetic background in seizure disorders through gene–gene interactions. For example, mutations in both the SCN1A and SCN2A genes are thought to be required for the expression of certain forms febrile seizures and Dravet syndrome–like phenotypes.35,36 In mice, the combination of specific mutations in the both Scn2a and Kcnq2 genes leads to more severe seizure phenotypes than either mutation individually.37 Conversely, such mutations can be compensatory. Background-specific gene–gene interactions can also exhibit protective effects. Mutations in mouse Scn8a that cause seizures can actually suppress the increased seizure susceptibility phenotype of Scn1a-deficient mice.38,39 That we find EFHC1 P77T-R221H haplotype to be as high as 8% in populations of West African ancestry is evidence that some EFHC1 coding mutations can exist in relatively benign states in some backgrounds (e.g., West Africans) but perhaps not in others (e.g., Hispanics in Los Angeles). Similarly, the EFHC1 F229L amino acid substitution can be found in Tuscan Italians (e.g., the 1,000 Genomes Project database), but like the P77T-R221H allele, F229L also segregated with JME in Hispanic JME pedigrees. Again this implies background-specific effects interacting with EFHC1 alleles in JME. Benign EFHC1 alleles that exist in two separate ethnicities (e.g., West African and Tuscan Italian) may increase susceptibility in a Mexican population from Los Angeles population. What must be considered is that the Mexican population is a group likely to have a large contribution from Native American/Mesoamerican ancestry. Because of the genetic background difference, it cannot be ruled out that the Mesoamerican population is more vulnerable to the effect of certain, perhaps deleterious, EFHC1 alleles.
In summary, the observation that (1) just as in the majority of 6p12-p11-linked JME families, we do not find pathogenic EFHC1 coding mutations associated with JME in our sample and (2) alleles originally reported to be causative for, and segregate with, JME in one population (Mexicans) are, in fact, common in other populations clearly demonstrates that, first, proving that a putative epilepsy gene is, in fact, the cause of a disease, even in families showing linkage to the region, can be a difficult task and, second, ethnicity must be taken into consideration in studies of epilepsy. We offer an updated view of the landscape of purportedly pathogenic EFHC1 coding mutations, taking distribution by ethnicity-based ancestry into account (Table2). Because we show that P77T-R221H exists in healthy individuals (perhaps depending on genetic background), the same possibility must also be considered for the F229L allele. Therefore, observations such as F229L segregating recessively with intractable epilepsy of infancy in a Moroccan Jewish family linked to 6p12-p1140 need to be carefully considered. Cell-based assays and studies of mouse Efhc1 demonstrate that that the gene is important in neuronal development and seizure susceptibility. Alteration of EFHC1 function is likely causative in some cases of JME, but given that the majority of linked families do not show mutations, that apparently benign variants are relatively common in other populations, and that no population level association of JME with markers in the region has been observed,17 the story is far from being understood. Not only may gene–gene interaction and genetic background play a role, but also noncoding variants or mutations in nearby loci may be behind most of the 6p12-p11-linked JME cases. Moreover, until the story is more completely understood, the prediction of epilepsy susceptibility in any given individual carrying reportedly pathogenic EFHC1 alleles must be handled with caution, as any overestimation of the effect of these alleles can negatively influence clinical decisions and patient care.
Table 2.
All purported pathogenic EFHC1 exonic mutations, the corresponding amino acid change, and the populations where the mutation has been seen
| EFHC1 allele | Mex | Hon | Jap | YRI | ASW | PUR | TSI | His | AA | Cau | His | AA | Cau |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JME (reported) | 1,000 Genomes | NYC patients | NYC controls | ||||||||||
| P77T | + | + | + | + | + | + | |||||||
| D210N | + | ||||||||||||
| R221H | + | + | + | + | + | + | |||||||
| F229L | + | + | + | ||||||||||
| D253Y | + | ||||||||||||
| T252K | + | ||||||||||||
| V264fsx280 | + | ||||||||||||
| Q277X | + | ||||||||||||
| T580R | + | ||||||||||||
His = New York Hispanic; AA = New York black/African American; Cau = New York Caucasian, all from this current study.
Acknowledgments
This work supported in part by grants NS27941, NS70323, and NS61829. We thank the reviewers for their insightful comments and suggestions.
Biography
Dr. Greenberg is a Professor of Neuroscience at Nationwide Children's Hospital and The Ohio State University.
Disclosure
The authors have no relevant conflicts of interest of financial disclosures to report. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- de Nijs L, Wolkoff N, Grisar T, et al. Juvenile myoclonic epilepsy as a possible neurodevelopmental disease: role of EFHC1 or Myoclonin1. Epilepsy Behav. 2013;28(Suppl. 1):S58–S60. doi: 10.1016/j.yebeh.2012.06.034. [DOI] [PubMed] [Google Scholar]
- Bai D, Alonso ME, Medina MT, et al. Juvenile myoclonic epilepsy: linkage to chromosome 6p12 in Mexico families. Am J Med Genet. 2002;113:268–274. doi: 10.1002/ajmg.10724. [DOI] [PubMed] [Google Scholar]
- Liu AW, Delgado-Escueta AV, Gee MN, et al. Juvenile myoclonic epilepsy in chromosome 6p12-p11: locus heterogeneity and recombinations. Am J Med Genet. 1996;63:438–446. doi: 10.1002/(SICI)1096-8628(19960614)63:3<438::AID-AJMG5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Liu AW, Delgado-Escueta AV, Serratosa JM, et al. Juvenile myoclonic epilepsy locus in chromosome 6p21.2-p11: linkage to convulsions and electroencephalography trait. Am J Hum Genet. 1995;57:368–381. [PMC free article] [PubMed] [Google Scholar]
- Pinto D, de Haan GJ, Janssen GA, et al. Evidence for linkage between juvenile myoclonic epilepsy-related idiopathic generalized epilepsy and 6p11-12 in Dutch families. Epilepsia. 2004;45:211–217. doi: 10.1111/j.0013-9580.2004.36003.x. [DOI] [PubMed] [Google Scholar]
- Serratosa JM, Delgado-Escueta AV, Medina MT, et al. Clinical and genetic analysis of a large pedigree with juvenile myoclonic epilepsy. Ann Neurol. 1996;39:187–195. doi: 10.1002/ana.410390208. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Delgado-Escueta AV, Aguan K, et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842–849. doi: 10.1038/ng1393. [DOI] [PubMed] [Google Scholar]
- Medina MT, Suzuki T, Alonso ME, et al. Novel mutations in Myoclonin1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology. 2008;70:2137–2144. doi: 10.1212/01.wnl.0000313149.73035.99. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Miyamoto H, Nakahari T, et al. Efhc1 deficiency causes spontaneous myoclonus and increased seizure susceptibility. Hum Mol Genet. 2009;18:1099–1109. doi: 10.1093/hmg/ddp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nijs L, Leon C, Nguyen L, et al. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat Neurosci. 2009;12:1266–1274. doi: 10.1038/nn.2390. [DOI] [PubMed] [Google Scholar]
- de Nijs L, Wolkoff N, Coumans B, et al. Mutations of EFHC1, linked to juvenile myoclonic epilepsy, disrupt radial and tangential migrations during brain development. Hum Mol Genet. 2012;21:5106–5117. doi: 10.1093/hmg/dds356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Louwaars S, Westland B, et al. Heterogeneity at the JME 6p11-12 locus: absence of mutations in the EFHC1 gene in linked Dutch families. Epilepsia. 2006;47:1743–1746. doi: 10.1111/j.1528-1167.2006.00676.x. [DOI] [PubMed] [Google Scholar]
- Beck C, Moulard B, Steinlein O, et al. A nonsense mutation in the alpha4 subunit of the nicotinic acetylcholine receptor (CHRNA4) cosegregates with 20q-linked benign neonatal familial convulsions (EBNI) Neurobiol Dis. 1994;1:95–99. doi: 10.1006/nbdi.1994.0012. [DOI] [PubMed] [Google Scholar]
- Stogmann E, Lichtner P, Baumgartner C, et al. Idiopathic generalized epilepsy phenotypes associated with different EFHC1 mutations. Neurology. 2006;67:2029–2031. doi: 10.1212/01.wnl.0000250254.67042.1b. [DOI] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Bai D, Bailey JN, Duron RM, et al. DNA variants in coding region of EFHC1: SNPs do not associate with juvenile myoclonic epilepsy. Epilepsia. 2009;50:1184–1190. doi: 10.1111/j.1528-1167.2008.01762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Delgado-Escueta AV, Alonso ME, et al. Mutation analyses of genes on 6p12-p11 in patients with juvenile myoclonic epilepsy. Neurosci Lett. 2006;405:126–131. doi: 10.1016/j.neulet.2006.06.038. [DOI] [PubMed] [Google Scholar]
- Cherepanova NS, Leslie E, Ferguson PJ, et al. Presence of epilepsy-associated variants in large exome databases. J Neurogenet. 2013;27:1–4. doi: 10.3109/01677063.2013.772176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman R, Hirose S, Jain S, et al. Genetic testing in the epilepsies–report of the ILAE Genetics Commission. Epilepsia. 2010;51:655–670. doi: 10.1111/j.1528-1167.2009.02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulley JC, Scheffer IE, Petrou S, et al. SCN1A mutations and epilepsy. Hum Mutat. 2005;25:535–542. doi: 10.1002/humu.20178. [DOI] [PubMed] [Google Scholar]
- Grisar T, Lakaye B, de Nijs L, et al. Myoclonin1/EFHC1 in cell division, neuroblast migration, synapse/dendrite formation in juvenile myoclonic epilepsy. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper's basic mechanisms of the epilepsies. 4th. Bethesda, MD: [Internet] Accessed November 13, 2014. [Google Scholar]
- Leon C, de Nijs L, Chanas G, et al. Distribution of EFHC1 or Myoclonin 1 in mouse neural structures. Epilepsy Res. 2010;88:196–207. doi: 10.1016/j.eplepsyres.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Subaran R. Blinders, phenotype, and fashionable genetic analysis: a critical examination of the current state of epilepsy genetic studies. Epilepsia. 2011;52:1–9. doi: 10.1111/j.1528-1167.2010.02904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Bigham AW, Mei R, et al. A genomewide admixture mapping panel for Hispanic/Latino populations. Am J Hum Genet. 2007;80:1171–1178. doi: 10.1086/518564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg DA, Durner M, Keddache M, et al. Reproducibility and complications in gene searches: linkage on chromosome 6, heterogeneity, association, and maternal inheritance in juvenile myoclonic epilepsy. Am J Hum Genet. 2000;66:508–516. doi: 10.1086/302763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Leal SM. Discovery of rare variants via sequencing: implications for the design of complex trait association studies. PLoS Genet. 2009;5:e1000481. doi: 10.1371/journal.pgen.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelak K, Shianna KV, Ge D, et al. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6:e1001111. doi: 10.1371/journal.pgen.1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NA, Coram MA, Shriver MD, et al. Ancestral components of admixed genomes in a Mexican cohort. PLoS Genet. 2011;7:e1002410. doi: 10.1371/journal.pgen.1002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Estrada A, Gignoux CR, Fernandez-Lopez JC, et al. Human genetics. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science. 2014;344:1280–1285. doi: 10.1126/science.1251688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser WA, Kurland LT. The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia. 1975;16:1–66. doi: 10.1111/j.1528-1157.1975.tb04721.x. [DOI] [PubMed] [Google Scholar]
- Ratnapriya R, Vijai J, Kadandale JS, et al. A locus for juvenile myoclonic epilepsy maps to 2q33-q36. Hum Genet. 2010;128:123–130. doi: 10.1007/s00439-010-0831-6. [DOI] [PubMed] [Google Scholar]
- Peiffer A, Thompson J, Charlier C, et al. A locus for febrile seizures (FEB3) maps to chromosome 2q23-24. Ann Neurol. 1999;46:671–678. doi: 10.1002/1531-8249(199910)46:4<671::aid-ana20>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Singh NA, Pappas C, Dahle EJ, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5:e1000649. doi: 10.1371/journal.pgen.1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JA, Yang Y, Beyer B, et al. Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Hum Mol Genet. 2006;15:1043–1048. doi: 10.1093/hmg/ddl019. [DOI] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, et al. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- Hawkins NA, Martin MS, Frankel WN, et al. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis. 2011;41:655–660. doi: 10.1016/j.nbd.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger I, Dor T, Halvardson J, et al. Intractable epilepsy of infancy due to homozygous mutation in the EFHC1 gene. Epilepsia. 2012;53:1436–1440. doi: 10.1111/j.1528-1167.2012.03536.x. [DOI] [PubMed] [Google Scholar]