

Abstract
Despite the fact that mitochondrial dysfunctions are increasingly recognized as key components in stress-related mental disorders, very little is known about the association between posttraumatic stress disorder (PTSD) and mitochondrial variants. To identify susceptibility mitochondrial genes for PTSD, we analyzed a total number of 978 mitochondrial single-nucleotide polymorphisms (mtSNPs) in a sample of 1238 individuals participating in the KORA (Cooperative Health Research in the Region of Augsburg) study. Participants were classified with ‘no PTSD', ‘partial PTSD' or ‘full PTSD' by applying the Posttraumatic Diagnostic Scale and the Impact of Event Scale. To assess PTSD–mtSNP association while taking heteroplasmy into account, we used the raw signal intensity values measured on the microarray and applied linear regression. Significant associations were obtained between full versus no PTSD and two mtSNPs; mt8414C→T (β=−0.954±0.06, Padjusted=0.037) located in adenosine triphosphate (ATP) synthase subunit 8 (MT-ATP8) and mt12501G→A (β=−1.782±0.40, Padjusted=0.015) located in the NADH dehydrogenase subunits 5 (MT-ND5). Heteroplasmy for the two variants towards a larger number of the respective minor alleles increases the risk of having PTSD. NADH dehydrogenase and ATP synthase are both linked to the regulation of reactive oxygen species. Our results highlight the important role of the mitochondrial genome among the factors that contribute to the risk of PTSD. Mitochondrial genetic variants may be more important than has previously been assumed, leading to further insights regarding effects of existing medications, or even to the development of innovative treatments. As this is the first mitochondrial genome-wide association study for PTSDs, further analyses are needed to follow up on the present findings.
Introduction
Posttraumatic stress disorder (PTSD) is a recognized psychiatric disorder that may develop after a person was exposed to one or more traumatic events, such as sexual assault, prolonged sexual abuse, witnessing violent deaths, military combat, being held hostage, terrorist attacks, natural disasters, serious injury or the threat of death. The diagnosis may be given when a group of symptoms such as intrusion, avoidance and startle continue for more than a month after the traumatic event.1 Despite being exposed to an extremely stressful life event, only a minority of trauma victims develop a sustained prolonged stress response syndrome. Lukaschek et al.2 recently showed that only 1.7% of all subjects who met the exposure event, subsequently developed a PTSD (with 8.8% developing partial PTSD). To date, it is not exactly clear why some people develop the condition and others do not. Risk of PTSD is likely to be influenced by characteristics of the trauma and of the individual, as well as genetic factors.3, 4 Association studies have implicated some genes to be directly associated with PTSD5, 6, 7, 8, 9 or to interact with childhood abuse to regulate PTSD risk,9, 10 or to affect risk for PTSD and related symptoms in interaction with other environmental factors.11, 12, 13 However, these studies have failed to identify conclusively a genetic variant that exerts a main effect on risk of PTSD.14 The unexplained portion of the genetic variance has been suggested to be related to telomere shortening,15 epigenetic modifications16 and mitochondrial dysfunctions.17, 18
Cells have a limited number of ways in which they can respond to threat. The cell danger response (CDR) is a cellular metabolic response regulated and coordinated in the brain that is activated when a cell encounters a chemical (mercury, cadmium, arsenic and nickel, as well as the plasticizer bisphenol A and pesticides), physical (heat, salt, pH shock, and UV or ionizing radiation) or microbial threat (virus, bacteria, fungi and parasites) that could injure or kill the cell.19 Psychological trauma can activate the CDR, produce chronic inflammation and increase the risk of many disorders.20 Mitochondria have evolved to sense all of these threats according to the induced changes in electron flow available for normal metabolism. Abnormal persistence of the CDR ultimately leads to organ impairment and behavior, resulting in chronic disease. PTSD is one of the diseases that may result from a pathological persistence of the CDR that occurs after the inciting trauma is gone.19
The primary function of mitochondria is to generate large quantities of energy in the form of adenosine triphosphate (ATP). Mitochondria contain their own DNA (mtDNA), which is ~16.6 kb and codes for 13 subunits of the mitochondrial respiratory chain complexes, two ribosomal genes and 22 transfer RNA genes that are required for mitochondrial protein synthesis. Mitochondria consume oxygen and substrates to generate the vast majority of ATP while producing reactive oxygen species (ROS, also called free radicals) in the process. An excess of ROS may damage DNA, proteins and lipids is not rapidly quenched. This damaged is termed oxidative stress. The 13 structural genes are essential for energy production through the process of oxidative phosphorylation (OXPHOS), a process performed by the electron transport chain, and generation of ATP as a source of chemical energy. OXPHOS consists of five enzyme complexes (I–V) and is the primary source of energy for aerobic cells. However, the mitochondrial electron transport chain is also the major source of ROS. Oxidative stress has been reported in many neurological and neurodegenerative disorders.21, 22, 23 In addition to supplying cellular energy and involvement in oxidative stress, mitochondria also participate in a wide range of other cellular processes, including signal transduction, cell cycle regulation, thermogenesis and apoptosis.
Functional brain imaging studies have demonstrated increased function in the amygdala and decreased activity in the hippocampus and prefrontal cortex (PFC) in PTSD patients24, 25, 26 and in animal models.27 Although the PFC, amygdala and hippocampus are the brain regions considered to be related to PTSD,24, 25 the underlying molecular mechanisms are unknown.26 One of the possibilities is that functional and structural changes in the brain may result from mitochondria-centered responses to repeated or chronic harmful stressors.17, 18 More generally, mitochondrial dysfunctions are increasingly recognized as key components in stress-related mental disorders.17, 18, 21, 22, 23, 26
Mitochondrial mutations can be both somatic and inherited through the maternal line.28 One peculiarity of mtDNA is the heteroplasmy effect, which was originally believed to be a rare phenomenon. As many mtDNA copies are present in a cell and because they have a high mutation rate, new mutations may arise among many of the other mtDNA; consequently mutant and wild-type mtDNA can co-exist.29 For this reason there is heterogeneity of mtDNA within an individual, and even within the same cell. The clinical expression of some phenotypes is determined by the relative proportion of wild-type and mutant mitochondrial genetic variants in different tissues. Mitochondria are highly susceptible to damage due to their finite DNA and protein repair mechanisms. Failing mitochondria contribute to CDR by inducing a bioenergetic deficit, oxidative stress and a proinflammatory state. Mitochondrial genes have been implicated in psychiatric disorders including schizophrenia,30 bipolar disorder,31 major depression32 and anxiety disorder.33
To date, mitochondrial genome-wide association studies for identifying mitochondrial genes underlying the pathogenesis in PTSD have not been documented. The purpose of the current study was to conduct a mitochondrial genome-wide association study to identify genetic variants influencing PTSD. In particular, we tested 976 mitochondrial single-nucleotide polymorphisms (mtSNPs) in a sample of 1238 individuals, aged 31–72 years.
Materials and methods
Study design and population
The KORA (Cooperative Health Research in the Region of Augsburg) study is a series of independent population-based epidemiological surveys and follow-up studies of participants living in the region of Augsburg in southern Germany, an area with demographic and socioeconomic characteristics roughly reflecting those of an average central European population. All the participants are residents of German nationality identified through the registration office and written informed consent was obtained from each participant.34
The study was approved by the local ethics committee. The study design, sampling method and data collection have been described in detail elsewhere.35 The present study includes data of the follow-up study KORA F4 (2006–2008) including a total number of 1238 unrelated individuals.
Sample size needed for detecting causal genetic variants in the mitochondrial genome
MacRae et al.36 compared the power for detecting causal genetic variants in the mitochondrial genome with that for detecting a locus in the nuclear genome given equal effect sizes. Although their analysis was done with respect to quantitative phenotypes, it should be equally valid for dichotomous phenotypes, that is, when using penetrance parameters instead of quantitative allelic effects. The result of the comparison depends on the number of tests that were performed in both scenarios and also on the variance explained by the causal genetic variant. The number of tests is significantly reduced for mtDNA because it is so much shorter than the nuclear genome. For a fixed amount of phenotypic variance explained by the causal locus and a given power, this has the effect that the sample size required for a mitochondrial association scan is roughly half the size required for an association scan on the nuclear genome. It could be argued that effect size at the mitochondrial locus should be compared with the additive effect of one allele at a locus in the nuclear genome (a) or to the difference between the two homozygous genotypes of the nuclear locus (2a). If the effect size for the nuclear locus is defined as the additive effect size, then the genetic variance explained by the autosomal locus is twice that explained by the mitochondrial locus with the same allele frequency and effect size. However, if the mitochondrial effect size is equated to the difference between the two homozygous genotypes, then the variance explained by the autosomal locus is only half of that explained by the mitochondrial locus. To sum up, sample sizes are related as follows: under the first effect size definition, the sample size required for the mitochondrial genome scan is roughly the same as that needed for an autosomal genome scan, whereas under the second definition, a mitochondrial genome scan requires only one-fourth the sample size needed by an autosomal genome scan.
PTSD phenotype
According to ICD-10,37 the first criterion for a diagnosis of PTSD requires that an individual be exposed to a traumatic event (criterion A). To assess criterion A in KORA F4, the Posttraumatic Diagnostic Scale38 was given to the participants as well as an open question about other traumatic events. Core PTSD symptoms were then classified on the basis of the re-experiencing of the trauma in intrusive memories, flashbacks or nightmares (criterion B), avoidance of activities and situations reminiscent of the trauma (criterion C) and a state of hyperarousal with hypervigilance, an enhanced startle reaction, and insomnia (criterion D). Subjects who met criteria A–D were counted as ‘full PTSD' cases. Subjects who met criterion A and any one or two of the criteria B–D were counted as ‘partial PTSD' cases.2 Subjects who met criterion A but having no symptoms were defined as ‘no PTSD' subjects.
Genotyping
DNA was extracted from full blood after the blood draw and then stored at −80 °C. Only SNPs located in the mitochondrial genome (mtSNPs) were considered in this study. Genotyping was performed using the following platforms: Affymetrix 6.0 GeneChip array (Santa Clara, CA, USA; 465 mtSNPs), Affymetrix Axiom chip array (252 mtSNPs), Illumina Human Exome Beadchip array (San Diego, CA, USA; 226 mtSNPs) and Illumina MetaboChip 200K (135 mtSNPs). Some of the 1238 genotyped individuals were present on more than one chip. Some of them (601) were included on all chips. An overview of the genotyping data and the study population is given in Table 1. Most of the covered mtSNPs have distinct positions identified by different chips. Although the Affymetrix 6.0 is the one containing the largest number of mtSNPs, some regions are not well covered. The Illumina Metabochip contains the smallest number of mtSNPs and many regions are uncovered, especially the hypervariable regions (HVR I and HVR II) as well as the CO1 and CO2 genes. However, when all chips are considered together, good overall coverage of the mitochondrial genome is obtained.39 Genotype calling algorithms may be controversial when applied to mtSNPs due to the heteroplasmy effect. The mtDNA tend to be heterogenous in the sense that different mitochondria of an individual may have different genotypes, such that genotype at an mtSNP may not be restricted to zero, one or two minor alleles. This issue affects the possibility of estimating genotypes and makes calling algorithms useless. Therefore, whenever one intends to identify susceptibility genes located in the mtDNA, we recommend accounting for heteroplasmy using individual-level allele frequencies obtained from the intensity values39 or sequencing data rather than genotypes calls obtained by algorithms that were designed for nuclear SNPs.
Table 1. Distribution of characteristics of the study population by PTSD status.
| Total | No PTSD | Partial PTSD | Full PTSD | |
|---|---|---|---|---|
| Sample size | 1238 | 875 (70,7%) | 312 (25.2%) | 51 (4.1%) |
| Males (%) | 616 (49.8%) | 472 (53.9%) | 128 (41.0%) | 16 (31.4%) |
| Mean age males | 52.9 | 52.3 | 54.2 | 58.6 |
| Mean age females | 51.2 | 50.8 | 51.8 | 52.3 |
| Affy. 6.0 (465 mtSNPs) | 739 | 502 | 203 | 34 |
| Affy. Axiom (252 mtSNPs) | 1185 | 834 | 300 | 51 |
| Illum. Exome (226 mtSNPs) | 1177 | 828 | 298 | 51 |
| Illum. Metabo (135 mtSNPs) | 1216 | 855 | 310 | 51 |
Abbreviations: Affy., Affymetrix; Illum., Illumina; mtSNP, mitochondrial single-nucleotide polymorphism; PTSD, posttraumatic stress disorder.
Quality control
Quality control was performed for each genotyping chip as described
elsewhere.39 An attempt to
remove the chip-specific global background intensity was made by computing,
separately for each individual, the 5% quantile intensity and
subtracting it from all intensities. In a second step, the intensities were
quantile normalized applying the method proposed by Bolstad et
al.40 and implemented
in the limma R package.41 After
quantile normalization, log2 intensity ratios,  , were computed for each
individual and an iterative outlier detection procedure was
applied.39 A summary of
the quality control results is given in Table 2.
From the original number of mtSNPs, 63 (5.8%) were removed because
their position could not be placed in Build 38. For the Axiom chip, 39
mtSNPs (18%) were removed due to an upper bound cutoff that has been
described in detail in our previous paper.39 A total number of 446 (<0.05%)
intensity ratios were considered to be outliers and removed from the
analysis.
, were computed for each
individual and an iterative outlier detection procedure was
applied.39 A summary of
the quality control results is given in Table 2.
From the original number of mtSNPs, 63 (5.8%) were removed because
their position could not be placed in Build 38. For the Axiom chip, 39
mtSNPs (18%) were removed due to an upper bound cutoff that has been
described in detail in our previous paper.39 A total number of 446 (<0.05%)
intensity ratios were considered to be outliers and removed from the
analysis.
Table 2. Summary of the quality control.
| Chip | mtSNPs |
mtSNPs excluded
|
Sample size | ISNP | Itot | Intensity ratio outliers | |
|---|---|---|---|---|---|---|---|
| UB | no_B38 | ||||||
| Affy. 6.0 | 411 | 0 | 54 | 739 | 3 | 1 822 374 | 194 (<0.05%) |
| Affy. Axiom | 213 | 39 | 0 | 1185 | 4 | 2 019 240 | 33 (<0.05%) |
| Illum. Exome | 226 | 0 | 0 | 1177 | 1 | 5 32 004 | 146 (<0.05%) |
| Illum. Metabo | 126 | 0 | 9 | 1216 | 1 | 306 432 | 73 (<0.05%) |
The number of mitochondrial single-nucleotide polymorphisms (mtSNPs) refers to the SNPs that passed quality control. Several mtSNPs were excluded due to the upper bound cutoff (UB)39 or because the basepair position was not available in Build 38 (no_B38). Sample size is based on the particular chip. One person may be present on more than one chip. ISNP stands for the number of intensity measures per allele. Itot represents the total number of intensity measures in the sample (ISNP × 2 × sample size × mtSNPs). Each mtSNP contains two intensities (allele A and allele B) and one intensity ratio (A/B). For example, for the Illumina (Illum.) Metabo we have 1216 individuals resulting in 2432 intensities (1216 ratios) per mtSNP. For all 126 mtSNPs (306 432 intensities and 153 216 ratios), only 73 intensity ratios were detected as outliers corresponding to 0.04%. Only the outlier (intensity ratio for that particular person) is removed and the mtSNP remains in the analysis. Affy., Affymetrix.
Statistical methods
To approach the heteroplasmy present in the mitochondria, we used the raw
signals of luminous intensity, where every measurement is associated with a
specific mtSNP and represents one of its alleles.42, 43 The number of
measures n per mtSNP depends on the vendor-specific technology used
on the genotyping chip. At the very least, there have to be two signals, one
for each of the two alleles. Often, however, the chip design includes more
than one measurement per SNP and allele. That is, for every individual and
SNP, we have intensity measurements (A1, B1),...,
(An, Bn) with n⩾1, where
Ai and Bi represent the intensities of the two
alleles A and B. The best way to assess the association of PTSD with the
mtSNP intensities is to apply linear regression analysis using PTSD (no,
partial, full PTSD) as independent variable with no PTSD as reference
category and mtSNP as outcome variable. The mtSNP enters the model as
outcome via the log2-transformed intensity ratio, 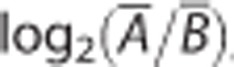 , where
, where  and
and  denote the mean
intensity, or single measure in case of n=1, for the A
allele and B allele, respectively. The quantitative covariate age at
examination was centered, to improve the convergence properties of the model
estimates. Sex is also introduced in the model as a covariate. Each type of
genotyping chip needs to be analyzed separately because different chips make
use of different technologies, even between chips of the same manufacturer.
P-values are obtained from a Wald test, which is based on the
asymptotic normality of the regression coefficient estimates, and corrected
for multiple comparisons applying Bonferroni with the correction factor
being equal to the number of mtSNPs used in the analysis multiplied for the
two outcomes. The two Affymetrix chips, Affy 6.0 and Axiom, share 170
positions and Illumina chips, Exome and Metabochip share 44 positions. Only
nine positions are common to all the four chips. Overlapping mtSNPs were
analyzed separately for each respective genotyping chip, giving in this way,
the opportunity of validation in the case of significant results. All the
analyses were performed with the statistical software R.44
denote the mean
intensity, or single measure in case of n=1, for the A
allele and B allele, respectively. The quantitative covariate age at
examination was centered, to improve the convergence properties of the model
estimates. Sex is also introduced in the model as a covariate. Each type of
genotyping chip needs to be analyzed separately because different chips make
use of different technologies, even between chips of the same manufacturer.
P-values are obtained from a Wald test, which is based on the
asymptotic normality of the regression coefficient estimates, and corrected
for multiple comparisons applying Bonferroni with the correction factor
being equal to the number of mtSNPs used in the analysis multiplied for the
two outcomes. The two Affymetrix chips, Affy 6.0 and Axiom, share 170
positions and Illumina chips, Exome and Metabochip share 44 positions. Only
nine positions are common to all the four chips. Overlapping mtSNPs were
analyzed separately for each respective genotyping chip, giving in this way,
the opportunity of validation in the case of significant results. All the
analyses were performed with the statistical software R.44
Results
We performed mitochondrial genome-wide association analysis separately for each genotyping chip and corresponding sample (see Table 2). After quality control, a total number of 976 mtSNPs were included in the analysis. The resulting P-values are plotted in Figure 1 for each genotyping chip. The x axis represents the mitochondrial genome, showing the position and relative size of each of the 13 major mitochondrial genes. A mtSNP was considered significant when the adjusted P-value resulted to be <0.05 after Bonferroni correction.
Figure 1.

Mitochondrial genome-wide P-values. On the y axis, P-values of the regression coefficient for full PTSD transformed into the negative of the base 10 logarithm, −log10 (P-value), are shown. The x axis represents the mitochondrial genome, displaying the position and relative size of each of the 13 major mitochondrial genes, 12S and 16S rRNAs, hypervariable region 1 (HVR I), hypervariable region 2 (HVR II), as well as the position of the 22 tRNAs (gray). PTSD, posttraumatic stress disorder; rRNA, ribosomal RNA; tRNA, transfer RNA.
Two significant mtSNPs were found to be associated with full PTSD (mt8414C→T and mt12501G→A). No mtSNPs were significant for partial PTSD, after adjustment for multiple testing (results not shown). The mt8414 is included on the Illumina Exome chip and mt12501 on the Affymetrix 6.0. No significant mtSNPs were observed when analyzing the Axiom and Metabochip. The most significant variant, mt12501G→A, associated with PTSD (β=−1.78±0.40, Pnominal=7.4 × 10−6, Padjusted=0.015) is located in the NADH dehydrogenase (complex I) subunits 5 (MT-ND5). The other variant mt8414C→T (β=−0.954±0.06, Pnominal=1.9 × 10−5, Padjusted=0.037), is located in ATP synthase (complex V) subunit 8 (MT-ATP8). More details about the estimated model parameters for each significant mtSNP are given in Table 3. A negative parameter estimate (β<0) indicates that the risk allele is the minor allele, whereas β>0 indicates that the risk allele is the major allele.
Table 3. Significant results of the association analysis between PTSD and mtSNPs by linear regression.
| mt8414C→T | mt12501G→A | |
|---|---|---|
| Chip | Illumina Exome | Affymetrix 6.0 |
| rs_name | rs28358884 | rs28397767 |
| Position (bp) | 8 414 | 12 501 |
| Alleles | C→T | G→A |
| Minor allele | T | A |
| Functional consequence | Missense | Synonymous |
| Protein | ATP8 | ND5 |
| Gene | MT-ATP8; subunit of ATP synthase, complex V | MT-ND5; subunit of NADH dehydrogenase, complex I |
| β 0 | 0.011±0.02, P=0.65 | 0.1671±0.123, P=0.17 |
| β part_PTSD | 0.041±0.03, P=0.20 | −0.117±0.1857, P=0.51 |
| β full_PTSD | −0.954±0.06, P=1.9 × 10−5 | −1.782±0.395, P=7.4 × 10−6 |
| β sex | −0.017±0.03, P=0.52 | −0.108±0.165, P=0.51 |
| β age | −0.018±0.001, P=0.18 | 0.011±0.011, P=0.313 |
Abbreviations: ATP, adenosine triphosphate; mtSNP, mitochondrial single-nucleotide polymorphism; PTSD, posttraumatic stress disorder.
Genomic position in base pairs (bp), alleles, rs_number and point
mutation are based on the Mitomap (http://mitomap.org) GRCh38 human genome assembly
(rCRS, GeneBank ID J01415.2). An estimated effect size
(βSNP) <0 indicates that the risk
allele is the minor allele. Nominal P-values are provided
for each β. The response variable is  with B
being the minor allele. Covariate sex baseline: male.
with B
being the minor allele. Covariate sex baseline: male.
These findings indicate that participants with full PTSD had a significantly lower ratio of C/T alleles for the mt8414 locus than participants with no PTSD, that is, a man affected with full PTSD is expected to have more T alleles than a man with no PTSD, making the T allele a risk factor of developing PTSD. Likewise at the mt12501 locus, a low ratio of G/A alleles is a risk factor for developing PTSD making the G allele protective against PTSD. In other words, a larger number of the respective minor allele for the mt8414 and mt12501 loci increases the risk for developing PTSD. The effect estimate for sex and age (βsex and βage) were not significant in our model, indicating no differences between age, women and men in the ratio of alleles at these two variants.
Discussion
It is well known that mitochondria strongly affect many intracellular processes coupled to signal transduction, neuron survival and plasticity. It has been suggested that impaired mitochondrial function may manifest itself in various ways, and may be related to many psychiatric and neurodegenerative diseases (including bipolar disorder, major depressive disorder, schizophrenia, autism, PTSD and anxiety).17, 18, 21, 22, 23, 26, 32, 45, 46 In the current study, in which we examined associations for partial and full PTSD and mtSNPs, we identified two mitochondrial variants, mt8414C→T located in the ATP synthase subunit 8 (MT-ATP8) and mt12501G→A located in NADH subunit 5 (MT-ND5), that were significantly associated with full PTSD after adjusting for multiple testing. Although mtDNA is transmitted from mother to offspring unchanged (lack of recombination), due to the higher mutation rate in mtDNA compared with nuclear DNA, many mutations have occurred only recently rather than many generations ago, and even in the current generation. For that reason, pairwise LD is reduced, and when one mtSNP is significant it is not expected that the neighboring mtSNPs are also significant.
MT-ATP8mt8414C→T
Mitochondrially encoded ATP synthase, complex V, is an important enzyme that provides energy to be used by the cell through the synthesis of ATP. In mammals, it produces most of the cellular ATP. Alteration of ATP synthase biogenesis may cause two types of isolated defects: a qualitative effect when the enzyme is structurally modified and does not function properly and a quantitative effect when it is present in abnormal amounts. In both cases, the cellular energy provision is impaired, which leads to a dysregulation of ROS production by the mitochondrial respiratory chain.47 The mt-ATP synthase subunits 6 and 8 variations have been suggested in spinocerebellar ataxias.48 To date, no association between ATP synthase and PTSD has been reported.
MT-ND5mt12501G→A
Mitochondrially encoded NADH dehydrogenase subunit, complex I, is the first enzyme in the mitochondrial electron transport chain. It extracts energy from NADH, produced by the oxidation of sugars and fats, and traps the energy in a potential difference or voltage across the mitochondrial inner membrane. The potential difference is used to power the synthesis of ATP. Because complex I is central to energy production in the cell, its malfunction may result in a wide range of metabolic disorders. Some of them are due to mutations in the mitochondrial genome, while others, which result from a decrease in the activity of complex I, or an increase in the production of ROS, are not yet well understood. Increased ROS production may increase the spectrum of somatic mutations produced by oxidative damage. Thus, brain regions that are involved in dopamine metabolism, such as the PFC and the caudate nucleus may be particularly vulnerable to oxidative damage. Impairment of complex I has been associated with the regulation of proteins in the PFC of patients with bipolar disorder.49 PTSD has been associated with decreased activity in the dorsolateral PFC, the brain region that regulates working memory and preparation and selection of fear responses.26
In the models for the two significant mtSNPs identified in this study, no significant differences were found in the allele ratios between men and women. Despite the fact that sex differences in the incidence of heteroplasmy have been observed in mussels50 and plants,51 to date, no differences have been reported in humans.52, 53 Nevertheless, the variant MT-ATP8mt8414C→T is a missense mutation which leads to an amino acid change, thus being a non-synonymous variant. So, individuals with an excess of missense mutations may carry an appreciable fraction of an altered protein that is responsible for developing PTSD. The other variant MT-ND5mt12501G→A is synonymous, that is, it codes for the same amino acid. How an excess of synonymous mutations at this locus could impact PTSD needs further investigation, since the single-nucleotide change leads to an unchanged protein. However different codons might lead to different protein expression levels.
The possible role of mitochondria in the development of PTSD might be concerned with a massive ROS overload, which may have two effects. First, excessive ROS concentrations increase the spectrum of somatic mutations produced by oxidative damage. Thus, brain regions may be particularly vulnerable to oxidative damage due to its high demand for oxygen, and its abundance of highly peroxidisable substrates.54 Second, ATP synthase activity may cause depletion of mitochondrial ATP levels and significant stimulation of ROS production, followed by depolarization of mitochondrial membrane potential.55 The loss of mitochondrial membrane potential leads to release of cytochrome c provoking upregulation of proinflammatory cytokines.56
The significant mtSNPs found to be associated with PTSD in this study, are located in ATP synthase and NADH dehydrogenase. Both complexes are involved in the regulation of mitochondrial ROS. However, the complexity of mitochondrial ROS metabolism suggests that interventions such as the administration of one or a few antioxidants may be too simplistic. A more complete approach to antioxidant therapy might be to decrease ROS generation (for example, by expressing uncoupling proteins) and to upregulate the multilayered endogenous mitochondrial and intracellular antioxidant defense network.22 However, this will require a considerably better understanding of ROS biology than we have at present. Although a wide range of pharmacologically distinct antidepressants and mood stabilizers are available, the molecular mechanisms of their therapeutic effects have not yet been sufficiently clarified.
The major advantage of the present study is the unselected access to a large-scale population-based study. However, longitudinal analysis taking subjects without PTSD at baseline remains a challenge (among others due to low incidence rates of PTSD after experiencing a traumatic event2), because most of the people who experience a traumatic event will not develop PTSD.57 No data are available on the exposure time of suffering from the consequences of a traumatic event. The distinct response pattern of these two mtSNPs in partial and full PTSD cases also confirms the clinical relevance of distinguishing the subdivision of PTSD.58, 59
In summary, our study detected two novel variants located in the MT-ATP8 and MT-ND5 genes that are associated with full PTSD. Heteroplasmy in these variants toward a larger number of the respective minor allele increases the risk of developing PTSD. Although further analyses are needed to follow up on the present findings, our results highlight the important role of the mitochondrial genome among the factors that contribute to the risk of PTSD and suggest that mitochondrial genetic variants may be more important than has previously been assumed. Focusing on mitochondrial variants may lead to further insights regarding effects of existing medications, or even to the development of innovative treatments.
Acknowledgments
We thank all members of the field staff involved in the KORA study and we express our appreciation to all study participants. The KORA research platform (KORA, Cooperative Research in the Region of Augsburg) was initiated and financed by the Helmholtz Zentrum München—German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ.
The authors declare no conflict of interest.
References
- Association AP Diagnostic and Statistical Manual of Mental Disorders5th edn. American Psychiatric Association: Arlington, VA, USA; 2013 [Google Scholar]
- Lukaschek K, Kruse J, Emeny RT, Lacruz ME, von Eisenhart Rothe A, Ladwig KH. Lifetime traumatic experiences and their impact on PTSD: a general population study. Soc Psychiatry Psychiatr Epidemiol. 2013;48:525–532. doi: 10.1007/s00127-012-0585-7. [DOI] [PubMed] [Google Scholar]
- Stein MB, Jang KL, Taylor S, Vernon PA, Livesley WJ. Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: a twin study. Am J Psychiatry. 2002;159:1675–1681. doi: 10.1176/appi.ajp.159.10.1675. [DOI] [PubMed] [Google Scholar]
- True WR, Rice J, Eisen SA, Heath AC, Goldberg J, Lyons MJ, et al. A twin study of genetic and environmental contributions to liability for posttraumatic stress symptoms. Arch Gen Psychiatry. 1993;50:257–264. doi: 10.1001/archpsyc.1993.01820160019002. [DOI] [PubMed] [Google Scholar]
- Amstadter AB, Koenen KC, Ruggiero KJ, Acierno R, Galea S, Kilpatrick DG, et al. Variant in RGS2 moderates posttraumatic stress symptoms following potentially traumatic event exposure. J Anxiety Disord. 2009;23:369–373. doi: 10.1016/j.janxdis.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolassa IT, Kolassa S, Ertl V, Papassotiropoulos A, De Quervain DJ. The risk of posttraumatic stress disorder after trauma depends on traumatic load and the catechol-o-methyltransferase Val(158)Met polymorphism. Biol Psychiatry. 2010;67:304–308. doi: 10.1016/j.biopsych.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Lee MS, Kang RH, Kim H, Kim SD, Kee BS, et al. Influence of the serotonin transporter promoter gene polymorphism on susceptibility to posttraumatic stress disorder. Depress Anxiety. 2005;21:135–139. doi: 10.1002/da.20064. [DOI] [PubMed] [Google Scholar]
- Segman RH, Cooper-Kazaz R, Macciardi F, Goltser T, Halfon Y, Dobroborski T, et al. Association between the dopamine transporter gene and posttraumatic stress disorder. Mol Psychiatry. 2002;7:903–907. doi: 10.1038/sj.mp.4001085. [DOI] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Yang C, Zhao H, Farrer LA, Gelernter J. Genome-wide association study identifies new susceptibility loci for posttraumatic stress disorder. Biol Psychiatry. 2013;74:656–663. doi: 10.1016/j.biopsych.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick DG, Koenen KC, Ruggiero KJ, Acierno R, Galea S, Resnick HS, et al. The serotonin transporter genotype and social support and moderation of posttraumatic stress disorder and depression in hurricane-exposed adults. Am J Psychiatry. 2007;164:1693–1699. doi: 10.1176/appi.ajp.2007.06122007. [DOI] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Farrer L, Gelernter J. Serotonin transporter 5-HTTLPR genotype moderates the effects of childhood adversity on posttraumatic stress disorder risk: a replication study. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:644–652. doi: 10.1002/ajmg.b.32068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, Brady K, et al. Interactive effect of stressful life events and the serotonin transporter 5-HTTLPR genotype on posttraumatic stress disorder diagnosis in 2 independent populations. Arch Gen Psychiatry. 2009;66:1201–1209. doi: 10.1001/archgenpsychiatry.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis MC, Nugent NR, Amstadter AB, Koenen KC. Genetics of post-traumatic stress disorder: review and recommendations for genome-wide association studies. Curr Psychiatry Rep. 2010;12:313–326. doi: 10.1007/s11920-010-0126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladwig KH, Brockhaus AC, Baumert J, Lukaschek K, Emeny RT, Kruse J, et al. Posttraumatic stress disorder and not depression is associated with shorter leukocyte telomere length: findings from 3,000 participants in the population-based KORA F4 study. PLoS One. 2013;8:e64762. doi: 10.1371/journal.pone.0064762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. 2013;16:33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab. 2007;18:190–198. doi: 10.1016/j.tem.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhou R, Li X, Ursano RJ, Li H. Stress-induced change of mitochondria membrane potential regulated by genomic and non-genomic GR signaling: a possible mechanism for hippocampus atrophy in PTSD. Med Hypotheses. 2006;66:1205–1208. doi: 10.1016/j.mehy.2005.11.041. [DOI] [PubMed] [Google Scholar]
- Naviaux RK. Oxidative shielding or oxidative stress? J Pharmacol Exp Ther. 2012;342:608–618. doi: 10.1124/jpet.112.192120. [DOI] [PubMed] [Google Scholar]
- Ehlert U. Enduring psychobiological effects of childhood adversity. Psychoneuroendocrinology. 2013;38:1850–1857. doi: 10.1016/j.psyneuen.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145:1233–1248. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- Bremner JD. Functional neuroimaging in post-traumatic stress disorder. Exp Rev Neurother. 2007;7:393–405. doi: 10.1586/14737175.7.4.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79. doi: 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- Su YA, Wu J, Zhang L, Zhang Q, Su DM, He P, et al. Dysregulated mitochondrial genes and networks with drug targets in postmortem brain of patients with posttraumatic stress disorder (PTSD) revealed by human mitochondria-focused cDNA microarrays. Int J Biol Sci. 2008;4:223–235. doi: 10.7150/ijbs.4.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci. 2004;1032:1–7. doi: 10.1196/annals.1314.001. [DOI] [PubMed] [Google Scholar]
- Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci USA. 1980;77:6715–6719. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Med. 2013;3:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar D, Laifenfeld D. Mitochondria, synaptic plasticity, and schizophrenia. Int Rev Neurobiol. 2004;59:273–296. doi: 10.1016/S0074-7742(04)59011-6. [DOI] [PubMed] [Google Scholar]
- Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry. 2004;61:300–308. doi: 10.1001/archpsyc.61.3.300. [DOI] [PubMed] [Google Scholar]
- Shao L, Martin MV, Watson SJ, Schatzberg A, Akil H, Myers RM, et al. Mitochondrial involvement in psychiatric disorders. Ann Med. 2008;40:281–295. doi: 10.1080/07853890801923753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einat H, Yuan P, Manji HK. Increased anxiety-like behaviors and mitochondrial dysfunction in mice with targeted mutation of the Bcl-2 gene: further support for the involvement of mitochondrial function in anxiety disorders. Behav Brain Res. 2005;165:172–180. doi: 10.1016/j.bbr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Wichmann HE, Gieger C, Illig T. KORA-gen—resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67 (Suppl 1):S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- Holle R, Happich M, Lowel H, Wichmann HE. KORA—a research platform for population based health research. Gesundheitswesen. 2005;67 (Suppl 1):S19–S25. doi: 10.1055/s-2005-858235. [DOI] [PubMed] [Google Scholar]
- McRae AF, Byrne EM, Zhao ZZ, Montgomery GW, Visscher PM. Power and SNP tagging in whole mitochondrial genome association studies. Genome Res. 2008;18:911–917. doi: 10.1101/gr.074872.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization International Statistical Classification of Diseases and Health Related Problems10th edn. World Health Organization: Geneva,Switzerland; 2010 [Google Scholar]
- Foa EBC, Jaycox L, Perry K. The validation of a self-report measure of posttraumatic stress disorder: The Posttraumatic Diagnostic Scale. Psychol Assess. 1997;9:445–451. [Google Scholar]
- Flaquer A, Baumbach C, Kriebel J, Meitinger T, Peters A, Waldenberger M, et al. Mitochondrial genetic variants identified to be associated with BMI in adults. PLoS One. 2014;9:e105116. doi: 10.1371/journal.pone.0105116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Smyth GK.limma: linear models for microarray dataIn: Gentleman R (ed)Bioinformatics and Computational Biology Solutions Using R and BioconductorStatistics for Biology and Health Springer Science+Business Media: New York, NY, USA; 2005397–420. [Google Scholar]
- Macgregor S, Visscher PM, Montgomery G. Analysis of pooled DNA samples on high density arrays without prior knowledge of differential hybridization rates. Nucleic Acids Res. 2006;34:e55. doi: 10.1093/nar/gkl136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaquer A, Heinzmann A, Rospleszcz S, Mailaparambil B, Dietrich H, Strauch K, et al. Association study of mitochondrial genetic polymorphisms in asthmatic children. Mitochondrion. 2014;14:49–53. doi: 10.1016/j.mito.2013.11.002. [DOI] [PubMed] [Google Scholar]
- R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria; 2013. [Google Scholar]
- Jou SH, Chiu NY, Liu CS. Mitochondrial dysfunction and psychiatric disorders. Chang Gung Med J. 2009;32:370–379. [PubMed] [Google Scholar]
- Rezin GT, Amboni G, Zugno AI, Quevedo J, Streck EL. Mitochondrial dysfunction and psychiatric disorders. Neurochem Res. 2009;34:1021–1029. doi: 10.1007/s11064-008-9865-8. [DOI] [PubMed] [Google Scholar]
- Houstek J, Pickova A, Vojtiskova A, Mracek T, Pecina P, Jesina P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim Biophys Acta. 2006;1757:1400–1405. doi: 10.1016/j.bbabio.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Safaei S, Houshmand M, Banoei MM, Panahi MS, Nafisi S, Parivar K, et al. Mitochondrial tRNALeu/Lys and ATPase 6/8 gene variations in spinocerebellar ataxias. Neurodegener Dis. 2009;6:16–22. doi: 10.1159/000170885. [DOI] [PubMed] [Google Scholar]
- Andreazza AC, Shao L, Wang JF, Young LT. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry. 2010;67:360–368. doi: 10.1001/archgenpsychiatry.2010.22. [DOI] [PubMed] [Google Scholar]
- Quesada H, Skibinski DA, Skibinski DO. Sex-biased heteroplasmy and mitochondrial DNA inheritance in the mussel Mytilus galloprovincialis Lmk. Curr Genet. 1996;29:423–426. doi: 10.1007/BF02221509. [DOI] [PubMed] [Google Scholar]
- Woloszynska M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes—though this be madness, yet there's method in't. J Exp Botany. 2010;61:657–671. doi: 10.1093/jxb/erp361. [DOI] [PubMed] [Google Scholar]
- de Camargo MA, Paneto GG, de Mello AC, Martins JA, Barcellos W, Cicarelli RM. No relationship found between point heteroplasmy in mitochondrial DNA control region and age range, sex and haplogroup in human hairs. Mol Biol Rep. 2011;38:1219–1223. doi: 10.1007/s11033-010-0220-1. [DOI] [PubMed] [Google Scholar]
- Ramos A, Santos C, Mateiu L, Gonzalez Mdel M, Alvarez L, Azevedo L, et al. Frequency and pattern of heteroplasmy in the complete human mitochondrial genome. PLoS One. 2013;8:e74636. doi: 10.1371/journal.pone.0074636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev. 2012;2012:428010. doi: 10.1155/2012/428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Ganguly A, BoseDasgupta S, Das BB, Pal C, Jaisankar P, et al. Mitochondria-dependent reactive oxygen species-mediated programmed cell death induced by 3,3'-diindolylmethane through inhibition of F0F1-ATP synthase in unicellular protozoan parasite Leishmania donovani. Mol Pharmacol. 2008;74:1292–1307. doi: 10.1124/mol.108.050161. [DOI] [PubMed] [Google Scholar]
- Barbu A, Welsh N, Saldeen J. Cytokine-induced apoptosis and necrosis are preceded by disruption of the mitochondrial membrane potential (Deltapsi(m)) in pancreatic RINm5F cells: prevention by Bcl-2. Mol Cell Endocrinol. 2002;190:75–82. doi: 10.1016/s0303-7207(02)00009-6. [DOI] [PubMed] [Google Scholar]
- National Center for PTSD. What is PTSD?Available from http://www.ptsd.va.gov/public/pages/what-is-ptsd.asp .
- Breslau N, Lucia VC, Davis GC. Partial PTSD versus full PTSD: an empirical examination of associated impairment. Psychol Med. 2004;34:1205–1214. doi: 10.1017/s0033291704002594. [DOI] [PubMed] [Google Scholar]
- Mylle J, Maes M. Partial posttraumatic stress disorder revisited. J Affect Disord. 2004;78:37–48. doi: 10.1016/s0165-0327(02)00218-5. [DOI] [PubMed] [Google Scholar]