

Abstract
The goal of fluorometric analysis is to serve as an efficient, cost effective, high throughput method of analyzing phagocytosis and other cellular processes. This technique can be used on a variety of cell types, both adherent and non-adherent, to examine a variety of cellular properties. When studying phagocytosis, fluorometric technique utilizes phagocytic cell types such as macrophages, and fluorescently labeled opsonized particles whose fluorescence can be extinguished in the presence of trypan blue. Following plating of adherent macrophages in 96-well plates, fluorescent particles (green or red) are administered and cells are allowed to phagocytose for varied amounts of time. Following internalization of fluorescent particles, cells are washed with trypan blue, which facilitates extinction of fluorescent signal from bacteria which are not internalized, or are merely adhering to the cell surface. Following the trypan wash, cells are washed with PBS, fixed, and stained with DAPI (nuclear blue fluorescent label), which serves to label nuclei of cells. By a simple fluorometric quantification through plate reading of nuclear (blue) or particle (red/green) fluorescence we can examine the ratio of relative fluorescence units of green:blue and determine a phagocytic index indicative of amount of fluorescent bacteria internalized per cell. The duration of assay using a 96-well method and multichannel pipettes for washing, from end of phagocytosis to end of data acquisition, is less than 45 min. Flow cytometry could be used in a similar manner but the advantage of fluorometry is its high throughput, rapid method of assessment with minimal manipulation of samples and quick quantification of fluorescent intensity per cell. Similar strategies can be applied to non adherent cells, live labeled bacteria, actin polymerization, and essentially any process utilizing fluorescence. Therefore, fluorometry is a promising method for its low cost, high throughput capabilities in the study of cellular processes.
Keywords: Immunology, Issue 93, Fluorometry, phagocytosis, high throughput assay, actin polymerization, immunology
Introduction
Quantification of fluorescent signal has been widely used in a multitude of scientific methods ranging from PCR, flow cytometry, confocal microscopy, and FRET analysis to multiplex ELISA. Fluorescence imaging and quantification has a broad application and can be a great tool for quantitative analysis of various cellular processes. Use of fluorescent markers and their signal has been revolutionized in the last decade, and emergence of fluorescent plate readers has facilitated the high throughput quantification of fluorescence emitted during cellular processes.
Fluorometric analysis can serve as a great tool in quantification of phagocytosis. Phagocytosis has been studied since the discovery of phagocytes by Metchnikoff in 18001. Over the years, a variety of methods have been utilized to examine this important process essential for innate immune defense against invading bacterial, viral, fungal and parasitic pathogens2-5. Previous methods of quantification utilized microscopy and stereological techniques in order to visualize cells that are phagocytosing, which were then quantified by counting of internalized particles (manually or with use of software)6-8. Some disadvantages to using microscopy alone for quantitative analysis are that manual counting of bacteria is labor intensive and more prone to observer bias. Another method used in quantification of phagocytosis is the microbiological technique of plating of bacteria from cell lysates (following phagocytosis) on bacterial culture plates, but this method can fail to account for bactericidal mechanisms and presence of partially internalized bacteria. This method is even more labor intensive compared to microscopy and takes several days to analyze. Flow cytometry seems to be the quickest, most effective way to quantify phagocytosis and has been utilized by many groups9-11, but the high cost commonly associated with the instrument needed for the analysis makes it the most expensive method when compared to the previously mentioned assays.
The fluorometric method is a good alternative to flow cytometry for analysis of particle internalization since it offers unbiased quantification of fluorescence using equipment that is not as cost prohibitive. Other added benefits of fluorometry are high efficiency, and high throughput capabilities for quantifying fluorescence emitted by the labeled internalized particles.
Benefits of fluorometry can be extrapolated to quantification of processes other than phagocytosis. For example, fluorometric analysis can be applied to study any process leading to changes in expression of intracellular or membrane-bound receptors, changes in cell permeability/viability, transfection efficacy, and modulation in actin polymerization. One downside of fluorometric technique is that, depending on the labels used, there may be a high experiment to experiment variability which can usually be resolved by demonstrating data through relative quantification, such as fold change or percent increase from control.
Protocol
NOTE: This protocol can be used for multiple applications such as quantification of phagocytosis and actin polymerization as used in our previously published work12. The protocol lends itself to a variety of modification, and Table 1 lists cell types and particles successfully utilized in past studies. Standard use of this protocol for quantification of phagocytosis or actin polymerization is illustrated in Diagram 1.
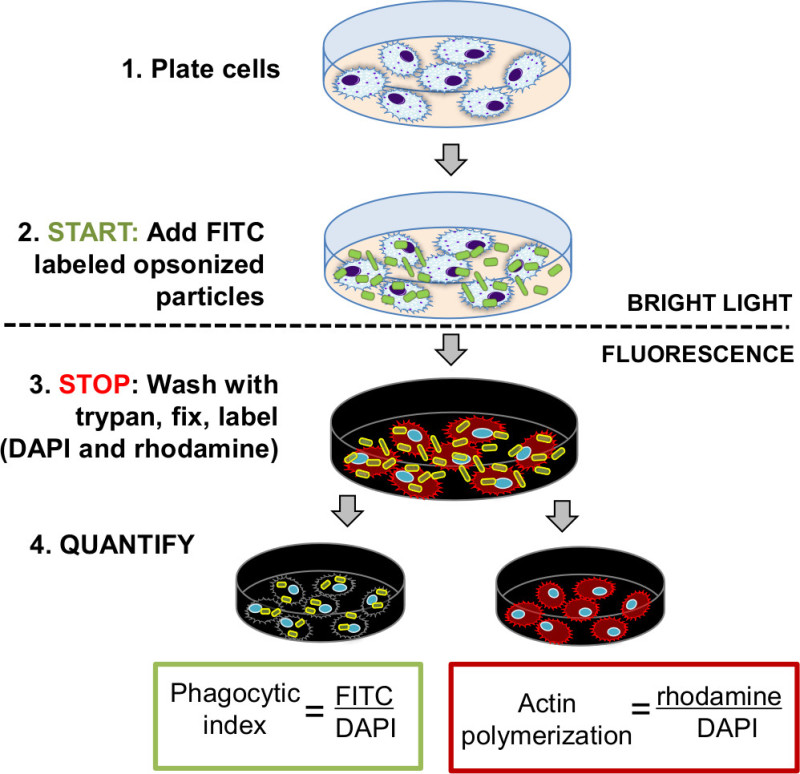
Diagram 1: Diagram of fluorometric assay for quantification of phagocytosis and actin polymerization. After the cells are plated, treated, and allowed to adhere, particles labeled with green fluorescence (FITC) are added to the cells for phagocytosis. Reaction is stopped by trypan or antibiotic wash (to eliminate non-internalized particles) and fixation is performed with 4% paraformaldehyde. Cells are subsequently stained with red fluorescent actin label (rhodamine phalloidin) and blue fluorescent label (DAPI). Indexes of phagocytosis and actin polymerization are quantified as a ratio of relative fluorescence units of green/blue (FITC/DAPI), or red/blue (rhodamine/DAPI) fluorescence.
1. Plating and Treating Cells
NOTE: Before beginning, consider running the treatments with at least 4 technical replicates, and include the following controls in the plate layout: cells only (unstained, with DAPI alone or rhodamine alone) and particle only.
Conduct all addition of reagents in steps 1-4 in a sterile cell culture hood in order to protect the samples from contamination.
- Plate macrophages (murine J774 cell line) in a 96-well plate at 1-5 x 104 cells in 50 μl (per well) of supplemented preheated media. Ideal plates for this method are black 96-well plates with clear or black bottoms, depending on reader capabilities.
- For supplemented media, use 10% heat inactivated FBS to avoid interference of complement cascade, and 1% penicillin/streptomycin (if not using live bacteria for phagocytosis). If using adherent cells allow 1-2 hr for adhesion, and for non-adherent cells, spin down the plate with cells for 10 min at 500 x g.
Add desired agonists/antagonists or vehicle control at 2x concentration (resuspended in PBS or more preferably, in supplemented media) in 50 μl for a final volume of 100 μl (1x) and incubate for a desired amount of time. Multichannel pipette and reagent reservoirs will facilitate fastest processing.
2. Opsonization of Fluorescent Particles
 Table 1: Tested cell types and fluorescent particles in fluorometric analysis of phagocytosis and actin polymerization.Table 1 illustrates combinations of cell types (cell lines, and primary cells) that our group has used via the fluorometric method. Other than looking at phagocytosis and actin polymerization by wild type cells, we have also utilized some of these particles for examination of phagocytosis by transfected cells expressing a GFP (green fluorescent protein). Fluorescence of the cells transfected with plasmids containing fluorescent reporters can also be used as a cellular marker in addition to DAPI. Table legend: A - actin polymerization; P - phagocytosis; PT - phagocytosis by GFP labeled transfected cells; OPDex - opsonized dextran bead; HKOP - heat killed opsonized.
Table 1: Tested cell types and fluorescent particles in fluorometric analysis of phagocytosis and actin polymerization.Table 1 illustrates combinations of cell types (cell lines, and primary cells) that our group has used via the fluorometric method. Other than looking at phagocytosis and actin polymerization by wild type cells, we have also utilized some of these particles for examination of phagocytosis by transfected cells expressing a GFP (green fluorescent protein). Fluorescence of the cells transfected with plasmids containing fluorescent reporters can also be used as a cellular marker in addition to DAPI. Table legend: A - actin polymerization; P - phagocytosis; PT - phagocytosis by GFP labeled transfected cells; OPDex - opsonized dextran bead; HKOP - heat killed opsonized.
- Ensure that fluorescent particles and labels are protected from light at all times, during processing, washing, as well as during the incubation in order to prevent photobleaching
- For dry particles, weigh out fluorescent heat killed opsonized (HKOP) bacteria (E2861: E. coli K-12 strain), using manufacturer’s specifications. Take care to make sure the mass accounts for 10:1 - 20:1 bacteria:cell ratio (or at least a 10:1 ratio - commonly used in quantification of phagocytosis)13,14. Number of particles can be provided by the manufacturer or determined via couting serial dilutions of 1 mg/ml stock solution.
- Resuspend in 50 μl PBS and vortex for 1 minute at highest setting. Ensure proper vortexing (here and throughout the protocol) as the particles tend to aggregate which may affect efficacy of opsonization.
- For particles in suspension such as fluorescently labeled dextran beads (DEX beads), vortex the stock solution for 1 min, take 50 μl of suspension or depending on the bead concentration in the volume determine mount of beads to facilitate 10:1 bead:cell ratio.
Add an equal volume of the opsonizing reagent to the particles and vigorously vortex for another minute.
Incubate the particles with opsonizing reagent at 37 °C for 1 hr.
- Following the incubation, centrifuge the particles for 5 min at 500 x g, and remove the supernant containing excess opsonizing reagent. Wash the particles by adding 100 μl of PBS, vortex to resuspend the pellet, and centrifugate for 5 min at 500 x g.
- Repeat these steps two times to assure proper opsonization and removal of unbound antibody.
Prepare the working solution of opsonized particles by resuspending them in 5 ml of media (sufficient for one 96-well plate) and store away from light until addition to the cells.
3. Phagocytosis
Place the working solution of particles in a sterile reagent reservoir. From here, add 50 μl of opsonized particles to the cells prepared in step 1 (for a final concentration of 50 ng/ml) and allow phagocytosis to occur at standard cell culture conditions. Illustrated below are two methods of phagocytosis which can be assessed using this method (Diagram 2).
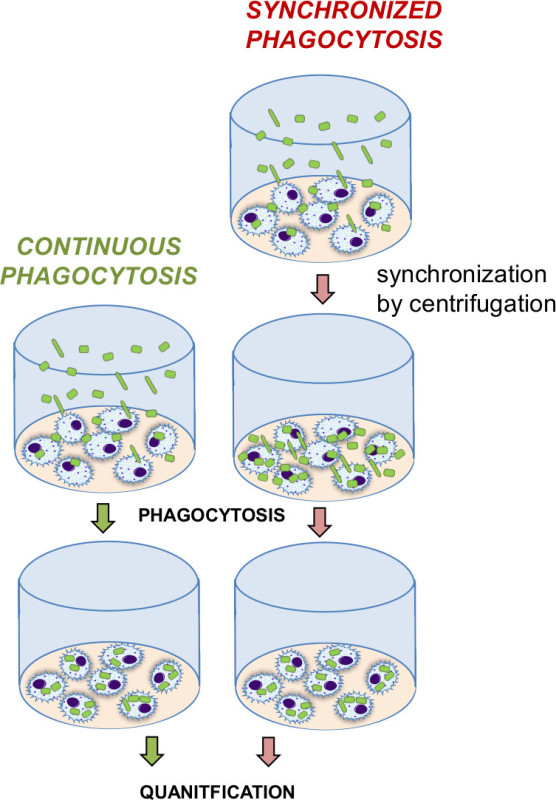 Diagram 2: Comparison of continuous versus synchronize phagocytosis. Continuous phagocytosis indicates continuous internalization of particles over time as they slowly reach the cells on the bottom of the well. A contrasting synchronization step (centrifugation) forces the particles to sink to the bottom, enhancing the particle contact with the cell, and leading to immediate internalization by the cells. Synchronized phagocytosis is a quicker process which more rapidly internalizes particles due to the increased cell:particle contact.
Diagram 2: Comparison of continuous versus synchronize phagocytosis. Continuous phagocytosis indicates continuous internalization of particles over time as they slowly reach the cells on the bottom of the well. A contrasting synchronization step (centrifugation) forces the particles to sink to the bottom, enhancing the particle contact with the cell, and leading to immediate internalization by the cells. Synchronized phagocytosis is a quicker process which more rapidly internalizes particles due to the increased cell:particle contact.
- For the continuous phagocytosis method (Diagram 2) that allow cells to continuously phagocytose opsonized bacteria as they slowly sink to the bottom and come in contact with cells, utilize longer time frames to allow for the bacteria to reach the bottom of the well and get in touch with the cells. If conducting a timecourse experiment, add bacteria at different time intervals and stop the entire plate of reactions at the same time to enhance the speed of processing.
- For the synchronized phagocytosis method (Diagram 2) that enhances particle: cell contact via centrifugation, spin down the entire plate containing cells and particles (5 min at 500 x g) as soon as the particles are added, to facilitate synchronization of all time points and to expedite the particle: cell interaction.
- Measure the time course starting right after the completion of the centrifugation step. If conducting a time course experiment, stop each time point independently at different time intervals as described below.
NOTE: The synchronized phagocytosis method is somewhat more laborious from the previous one, but provides higher phagocytic index in a shorter time.
4. Stopping Phagocytosis
For adherent cells, remove the supernatant and add 50 μl of diluted trypan blue with PBS (1:2 dilution), in order to extinguish fluorescence of non-internalized bacteria.
For non-adherent cells, spin down the plate and remove supernatant. Add 50 μl of diluted trypan blue. Ensure to centrifuge the plate at (5-10 min at 500 x g) between each wash in order to minimize the cell loss and enable adequate washing.
Following trypan incubation, wash cells two times with PBS to remove any residual trypan (until the PBS removed from the well turns clear). Trypan has been shown to quench fluorescence15,16 of non-internalized fluorescently labeled, heat killed, bacterial particles, but not for live bacteria or OPDex beads. For live bacteria, include a wash with antibiotics such as penicillin, streptomycin, or gentamicin instead of the trypan wash to avoid confounding effects of fluorescence from non-internalized particles.
Fix cells with 100 μl of 4% paraformaldehyde, and incubate for 15 min at room temperature, then wash with PBS (at 100 μl per well for each wash). At this step, save the cells at 4 °C for up to a week, or proceed directly to staining and quantification. NOTE: All the subsequent steps form this point foreword can be done outside of the cell culture hood.
Remove all liquid and add 50 μl of DAPI at 5 ng/ml reconstituted in PBS. Allow 5 min for staining, and then wash twice with PBS. Following the wash step, add 50 μl of PBS to each well and record phagocytosis.
5. Actin Staining
NOTE: In addition to phagocytosis, fluorometric method will enable quantification of actin polymerization by measuring intensity of actin, which is reduced during inhibition of lamellipodia, pseudopodiae, and membrane ruffling as a result of treatment with an inhibitor (Figure 1A). This is an optional step if interested in quantification of actin polymerization in addition to phagocytosis. If actin polymerization is the final goal, using fluorescent particles for phagocytosis is optional.
Following paraformaldehyde fixation (step 4.3), wash the plate twice with PBS at 100 μl per well for each wash. Add 50 μl of 0.1% Triton X-100 in PBS for permeabilization and incubate for -5 min at RT.
Wash out 0.1% Triton X-100 2 times with PBS (as above), and block with 1% bovine serum albumin (BSA) for 20 min to avoid non-specific binding of the label.
Following blocking, remove the BSA solution and immediately add rhodamine phalloidin. To prepare the working solution of rhodamine stain, use 100 μl of methanoic solution (provided by the manufacturer) in 5 ml of PBS (sufficient for one plate). From the working solution add 50 μl per well and incubate for 20 min at room temperature. NOTE: Following the washing steps and DAPI staining as in step 4.4 cells will be ready for quantification.
6. Quantification
To quantify the phagocytosis using a fluorescent plate reader, record green fluorescence using excitation filter at λ = 488 nm and emission filter at λ = 518 nm, and blue fluorescence using excitation filter at λ = 355 nm and emission filter at λ = 460 nm.
To quantify the actin polymerization using a fluorescent plate reader, record blue fluorescence via λ = 355 nm and λ = 460 nm (as above), and red fluorescence via excitation filter at λ = 584 nm and emission filter λ = 620 nm. NOTE: Orbital or central detection is acceptable for recording. Some cell types gather more to the edge of the well than the center, in which case orbital recording can be of advantage. In particular, average of three points around the orbit could also be used to obtain more accurate results.
Divide the total RFU of green or red fluorescence by blue fluorescence readout (from DAPI), depending on the particle label or phagocytic vs. actin quantification (Diagram 1). Due to high plate-to-plate variance, use relative indexes (such as relative to t = 0 control), to best illustrate trends observed and to facilitate more reliable statistical analysis.
Representative Results
Two major ways of quantifying phagocytosis and the subsequent actin polymerization through the use of this protocol is to observe continuous or synchronized phagocytosis.
Microscopic analysis (Figure 1B) illustrates fluorescent images comparable to what the fluorometer is recording (also see Diagram 1). In Figure 1B are red fluorescence of actin, green 268 fluorescence of FITC labels HKOP E. coli, nuclear DAPI stain, and the merged image of all three 269 fluorophores together. This image indicates effective phagocytosis, actin polymerization, and 270 effective trypan quenching of non-internalized particles.
Continuous phagocytosis of opsonized and unopsonized particles is quite comparable as seen in Figure 2A and C respectively, where J774 cells are internalizing dextran beads. Interestingly, actin polymerization following opsonophagocytosis increases, slowing down at later time points (Figure 2B), while the actin polymerization following internalization of unopsonized particles does not increase (Figure 2D). This is important to consider for future studies examining these processes since opsonophagocytosis leads to much greater actin polymerization than internalization of particles that are not opsonized. Neither of the phagocytic methods using dextran particles result in changes in cell viability as was confirmed via the MTT analysis (Figure 2E).
Unlike continuously increasing trends observed during continuous phagocytosis (Figure 2), synchronized phagocytosis of opsonized dextran beads has a bell shaped trend (Figure 3A), showing reduction in internalization following 30 min. This is to be expected considering processing time and nature of the protocol. This trend is also confirmed by the actin polymerization data (Figure 3B) of these cells where the highest polymerization time point is early on in the timecourse, while at 30 min polymerization returns to baseline. Although more labor intensive, this method has a value of examining phagocytosis right after the synchronization, therefore examining immediate phagocytic effects.
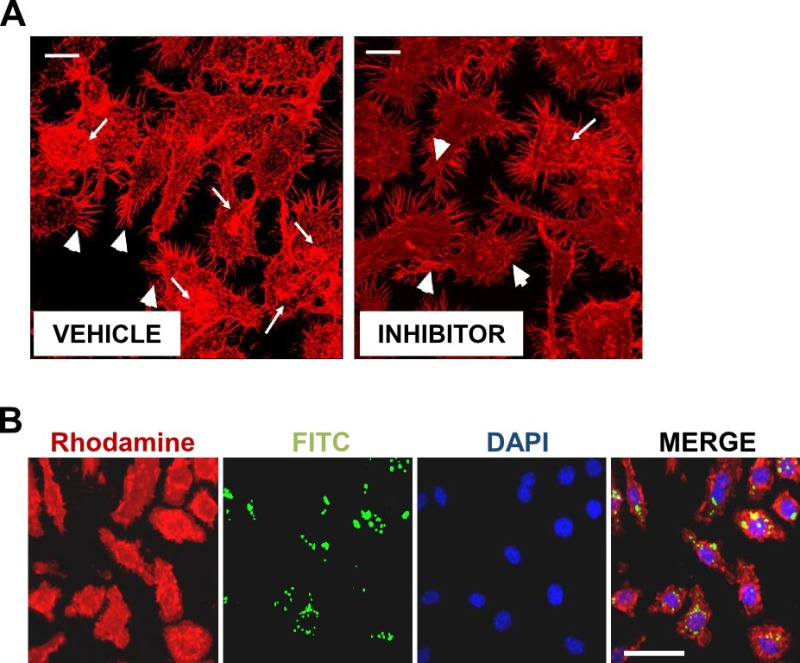 Figure 1: Confocal microscopy of phagocytosis and actin polymerization. Cells were allowed to internalize FITC conjugated OPDex bead (green) at 20:1 bead:cell ratio for 60 min, then cells were washed with trypan and PBS, fixed with paraforlmaldehyde, permeabilised with acetone, and stained for f-actin using rhodamine phalloidin (red), and DAPI (blue). Images illustrate confocal microscopic analysis at 60X with additional digital magnification; (A) white bar = 10 µm (B) white bar = 50 µm. (A) Illustrates changes in actin polymerization following addition of inhibitor. Arrowheads indicate pseudopodia formation, and arrows indicate changes in membrane ruffling. Figure 1A has been modified from Ninkovic and Roy12. Please click here to view a larger version of this figure.
Figure 1: Confocal microscopy of phagocytosis and actin polymerization. Cells were allowed to internalize FITC conjugated OPDex bead (green) at 20:1 bead:cell ratio for 60 min, then cells were washed with trypan and PBS, fixed with paraforlmaldehyde, permeabilised with acetone, and stained for f-actin using rhodamine phalloidin (red), and DAPI (blue). Images illustrate confocal microscopic analysis at 60X with additional digital magnification; (A) white bar = 10 µm (B) white bar = 50 µm. (A) Illustrates changes in actin polymerization following addition of inhibitor. Arrowheads indicate pseudopodia formation, and arrows indicate changes in membrane ruffling. Figure 1A has been modified from Ninkovic and Roy12. Please click here to view a larger version of this figure.
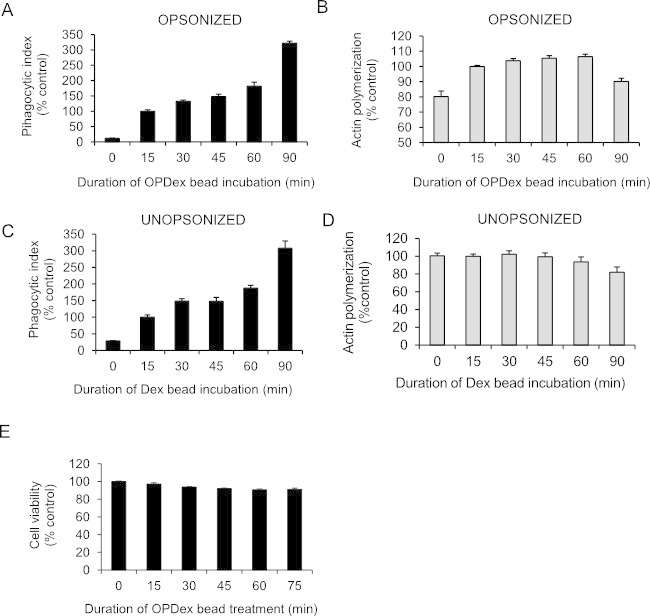 Figure 2: Fluorometric analysis following continuous phagocytosis. J774 murine macrophages were plated onto a 96-well plate and exposed to dextran beads at varying amounts of time, starting with 90, 60, 45, 30, and 15 min. Beads were added at different times and the phagocytic processes for all the reactions were stopped at the same time. Cells were washed with trypan, fixed, stained, and analyzed for phagocytosis of opsonized (A) or unopsonized (C) dextran particles and actin polymerization associated with each (B, D respectively). (E) MTT assay was conducted immediately following phagocytosis at different time points. This figure has been modified from Ninkovic and Roy12.
Figure 2: Fluorometric analysis following continuous phagocytosis. J774 murine macrophages were plated onto a 96-well plate and exposed to dextran beads at varying amounts of time, starting with 90, 60, 45, 30, and 15 min. Beads were added at different times and the phagocytic processes for all the reactions were stopped at the same time. Cells were washed with trypan, fixed, stained, and analyzed for phagocytosis of opsonized (A) or unopsonized (C) dextran particles and actin polymerization associated with each (B, D respectively). (E) MTT assay was conducted immediately following phagocytosis at different time points. This figure has been modified from Ninkovic and Roy12.
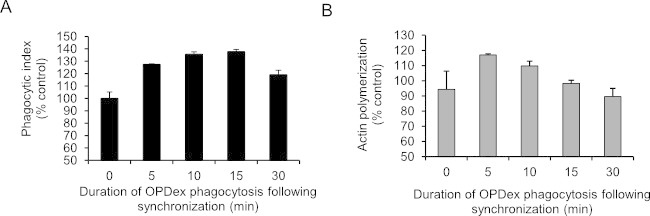 Figure 3: Fluorometric analysis following synchronized phagocytosis. FITC labeled OPDex beads were added to J774 murine macrophage cells plated in 96-well plates at a cell density of 104 cell/well. After a 30 min incubation the plate was centrifuged at 500 x g for 5 min. Phagocytosis was stopped one at a time, at time points stated in the x-axis by trypan wash, followed by PBS washes, paraformaldehyde fixation and actin staining. This figure has been modified from Ninkovic and Roy12.
Figure 3: Fluorometric analysis following synchronized phagocytosis. FITC labeled OPDex beads were added to J774 murine macrophage cells plated in 96-well plates at a cell density of 104 cell/well. After a 30 min incubation the plate was centrifuged at 500 x g for 5 min. Phagocytosis was stopped one at a time, at time points stated in the x-axis by trypan wash, followed by PBS washes, paraformaldehyde fixation and actin staining. This figure has been modified from Ninkovic and Roy12.
Discussion
Major limitations of the fluorometric technique are experimental variance as well as cell loss associated with extensive washing and use of non-adherent cells.
Variance is observed largely due to the variation in fluorescent particles as weighing and pipetting during preparation of the stock solution is not an exact way of maintaining identical numbers of particles from experiment to experiment. To address the problem of variance between experiments, data can be expressed in relative terms, for example as % phagocytosis or fold change from control (control – t = 0; particles added and removed immediately). This form of analysis generates reproducible trends, and values of % phagocytosis from control is similar from experiment to experiment. This kind of data analysis will facilitate statistical significance with 3-6 experimental replicates. Fluorometric technique can be used to effectively examine and reproduce observed trends despite of its variability if utilizing adequate data analysis. With mastering of the technique it is possible for a single scientist to run approximately six 96-well plates, or 600 treatments in a day.
Cell loss has been largely observed in studies with non-adherent cell types. Adherent cell types are the best to use in methods such as this one because they can withstand of the numerous and extensive washing steps.
Major critical steps should be taken to keep the fluorescent components away from direct light to avoid photo bleaching. Particles, labels, and the plate containing each or all components should be kept in dark (or simply wrapped in aluminum foil). Another essential component is the importance of technical replicates within a plate. Due to high well to well variance it is essential to mix all the reagents very well before pipetting and to have 6-8 technical replicates per treatment (equivalent to one row), in order to most accurately observe trends in the data.
Additional considerations important for use of plate formats such as these, is that the plate wells contain a relatively small volumes of reagents and are often prone to evaporation. If the treatments are longer than 24 hr, it is advisable to use the peripheral wells of the plate (rows and columns next to the edge) containing PBS alone, in order to serve as humidifiers and to reduce evaporation of cell culture media and agonists.
This technique is fairly simple to master and the troubleshooting involved is minimal. It is important to choose the right phagocytic method for the study and to be careful while washing (especially when using non-adherent cells) in order to avoid cell loss.
Major significance of the fluorometric technique is that it provides a high-throughput method for screening of fluorescent particle internalization, or actin polymerization. Most techniques used to date such as microscopy or microbiology are more labor intensive and more time consuming in comparison. Flow cytometry utilizes very similar principles as fluorometry, but takes hours to quantify compared to minutes required by fluorometry. Therefore, the fluorometric method is a good alternative to current methods of phagocytic quantification largely due to its high-throughput capabilities.
Disclosures
The authors have no competing financial interests.
Acknowledgments
The authors would like to thank the following funding sources: RO1 DA 12104, RO1 DA 022935, RO1 DA031202, K05DA033881, P50 DA 011806, 1R01DA034582 (to S.R) and F31 DA026264-01A1, T32 DA07097 (to J.N.).
References
- Murphy K. Janeway's Immunobiology. 8th edn. New York, NY: Garland Science; 2012. [Google Scholar]
- Burke DW, O'Connor DO, Zalenski EB, Jasty M, Harris WH. Micromotion of cemented and uncemented femoral components. The Journal Of Bone And Joint Surgery. British Volume. 1991;73(1):33–37. doi: 10.1302/0301-620X.73B1.1991771. [DOI] [PubMed] [Google Scholar]
- Duan X, et al. Resistance to malaria by enhanced phagocytosis of erythrocytes in LMP7-deficient mice. PloS One. 2013;8(3):59633. doi: 10.1371/journal.pone.0059633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dementhon K, El-Kirat-Chatel S, Noel T. Development of an in vitro model for the multi-parametric quantification of the cellular interactions between Candida yeasts and phagocytes. PloS One. 2012;7(3):32621. doi: 10.1371/journal.pone.0032621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Bader N, et al. Role of trehalose biosynthesis in Aspergillus fumigatus development, stress response, and virulence. Infection And Immunity. 2010;78(7):3007–3018. doi: 10.1128/IAI.00813-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Iborra B, et al. Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-gamma production through the HIF-1 alpha transcription factor. Journal Of Immunology. 2009;182(5):3155–3164. doi: 10.4049/jimmunol.0801710. [DOI] [PubMed] [Google Scholar]
- Ojielo CI, et al. Defective phagocytosis and clearance of Pseudomonas aeruginosa in the lung following bone marrow transplantation. Journal Of Immunology. 2003;171(8):4416–4424. doi: 10.4049/jimmunol.171.8.4416. [DOI] [PubMed] [Google Scholar]
- Neu C, et al. CD14-dependent monocyte isolation enhances phagocytosis of listeria monocytogenes by proinflammatory, GM-CSF-derived macrophages. PloS One. 2013;8(6):66898. doi: 10.1371/journal.pone.0066898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolovska A, et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nature Immunology. 2013;14(6):543–553. doi: 10.1038/ni.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janko C, et al. CRP/anti-CRP antibodies assembly on the surfaces of cell remnants switches their phagocytic clearance toward inflammation. Frontiers in Immunology. 2011;2(2):70. doi: 10.3389/fimmu.2011.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatworthy MR, et al. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcgammaRIIb reduce susceptibility to malaria. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(17):7169–7174. doi: 10.1073/pnas.0608889104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkovic J, Roy S. Morphine decreases bacterial phagocytosis by inhibiting actin polymerization through cAMP-, Rac-1-, and p38 MAPK-dependent mechanisms. The American Journal Of Pathology. 2012;180(3):1068–1079. doi: 10.1016/j.ajpath.2011.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix RN, Altschuler SE, Henson PM, Detweiler CS. Hemophagocytic macrophages harbor Salmonella enterica during persistent infection. PLoS Pathogens. 2007;3(12):193. doi: 10.1371/journal.ppat.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, et al. Stable gene transfer and expression in cord blood-derived CD34+ hematopoietic stem and progenitor cells by a hyperactive Sleeping Beauty transposon system. Blood. 2009;114(7):1319–1330. doi: 10.1182/blood-2009-03-210005. [DOI] [PubMed] [Google Scholar]
- Hed J. Methods for distinguishing ingested from adhering particles. Methods in Enzymology. 132. 1986. pp. 198–204. [DOI] [PubMed]
- Scott AJ, Woods JP. Monitoring internalization of Histoplasma capsulatum by mammalian cell lines using a fluorometric microplate assay. Medical Mycology. 2000;38(1):15–22. doi: 10.1080/mmy.38.1.15.22. [DOI] [PubMed] [Google Scholar]