

Abstract
Lysine methylation is an emerging post-translation modification and it has been identified on several histone and non-histone proteins, where it plays crucial roles in cell development and many diseases. Approximately 5,000 lysine methylation sites were identified on different proteins, which are set by few dozens of protein lysine methyltransferases. This suggests that each PKMT methylates multiple proteins, however till now only one or two substrates have been identified for several of these enzymes. To approach this problem, we have introduced peptide array based substrate specificity analyses of PKMTs. Peptide arrays are powerful tools to characterize the specificity of PKMTs because methylation of several substrates with different sequences can be tested on one array. We synthesized peptide arrays on cellulose membrane using an Intavis SPOT synthesizer and analyzed the specificity of various PKMTs. Based on the results, for several of these enzymes, novel substrates could be identified. For example, for NSD1 by employing peptide arrays, we showed that it methylates K44 of H4 instead of the reported H4K20 and in addition H1.5K168 is the highly preferred substrate over the previously known H3K36. Hence, peptide arrays are powerful tools to biochemically characterize the PKMTs.
Keywords: Biochemistry, Issue 93, Peptide arrays, solid phase peptide synthesis, SPOT synthesis, protein lysine methyltransferases, substrate specificity profile analysis, lysine methylation
Introduction
In the last two decades, several reports demonstrated the importance of post-translational modifications (PTM) in cellular development and several diseases like cancer, but recently protein lysine methylation has emerged as an another vital PTM. While initially histone lysine methylation was found to be an essential chromatin mark, later work also showed lysine methylation of several non-histone proteins 1-4. The sequential transfer of methyl groups from S-adenosyl-L-methionine to the ε-amino group of lysine residues is catalyzed by a family of enzymes called Protein Lysine Methyltransferases (PKMTs) that contains over 60 proteins in the human genome. PKMTs were initially discovered as histone modifying enzymes that methylate specific lysine residue but later reports demonstrated that they could also methylate non-histone proteins 5. Up to now, approximately 5,000 lysine methylation sites were identified on different proteins 6, but the enzymes responsible for these modifications are often not identified. One reason for this is that the specificity of most PKMTs has not been studied extensively. Therefore, it recurrently occurs that novel substrates of PKMTs are discovered. The lack of a detailed knowledge of the substrate specificity of PKMTs hinders understanding of their biological function and role. To study the specificity of a PKMT in detail, the methylation rates of many peptide substrates that differ in one or few amino acids must be measured and compared, which is ideally done using peptide arrays. Based on the resulting specificity profiles, potential substrates of PKMTs can be identified that can be studied further.
Peptide arrays are widely used tools for the biochemical analysis of antibodies, peptide modifying enzymes and mapping of protein-protein interaction sites (antibody-antigen, receptor-ligand) 7-9. Several hundreds of peptides are needed for such applications. Different methods are available for peptide synthesis, among them peptide synthesis on resin is very commonly used, but it has limitations in throughput and it is relatively expensive. These issues were resolved with the introduction of the SPOT synthesis method by Frank and colleagues 10. The SPOT synthesis method allows synthesis of several hundred peptides in parallel and on average it is inexpensive compared to resin synthesis. The peptides synthesized on cellulose membrane can be used either directly for various applications or peptides can be cleaved from the membrane and used as free peptides for in solution assays or to prepare peptide microarrays 10-13.
SPOT synthesis is a variant of the solid phase peptide synthesis, which uses a cellulose membrane as a solid support and employs the standard Fmoc-chemistry 10-13. Hence, the synthesis of peptide chains starts at the C-terminal end and proceeds towards the N-terminal end in contrast to the biological synthesis in ribosomes. Cellulose membranes are functionalized for the attachment of the first activated amino acids (Fig. 1). The SPOT method is based on the sequential delivery of activated amino acids in a droplet of solvent to defined spots on the membrane using an automated pipetting system. The droplet of liquid is dispensed on the porous membrane where it forms a circular wet spot, which later acts as an open reactor for the chemical reactions in peptide synthesis. The spot size is determined by the volume dispensed and the absorptive capacity of the membrane, multiples of such spots are arranged as arrays. The scale of synthesis correlates with the spot size and the loading capacity of the membrane. The distance between the spots and the density of arrays are managed by varying the spot sizes. Cellulose membrane has several advantages as solid phase in peptide synthesis, it is inexpensive, tolerant to the chemicals used in peptide synthesis, stable in aqueous solutions and easy to handle. In addition, its hydrophilic nature makes it suitable for several biological assay systems. SPOT synthesis can be carried out manually or automated (for 1000s of peptides) depending on the required number of peptides. A fully automated SPOT synthesizer from Intavis (Köln, Germany) is used for our applications. It permits synthesis of peptides in different amounts and of different length. Linear peptides are regularly synthesized with 15 to 20 amino acid length, in addition peptides of up to 42 amino acid can also be prepared by step-wise synthesis 14,15. However, increasing the number of amino acids leads to reductions in the overall coupling yields, which affects the quality of the peptides. Because of the low amount of peptides per spot, the products are often difficult to purify and the quality of individual peptides cannot be easily assessed. Therefore, the results obtained from SPOT peptide arrays must be confirmed either with peptides synthesized by standard methods in larger scale, which can be purified and analyzed according to standards in peptide synthesis or by synthesizing the proteins containing the desired peptide sequences. Still, we found the SPOT synthesis to be highly reliable and results generally to be reproducible. SPOT synthesis is not restricted to proteinogenic amino acids, several commercially available modified amino acids also can be used for synthesis, allowing peptides to be modified before and after the final cleavage of the side-chain protection group and, furthermore, it also allows incorporation of phosphorylated, methylated or acetylated amino acids 11.
Immobilized peptide libraries synthesized by the SPOT method can be directly used for many biological and biochemical assays. We employed peptide arrays comprising 300-400 peptides to investigate the substrate specificity of PKMTs. For enzymatic modification, the peptide arrays are incubated with the respective PKMT and labeled [methyl-3H]-AdoMet in an appropriate buffer. The methylation of the respective substrate is analyzed by following the enzymatic transfer of the radioactively labelled methyl groups from AdoMet to the peptide substrate via autoradiography (Fig. 3). By this procedure, the peptide arrays allow the study of methylation of different peptide substrates at the same time. One important advantage of this method is that all the peptides are methylated in competition, such that during the linear phase of the methylation kinetics, the relative methylation of each peptide is proportional to the catalytic rate constant divided by the dissociation constant (kcat/KD) of the enzyme for the respective peptide substrate. Therefore, the amount of radioactivity incorporated into each spot is directly correlated with the enzymatic activity towards the particular peptide. Using the results of a peptide array methylation experiment, the specificity profile of the PKMT can be defined and based on this novel substrates can be predicated. Peptide arrays allow the rapid and cost efficient validation of the methylation of novel substrates at the peptide level. For this, arrays are prepared that contain the predicted novel substrates together with modified peptides containing an Ala instead of Lys at the target sites as well as positive and negative control peptides. Finally, the novel substrates can be prepared as proteins together with mutants, in which the target Lys is altered to Ala and the methylation can be confirmed at the protein level. Depending on the results, this is then followed by biological studies addressing potential roles of the methylation of the newly described protein substrates.
Protocol
1. Preparation of Peptide Arrays
- Programming of the Peptide Sequences
- Design peptide sequences for the synthesis using the MultiPep Spotter software. Enter the peptide sequences in single letter codes. Peptide 1: T A R K S T G G K A
- Generate alanine or arginine walk libraries with a specific command (.replace, A) to screen the important amino acids required for the recognition of a defined substrate. For instance in the following example the first line is the wild type sequence and from the second line each amino acid in a peptide is altered to alanine sequentially. replace, A T-A-R-K-S-T-G A-A-R-K-S-T-G T-A-R-K-S-T-G T-A-A-K-S-T-G T-A-R-A-S-T-G T-A-R-K-A-T-G T-A-R-K-S-A-G T-A-R-K-S-T-A
- Use similar commands (.replace R, .replace N etc.) as described in 1.1.2 with all the naturally available amino acid residues (eventually omitting cysteine, methionine and tryptophan, because these residues are prone to oxidation and sometimes lower synthesis yield) to synthesize a peptide array for determining the consensus substrate sequence motif for an enzyme. NOTE: As shown in Figure 4A, the horizontal axis represents the given peptide sequence and the vertical axis represents the amino acids that are selected for command. Similar to the sequence shown in 1.1.2, A on the vertical axis means that an Ala sequentially replaces each position of the given sequence in the first row in Fig. 4A. Similarly arginine sequentially replaces each amino acid in the second row and so on with the other amino acids. Complete peptide array libraries are synthesized by systematically replacing each residue in the provided peptide substrate sequence with each of the other naturally available amino acid residues, this helps to understand the influence of each residue at each position of the given sequence.
- Alternatively, methylation of known or predicted peptide substrates can be easily verified synthesizing peptide libraries with the target peptides of the PKMT together with positive and negative methylation controls (i.e. peptides known to methylated or known not to be methylated). Synthesize peptides by exchanging the putative target lysine to alanine to confirm methylation of the target lysine as shown below by manual programming. Wild type peptide: S T G G K P R Q F L Mutant peptide : S T G G A P R Q F L NOTE: The software also has inherent scripts to create various libraries, like epitope mapping with a desired frame shift of the sequence to map important amino acids required for an interaction.
- Preparation of Membrane, Amino Acids and Reagents
- Pre-swell the SPOT membrane in DMF (N,N-Dimethylformamide) for 10 min and then place the wet membrane on the SPOT synthesizer frame without air bubbles or wrinkles.
- Wash the membrane three times with 100% ethanol. Dry the membranes completely for at least 10 min before starting the synthesis program.
- In parallel, freshly prepare 0.5 M working concentrations of commercially available Fmoc-protected amino acids by dissolving them in NMP and also prepare the activator and base as described in Table 1.
- Make sure that all the waste solution containers of the SPOT synthesizer were emptied and refill the solvent reservoirs with the DMF, ethanol and piperidine. NOTE: Now the machine is ready for the synthesis.
- Spotting of the First Amino Acid
- To generate the amide bond with the free amino group on the membrane, activate the carboxy group of the incoming amino acid by adding the activator (N,N’-Diisopropylcarbodiimide) and base solution (Ethyl hydroxyimino cyanoacetate) to the amino acid derivative.
- Spot the desired volumes of activated amino acids on the Amino-PEG functionalized membrane using the programmable robot. NOTE: Thereby, the C-terminus of the amino acid is coupled to the amino group of the membrane.
- Repeat this step three times to ensure better coupling of the first activated amino acid. Incubate the membrane for 20 min and then wash with DMF to remove uncoupled amino acids.
- Blocking of Free Spaces on Non-spot Areas NOTE: After the coupling of the first C-terminal amino acid of all peptides to the amino group of the membrane, the amino groups between the spots and also some of the amino groups within the spot areas do not form bonds with the amino acids.
- Block these free amino groups by incubating with the capping solution (20% acetic anhydride in DMF) for 5 min. NOTE: This avoids the coupling of amino acids in the further cycles with the membrane instead of the growing peptide chain.
- Removal of Fmoc Group
- After the blocking, wash the membrane with DMF 4 times and then incubate with 20% piperidine in DMF for 20 min to remove the Fmoc protection group. NOTE: This step leads to removal of the amino-protecting Fmoc groups and it is called deprotection.
- Afterwards, wash the membrane twice with DMF and twice with ethanol and allow it to dry. NOTE: Now the membrane is ready for the coupling of the next amino acids.
- Chain Elongation NOTE: Addition of each amino acid is called a cycle. Apart from the coupling of first amino acid, the cycle begins with the deprotection of Fmoc group of the coupled amino acid and it is followed by the coupling of the activated carboxyl group of an incoming amino acid to the amino group of the growing peptide chain.
- Repeat steps 1.3 and 1.5 until the desired peptide length is achieved.
- Staining NOTE: After the final cycle, the peptide arrays are stained with bromophenol blue to confirm the successful synthesis of peptides. Bromophenol blue binds with the free amino groups of the peptides. Peptide spots show light blue color after the staining but the intensity of the spots varies depending on the peptide sequence. This staining allows confirmation of the complete removal of piperidine, because in presence of piperidine bromophenol blue does not stain the peptides. In addition, this also helps to mark the peptide arrays for further usage.
- After the Fmoc group deprotection in the last synthesis cycle, wash the membrane with DMF and 100% ethanol. Then, treat the membrane with bromophenol blue (0.02% bromophenol blue in DMF) for a minimum of 5 min until it completely turns blue.
- Afterwards, wash the membrane with DMF and ethanol followed by drying for 10 min. Now take out the membrane from the synthesizer and perform side chain deprotection (section 1.8) manually. If needed, photograph the membrane to document the results of the peptide synthesis (Fig. 2).
- Side Chain Deprotection
- Treat the membranes with 20 to 25 ml of side chain deprotection mixture consisting of 95% Trifluoroacetic acid (TFA) to cleave the side chain protection groups and scavenger reagents (2.5% water and 2.5% triisopropylsilane) to protect the side chains of amino acids from modification during this step. Take a sufficient volume of deprotection mixture to ensure that it covers the membrane completely in a tightly closed chemical resistant box and incubate it for 1 to 2 hr while shaking gently.
- Wash the membrane six times with 20-25 ml of DCM (Dichloromethane) for 2 min each and finally twice with 100% ethanol and dry it in a desiccator overnight. NOTE: Now the membrane can be used for experiments. A scheme of the peptide array synthesis is provided in Figure 1.
2. Protein Expression and Purification
- Protein Expression of PKMTs as GST Fusions
- Using standard techniques, clone the gene encoding the full length PKMT or its catalytic domain into a bacterial expression vector (like pGEX-6p2) to express it as GST-fusion protein.
- Transfer the vector with the desired PKMT insert into BL21 or BL21 codon plus E. coli cells by the heat shock method or any other method.
- Prepare a pre-culture with 30 ml of Luria-Bertani (LB) media on the day of expression and incubate at 37 °C for 7 to 8 hr with continuous shaking.
- Next, transfer 10 ml of the pre-culture into a 2 L big baffled flasks containing 1 L of LB media and incubate at 37°C in the incubator with continuous shaking until the culture reaches a defined optical density at 600 nm. NOTE: While this can be optimized for each protein, we routinely induce at an OD600nm of about 0.8. The induction temperature has to be optimized for each protein individually, some proteins show good expression at 22°C and some at higher temperatures.
- Shift the cells to the induction temperature for 15 min, then induce with 1 mM of isopropyl-beta-D-thiogalactopyranoside and allow the culture to grow for 10-12 hr at induction temperature.
- Afterwards harvest the cells by centrifugation at 5,000 x g for 15 min and finally wash the pellet with 30 ml of STE buffer (10 mM Tris pH 8.0, 100 mM NaCl and 0.1 mM EDTA) and store at -20 °C for further usage.
- Protein Purification
- Thaw the cell pellet and re-suspend in 30 ml of sonication buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM DTT and 5% glycerol) and disrupt by sonication. NOTE: This buffer needs to be optimized for each protein eventually.
- Centrifuge the cell lysate at 22,000 x g for 1 hr and, later collect the supernatant and pass through a column containing 600 µl of glutathione Sepharose 4B resin.
- Wash the column with 50 ml of sonication buffer and next with 100 ml of high salt buffer (50 mM Tris pH 7.5, 500 mM NaCl, 1 mM DTT and 5% glycerol) to remove unspecifically bound proteins. Elute the bound protein with 5 ml of high salt buffer containing 40 mM glutathione.
- Dialyze the eluted protein in 2 L of dialysis buffer with low glycerol (20 mM Tris pH 7.4, 100 mM KCl, 0.5 mM DTT and 10% glycerol) for 2 hr and later in 20 mM Tris pH 7.4, 100 mM KCl, 0.5 mM DTT and 70% glycerol for 8 hr or overnight. Make sure all the purification buffers are at 4 °C and perform the protein purification in the cold room to avoid protein denaturation.
3. Peptide Array Methylation
- Pre-incubation of Peptide Arrays
- Perform peptide array methylation either in a box of appropriate size or in a plastic bag to avoid the wastage of enzyme and expensive radioactively labelled AdoMet. Pre-incubate the peptide array membrane in sealed plastic bag containing the respective methylation buffer without enzyme and labeled [methyl-3H]-AdoMet for 10 min. The volume of buffer depends upon the size of the array, for example use 8 ml of methylation buffer for a full specificity profile peptide array. Optimize the amount and concentration of the AdoMet for each enzyme.
- Methylation of Peptide Arrays
- Discard the pre-incubation buffer and incubate the membrane in 8 ml of methylation buffer containing the respective PKMT and labeled [methyl-3H]-AdoMet for 1 to 2 hr. NOTE: The amount of enzyme required for a methylation reaction depends upon the activity of specific PKMT, we initiate our screening experiments typically with 50 nM enzyme final concentration.
- Washing of Peptide Arrays
- Afterwards discard the methylation buffer in a radioactive waste container and wash the peptide arrays for 5 times with 20 ml of buffer containing 100 mM ammonium bicarbonate and 1% SDS for 5 min to remove the bound protein. Discard the washing buffer in a radioactive waste container.
- Detection of the Radioactive Signal
- After the washes incubate the membrane for 5 min in 10 ml of amplify solution (NAMP100V).
- Then, discard the amplify solution into the radioactive waste container for organic solvents, seal the peptide array in a plastic bag and put it in an autoradiography cassette.
- Place the autoradiography film on the peptide array in the dark room. Close the cassette carefully and expose it at -80°C for the necessary time. Then, develop the film to analyze the results. Capture the image couple of times with different exposition times to avoid saturation of strongly methylated peptide substrates.
4. Data Processing and Analysis
- Densitometric Analysis of Spot Intensities
- Scan the film in a conventional scanner and analyze the spot intensities by densitometry. Do this using ImageJ (which is freeware) or a commercial program. NOTE: We use Phoretix software for this task.
- Normalization and Averaging of Results from Different Membranes
- Conduct the peptide array methylation experiments at least in triplicate. Normalize the scanned intensities for each experiment by background subtraction and setting the activity at a reference substrate as 1.0. Average the data for the individual spots and report them as mean values and standard deviations.
- Data Analysis and Display
- Plot the data in 2D using a grayscale or color scheme to indicate the relative activity. Do this with Excel or Sigmaplot as shown here. Also, prepare a plot of the distribution of the standard deviations of the repeated experiments to analyze the quality of the data.
- To compare and display the accuracy of the recognition of each residue in the substrate peptide quantitatively, calculate the relative preference of the PKMT for each amino acid i at position x by a discrimination factor D 16,17: DX;i = < Vj≠i>/Vi - 1 Where vi is the rate of methylation of the peptide variant carrying amino acid i at position x and <vj≠i> is the average rate of methylation of all 19 peptides carrying a different amino acid j≠i at position x (including the wild type sequence). NOTE: The discrimination factor defined by this depends on the background level. If only one residue is accepted at a certain position with a background of 3%, a discrimination factor of 32.3 is obtained. No preference at a position results in a discrimination factor of 0.
Representative Results
Peptide arrays were successfully used to biochemically characterize the specificity of PKMTs, and several novel histone and non-histone substrates of PKMT were identified by this approach 5,16-19. Defining the correct substrate spectrum of a PKMT (or any enzyme) is an essential step towards understanding of its molecular mechanism and cellular functions.
As an example of the application of the peptide SPOT array methylation method, we describe the results of the specificity analysis of the NSD1 PKMT 19. Previous to our work, this enzyme was described to methylate several substrates including histone H3 at K36 and histone H4 at K20 but also the NF-kB family transcription factor p65 at K218 and K221. Fig. 4A shows an example of a methylation reaction of a peptide array with NSD1. For this experiments, a large peptide array based on the H3 (31-49) sequence was synthesized that contains all possible single amino acid alterations of the original sequence to test the significance of each amino acid for NSD1 interaction and methylation. In total 380 peptides were synthesized (20 possible amino acids x 18 residues plus one original H3 sequence in each row). The horizontal axis represents the sequence of the peptide and in the vertical direction the amino acid which is altered in the corresponding peptide is indicated. For example, the spot at line 17 column 4 contains a threonine at third position instead of glycine that is present in the wild type sequence (Fig. 4A). In such a way, point mutations are generated to test the preference of NSD1 for each native amino acid at each position in the peptide substrate. Methylation with NSD1 showed that it specifically acts on H3K36 and it has preferences on either side of the target lysine from 34 to 38.
Figure 4B represents the consolidation of the three peptide array methylation experiments with NSD1. The quantitative information was acquired from the individual peptide arrays and the results were normalized and averaged as described above. The standard deviations of individual averages show that the data are highly reproducible. Overall around 85% of the peptides shows SDs smaller than 20% and more than 97% of the peptide substrates demonstrated SDs smaller than 30% (Fig. 4C). In addition, we also calculated the discrimination factor to precisely determine the contribution of each amino acid at the tested positions. As described it provides the quantitative description of the peptide read out and preference of amino acids at the specific position (Fig. 5A). Our data show that NSD1 prefers aromatic residues at the -2 position (considering K36 as position 0) (F > Y > G); hydrophobic residues at -1 (I > L > V); basic residues at +1 (R > QKNM), where it cannot tolerate hydrophobic or aromatic residues; and hydrophobic residues at +2 (V > IA > P). At other sites (like -3, +3, or +6), NSD1 prefers some amino acids, but no strong residue specific readout was detected.
Based on this profile, potential novel peptide substrates can be found by database searches like using Scansite 20 (Fig. 5B). These peptides were prepared on a SPOT array including positive and negative controls and incubated with NSD1 and radioactively labelled AdoMet to identify the subset of them that are methylated at peptide level (Fig. 5C). As follow ups, peptide arrays can be prepared which include peptide variants, in which the target lysine is replaced by alanine, to confirm that methylation takes place at the predicted site. Then, the target proteins or subdomains of them containing the target lysine can be produced recombinantly and the methylation also tested at protein level. Finally, depending on the results, follow up experiments can investigate if the methylation also occurs in cells and which biological role it has.
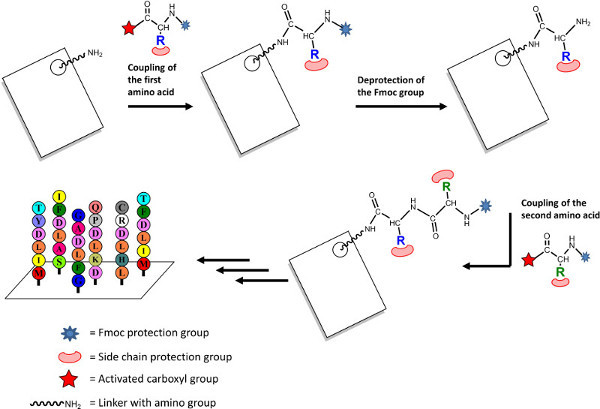 Figure 1: Scheme of peptide synthesis by the SPOT method on cellulose membrane. Please click here to view a larger version of this figure.
Figure 1: Scheme of peptide synthesis by the SPOT method on cellulose membrane. Please click here to view a larger version of this figure.
 Figure 2: Example of a peptide array stained with Bromophenol blue to confirm the synthesis of peptides. Please click here to view a larger version of this figure.
Figure 2: Example of a peptide array stained with Bromophenol blue to confirm the synthesis of peptides. Please click here to view a larger version of this figure.
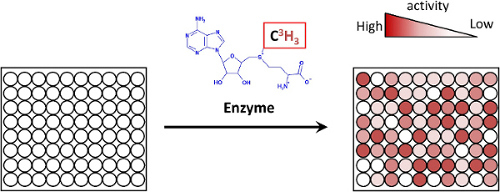 Figure 3: Scheme of the peptide array methylation experiment; peptide arrays were methylated by incubation with PKMT and labeled [methyl-3H]-AdoMet in an appropriate buffer. Afterwards the radioactivity transferred to each spot is detected by autoradiography. Please click here to view a larger version of this figure.
Figure 3: Scheme of the peptide array methylation experiment; peptide arrays were methylated by incubation with PKMT and labeled [methyl-3H]-AdoMet in an appropriate buffer. Afterwards the radioactivity transferred to each spot is detected by autoradiography. Please click here to view a larger version of this figure.
 Figure 4: Example results obtained with the NSD1 PKMT. A) Example of a substrate specificity peptide array for NSD1 using the H3 (31-49) sequence as template. The horizontal axis represents the H3 sequence and the vertical axis represents the amino acids by which the corresponding row is mutated. The first row contains peptides with original sequence. B) Consolidation of results from 3 independent peptide array methylation experiments with NSD1, the data were averaged results from all three experiments after normalization. C) Distribution of standard errors for the average methylation data shown in panel B. Reproduced from Kudithipudi et al. (2014) with some modifications 19. Please click here to view a larger version of this figure.
Figure 4: Example results obtained with the NSD1 PKMT. A) Example of a substrate specificity peptide array for NSD1 using the H3 (31-49) sequence as template. The horizontal axis represents the H3 sequence and the vertical axis represents the amino acids by which the corresponding row is mutated. The first row contains peptides with original sequence. B) Consolidation of results from 3 independent peptide array methylation experiments with NSD1, the data were averaged results from all three experiments after normalization. C) Distribution of standard errors for the average methylation data shown in panel B. Reproduced from Kudithipudi et al. (2014) with some modifications 19. Please click here to view a larger version of this figure.
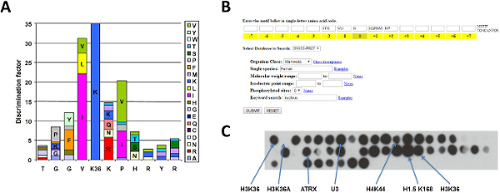 Figure 5: Discovery of novel PKMT substrates. A) Discrimination factors of NSD1 for the recognition of amino acid residues at the positions next to the target lysine (K36 in the experiment shown here). The data show that NSD1 prefers aromatic residues at the -2 position (considering K36 as position 0). Hydrophobic residues are recognized at the -1 and +2 positions, albeit with distinctive differences in details. At the +1 site basic residues and amides are preferred. At other sites some weak preferences, but no strong residue specific readout was detected. B) Screenshot of an example search at Scansite, using the specificity profile determined for NSD1. C) Peptide SPOT array containing several predicted novel NSD1 substrates together with positive (H3K36) and negative controls (H3K36A). Some of the predicated novel targets were strongly methylated (some of them annotated), in other cases the predictions could not be verified. Panel A and C is reproduced from Kudithipudi et al. (2014) with some modifications 19. Please click here to view a larger version of this figure.
Figure 5: Discovery of novel PKMT substrates. A) Discrimination factors of NSD1 for the recognition of amino acid residues at the positions next to the target lysine (K36 in the experiment shown here). The data show that NSD1 prefers aromatic residues at the -2 position (considering K36 as position 0). Hydrophobic residues are recognized at the -1 and +2 positions, albeit with distinctive differences in details. At the +1 site basic residues and amides are preferred. At other sites some weak preferences, but no strong residue specific readout was detected. B) Screenshot of an example search at Scansite, using the specificity profile determined for NSD1. C) Peptide SPOT array containing several predicted novel NSD1 substrates together with positive (H3K36) and negative controls (H3K36A). Some of the predicated novel targets were strongly methylated (some of them annotated), in other cases the predictions could not be verified. Panel A and C is reproduced from Kudithipudi et al. (2014) with some modifications 19. Please click here to view a larger version of this figure.
| Base | 1 M Oxyma Pure in NMP -> 2,13 g Oxyma Pure in 15 ml NMP |
| Activator | 2.4 ml N,N’-Diisopropylcarbodiimide in 15 ml NMP |
| Capping Mixture | 20% acetic anhydride in DMF -> 6 ml Acetic anhydride in 30 ml DMF |
| Fmoc Deprotection | 20% Piperidine in DMF -> 200 ml in 1,000 ml DMF |
| Sidechain Deprotection | 900 µl Triisopropylsilane + 600 µl ddH2O in 30 ml TFA |
| Staining | 0.02% bromophenol blue in DMF |
Table 1: Chemical mixtures and their composition used in the protocol.
Discussion
SPOT synthesis as described here is a powerful method to map protein-protein interaction sites and investigate the substrate recognition of peptide modifying enzymes. However, SPOT synthesis still has certain drawbacks, because although the peptides synthesized by the SPOT method are reported to have more than 90% purity 21 this is difficult to confirm in each instance. Therefore, results have to be reproduced by other methods for instance using protein domains or with purified peptides synthesized by standard methods. In addition, the other major drawback of SPOT arrays is that most time they cannot be reused to carry out several assays with one array.
To increase the efficiency of synthesis with amino acids of low stability, like the protected arginine, it is important to make them fresh for every 5 cycles. In enzymatic reactions or protein binding studies it is often observed that the periphery of the peptide spot has more signal intensity than the center (“ring spot effect”). This effect can be due to the high density of peptides in the central part of the spot and then it can be resolved by downscaling the peptide synthesis 22. For further trouble shooting of peptide synthesis the machine handbook and company service should be consulted. If methylation is not observed, positive control peptides (i.e. peptides known to be methylated by the enzyme under investigation) should be added to the membrane.
Alternate to SPOT arrays, high density peptide micro arrays 23 are available, but they are more expensive and require special equipment for usage. Furthermore the amount of peptide per spot is smaller, which requires more sensitive readout procedures. Protein arrays are also available to study these functions 24,25, but they are more difficult to prepare because each individual protein needs to be expressed and purified and protein folding on the array needs to be maintained. An additional advantage of peptide arrays is the possible introduction of post- translational modifications during the synthesis and the ease of mutational testing of the roles of individual amino acid residues.
It is an essential starting point for this protocol, to have at least one peptide identified, which is methylated by the enzyme under investigation. Methylation conditions and buffers have to be optimized, as well as the concentration of the methyltransferase. Exemplary time courses of methylation have to be determined to avoid complete methylation of the best substrate, which will cause loss of dynamic range.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work has been supported by the DFG grant JE 252/7.
References
- Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–488. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- Clarke SG. Protein methylation at the surface and buried deep: thinking outside the histone box. Trends in Biochemical Sciences. 2013;38:243–252. doi: 10.1016/j.tibs.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, et al. Protein lysine methyltransferase G9a acts on non-histone targets. Nature Chemical Biology. 2008;4:344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- Reineke U, Volkmer-Engert R, Schneider-Mergener J. Applications of peptide arrays prepared by the SPOT-technology. Current Opinion in Biotechnology. 2001;12:59–64. doi: 10.1016/s0958-1669(00)00178-6. [DOI] [PubMed] [Google Scholar]
- Winkler DF, Andresen H, Hilpert K. SPOT synthesis as a tool to study protein-protein interactions. Methods Mol. Biol. 2011;723:105–127. doi: 10.1007/978-1-61779-043-0_8. [DOI] [PubMed] [Google Scholar]
- Leung GC, Murphy JM, Briant D, Sicheri F. Characterization of kinase target phosphorylation consensus motifs using peptide SPOT arrays. Methods Mol. Biol. 2009;570:187–195. doi: 10.1007/978-1-60327-394-7_7. [DOI] [PubMed] [Google Scholar]
- Frank R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports--principles and applications. Journal of immunological methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- Hilpert K, Winkler DF, Hancock RE. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nature protocols. 2007;2:1333–1349. doi: 10.1038/nprot.2007.160. [DOI] [PubMed] [Google Scholar]
- Li SS, Wu C. Using peptide array to identify binding motifs and interaction networks for modular domains. Methods Mol. Biol. 2009;570:67–76. doi: 10.1007/978-1-60327-394-7_3. [DOI] [PubMed] [Google Scholar]
- Winkler DF, Hilpert K, Brandt O, Hancock RE. Synthesis of peptide arrays using SPOT-technology and the CelluSpots-method. Methods Mol. Biol. 2009;570:157–174. doi: 10.1007/978-1-60327-394-7_5. [DOI] [PubMed] [Google Scholar]
- Toepert F, et al. Combining SPOT synthesis and native peptide ligation to create large arrays of WW protein domains. Angew Chem. Int. Ed. Engl. 2003;42:1136–1140. doi: 10.1002/anie.200390298. [DOI] [PubMed] [Google Scholar]
- Volkmer R. Synthesis and application of peptide arrays: quo vadis SPOT technology. Chembiochem : a EuropeanJournal of Chemical Biology. 2009;10:1431–1442. doi: 10.1002/cbic.200900078. [DOI] [PubMed] [Google Scholar]
- Rathert P, Zhang X, Freund C, Cheng X, Jeltsch A. Analysis of the substrate specificity of the Dim-5 histone lysine methyltransferase using peptide arrays. Chemistr., & biology. 2008;15:5–11. doi: 10.1016/j.chembiol.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudithipudi S, Dhayalan A, Kebede AF, Jeltsch A. The SET8 H4K20 protein lysine methyltransferase has a long recognition sequence covering seven amino acid residues. Biochimie. 2012;94:2212–2218. doi: 10.1016/j.biochi.2012.04.024. [DOI] [PubMed] [Google Scholar]
- Dhayalan A, Kudithipudi S, Rathert P, Jeltsch A. Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chemistr., & Biology. 2011;18:111–120. doi: 10.1016/j.chembiol.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Kudithipudi S, Lungu C, Rathert P, Happel N, Jeltsch A. Substrate specificity analysis and novel substrates of the protein lysine methyltransferase NSD1. Chemistr., & Biology. 2014;21:226–237. doi: 10.1016/j.chembiol.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Research. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Ueno A, Mihara H. Peptide design based on an antibody complementarity-determining region (CDR): construction of porphyrin-binding peptides and their affinity maturation by a combinatorial method. Chemistry. 2000;6:3196–3203. doi: 10.1002/1521-3765(20000901)6:17<3196::aid-chem3196>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Kramer A, et al. Spot synthesis: observations and optimizations. J. Pept. Res. 1999;54:319–327. doi: 10.1034/j.1399-3011.1999.00108.x. [DOI] [PubMed] [Google Scholar]
- Stadler V, et al. Combinatorial synthesis of peptide arrays with a laser printer. Angew Chem. Int. Ed. Engl. 2008;47:7132–7135. doi: 10.1002/anie.200801616. [DOI] [PubMed] [Google Scholar]
- Schnack C, Hengerer B, Gillardon F. Identification of novel substrates for Cdk5 and new targets for Cdk5 inhibitors using high-density protein microarrays. Proteomics. 2008;8:1980–1986. doi: 10.1002/pmic.200701063. [DOI] [PubMed] [Google Scholar]
- Mah AS, et al. Substrate specificity analysis of protein kinase complex Dbf2-Mob1 by peptide library and proteome array screening. BMC Biochemistry. 2005;6 doi: 10.1186/1471-2091-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]