

Abstract
Anatomical path tracing is of pivotal importance to decipher the relationship between brain and behavior. Unraveling the formation of neural circuits during embryonic maturation of the brain however is technically challenging because most transsynaptic tracing methods developed to date depend on stereotaxic tracer injection. To overcome this problem, we developed a binary genetic strategy for conditional genetic transsynaptic tracing in the mouse brain. Towards this end we generated two complementary knock-in mouse strains to selectively express the bidirectional transsynaptic tracer barley lectin (BL) and the retrograde transsynaptic tracer Tetanus Toxin fragment C from the ROSA26 locus after Cre-mediated recombination. Cell-specific tracer production in these mice is genetically encoded and does not depend on mechanical tracer injection. Therefore our experimental approach is suitable to study neural circuit formation in the embryonic murine brain. Furthermore, because tracer transfer across synapses depends on synaptic activity, these mouse strains can be used to analyze the communication between genetically defined neuronal populations during brain development at a single cell resolution. Here we provide a detailed protocol for transsynaptic tracing in mouse embryos using the novel recombinant ROSA26 alleles. We have utilized this experimental technique in order to delineate the neural circuitry underlying maturation of the reproductive axis in the developing female mouse brain.
Keywords: Neuroscience, Issue 94, developmental biology, neural circuits, transsynaptic tracing, barley lectin, Tetanus Toxin fragment C, mouse embryo, gene targeting, ROSA26 locus, kisspeptin, GnRH, reproduction
Introduction
Anatomical path tracing is one of the most commonly utilized tools to decipher the relationship between brain and behavior1. Advancement in neural circuit tracing technologies has bestowed neuroscientists with the ability to trace neural circuits from genetically identified neuron populations in mice2. In spite of these technical advancements it remains challenging to unravel the formation of neural circuits especially during embryonic maturation. This is because most of the tracing methods developed to date are based upon stereotaxic injection of transsynaptic tracers or genetically modified neurotropic viruses (Figure 1)2,3. While these techniques achieve spatial and temporal resolution of connectivity, several inherent limitations such as technically challenging tracer injections into the developing brain, the reproducibility of the site of injection, potential inflammation at the injection site and most importantly cytotoxicity caused by neurotropic viruses limit their use4.
An alternative method is to express the transsynaptic tracers as transgenes in genetically altered mice. We have recently modified this technique and developed a binary genetic transsynaptic tracing system to map the neural circuits of any genetically identified neuronal population5. Our experimental strategy is based on two new knock-in mouse strains, which express either the bidirectional tracer barley lectin (BL)6 or the retrograde tracer Tetanus Toxin fragment C fused to GFP (GTT)7 from the ROSA26 locus after Cre-mediated recombination. Here we used these mouse strains to selectively express BL and GTT in neurons that produce kisspeptin, a neuropeptide that is implicated in regulating the maturation of the reproductive axis8,9. We demonstrate that this technique is suitable to visualize the development and maturation of kisspeptin neural circuitry during embryonic development of the female mouse brain5.
Breeding strategy
The R26-BL-IRES-τlacZ (BIZ) and the R26-GFP-TTC (GTT) tracer lines are knock-in strains5 that carry recombinant ROSA26 alleles. The R26-BIZ and the R26-GTT alleles are transcriptionally silent due to the presence of a strong transcriptional stop signal, which is flanked by two loxP sites5. Expression of the BIZ and GTT transgene is activated by Cre-mediated removal of the transcriptional stop signal. The R26-BIZ and R26-GTT alleles can be used independently by simply crossing with a Cre driver line. For analysis animals heterozygous for the respective Cre and R26 alleles can be used. Littermates carrying one Cre or one R26 allele, respectively, should be used as controls. Alternatively, it is also possible to generate triple knock-in animals carrying the Cre, R26-BIZ and R26-GTT alleles, however this will require one additional cross.
Protocol
NOTE: Ethics Statement: Procedures involving animal subjects were approved by the animal welfare committee of the University of Hamburg and the University of Saarland.
1. Preparation and Fixation of Embryonic Tissue
Arrange all the equipment needed to dissect out the embryos and prepare solutions for subsequent fixation of the tissue before sacrificing the animals. NOTE: Always prepare a fresh 4% paraformaldehyde (PFA) solution (4% PFA in 0.1 M phosphate buffered saline (PBS), adjust pH to 7.4).
Euthanize timed pregnant female mouse with an ethically approved procedure.
Transfer the mouse onto a platform. Wet the ventral side of the abdomen with 70% ethanol and make a vertical incision using sharp scissors to expose the abdominal cavity.
Identify the embryonic chain and pull it out carefully using blunt forceps and place it into ice-cold PBS.
Remove the individual embryos from the embryonic chain using fine scissors and forceps. Remove extra tissue around the embryos and wash them in ice-cold PBS.
Take a small tail biopsy (before transferring the embryo into PFA) and incubate it in tail lysis buffer for the gender identification and tracer allele genotyping PCR.
Start from the embryo at the most lateral side and transfer each embryo separately into a tube containing ice-cold 4% PFA on ice on a shaker.
Soak the embryos at the age of E13.5 or younger for 1.5 hr on ice with constant shaking. Soak the embryos at age E14.5 and E15.5 for 2.5 hr on ice with constant shaking.
For embryos age E16.5 or older, decapitate and remove the outer skin before soaking the head in PFA solution. Heads of E16.5 embryos can be soaked for 4.5 hr on ice with constant shaking.
After appropriate fixation, wash the embryos three times with ice-cold PBS. Transfer the embryos into 30% sucrose in PBS solution at 4 °C until they sink to the bottom.
2. Freezing
Arrange all the materials required before starting the freezing process: cryo-mold, cryo-gloves, featherweight entomology forceps, marker pen and optimal cutting temperature compound (O.C.T.)
Label the cryo-mold with a permanent alcohol-resistant marker (mention the orientation of tissue, date and genotype). Prepare a slush of crushed dry ice and 100% ethanol in an ice bucket. Place a glass beaker inside containing isopentane. Wait for 10 min for the isopentane to cool down.
Meanwhile remove the tissue from the sucrose solution, wipe off excess sucrose around the tissue using laboratory wipes. Transfer the tissue into a pre-labeled cryo-mold with sufficient O.C.T. to cover the tissue. Avoid any air bubbles in the O.C.T.; especially around the tissue.
Orient the tissue in O.C.T. using featherweight entomology forceps to avoid tissue damage. Start freezing by transferring the cryo-mold into the pre-cooled isopentane bath. Do not splash isopentane into the O.C.T. as it will not freeze properly. Wrap the samples in aluminum foil and store at -80 °C.
3. Cryosectioning
Cut 14 µm thin serial sections in series’ of five using a cryostat and collect on SuperFrost® Plus glass slides for immunofluorescence (IF) analysis. Store the slides at -80 °C until utilized.
4. Tracer visualization Using the Tyramide Signal Amplification (TSA) Protocol
- Prepare the buffers and reagents required for staining. Always freshly prepare the Tris-NaCl-Tween (TNT) buffer on the day of use.
- Prepare TNT buffer using 100 ml of 1 M Tris-HCl pH 7.5, 30 ml of 5 M NaCl and ddH2O up to a final volume of 1 L. Add 500 µl of Tween 20 using a 1 ml pipette. Add the Tween 20 slowly and flush the pipette up and down several times to ensure that all Tween 20 is in solution.
- Prepare Tris-NaCl-Blocking (TNB) buffer using 100 ml of 1 M Tris-HCl pH 7.5, 30 ml of 5 M NaCl and ddH2O up to a final volume of 1 L. Add 5 g Blocking Reagent (provided with the kit) slowly in small increments to the buffer while stirring. Heat the solution gradually to 55 °C with continuous stirring to completely dissolve the Blocking Reagent (this should not take longer than 30-60 min). To achieve homogenous heating use a water bath at 55 °C. The solution will appear milky. Bring the solution to room temperature before using. Aliquot and store at -20 °C for long-term storage.
- Reconstitute biotin tyramide reagent (provided with the kit) in Molecular Biology/HPLC-grade DMSO. Depending upon which kit is used the appropriate amount of DMSO may vary. Store the stock solution at 4 °C. NOTE: Sections from animals heterozygous for the respective Cre and R26 alleles can be used for circuit analysis. Use sections from littermates carrying one Cre or one R26 allele, respectively, as negative controls. Treat negative control slides exactly like slides from double heterozygous animals. In addition, include one control slide from a double heterozygous animal but leave out the TSA reagent (unamplified control).
Using a sharp fresh scalpel remove excess O.C.T. surrounding the tissue sections and mark the boundary around the sections with a PAP pen.
Dry the slides for 2-3 min at room temperature (RT). Wash the slides at RT 3x with TNT for 5 min each. Incubate the slides at RT for 30 min in 0.3% H2O2 solution in ice-cold methanol to quench endogenous peroxidase activity.
Wash 3x with PBS for 5 min each at RT. Incubate for 10 min in 0.5% Triton-X 100 in PBS for tissue permeabilization. Wash the slides at RT 3x with TNT for 5 min each.
Block with TNB (blocking solution, 200-300 µl per slide) for 30 min at RT in a humidified chamber. Make sure all the sections are completely covered by TNB. Do not let the slides dry out in between the steps.
Drain off excess blocking buffer and apply the primary antiserum recognizing the tracer (1:1,000 goat anti-WGA for BL, 1:15,000 rabbit anti-GFP for GTT) diluted in TNB for 2 hr at RT or overnight at 4 °C. Make sure to use enough volume to completely cover the tissue section (200-300 µl per slide). NOTE: The primary antisera dilutions recommended here may have to be adjusted.
Wash the slides at RT 3x with TNT for 5 min each with agitation.
Incubate the sections with biotinylated secondary antiserum recognizing the host of the first antiserum (1:500 horse anti-goat IgG for anti-WGA, 1:5,000 goat anti-rabbit IgG for anti-GFP in TNB for 1 hr at RT).
Wash 3x with TNT at RT for 5 min each with agitation. Incubate in 1:100 streptavidin (SA)-conjugated horseradish peroxidase (SA-HRP, provided with the kit) in TNB for 30 min at RT in a humidified chamber. Wash the slides at RT 3x with TNT for 5 min each. Meanwhile thaw the TSA.
Dilute TSA 1:50 in TSA amplification diluent (provided with the kit; use the TSA kit for the visualization of BL and the TSA+ kit for GTT). Incubate the tissue sections in diluted TSA for precisely 10 min at RT. Dilute TSA just before use, ideally during the previous washing step, do not use any remaining diluted TSA reagent for future experiments; always prepare fresh.
Wash the slides at RT 3x with TNT for 5 min each with agitation. Incubate with 1:500 Alexa Fluor® 546 streptavidin conjugate in TNB for 30 min at RT. Wash the slides at RT 3x with TNT for 5 min each with agitation.
Incubate for 10 min in 5% bisbenzimide solution in PBS at RT for nuclear staining. Wash the slides at RT 3x with TNT for 5 min each with agitation. Mount with Fluromount G and perform epifluorescence or confocal microscopy. Troubleshooting: Efficacy of tracer transfer will depend on the expression level of the ROSA26 locus in the producing cells (determined by the Cre driver line used) and on the neural circuit analyzed (e.g. number of neurons producing the tracer, convergence etc.). Therefore the primary antisera dilutions recommended here may have to be adjusted to prevent low or excess signal. Background should always be analyzed using appropriate control slides (see above).
5. τlacZ Staining for Embryonic Sections
- Prepare the buffers and reagents required for staining. Always freshly prepare the LacZ buffer C before use.
- Prepare 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) stock solution (40 mg/ml) by adding 40 g of X-gal to 1 ml 70% dimethylformamide (DMF). Cover with aluminium foil and store at -20 °C for long-term storage.
- Prepare nitro blue tetrazolium (NBT) stock solution (50 mg/ml) by adding 50 g of NBT to 1 ml 70% DMF. Cover with aluminium foil and store at -20 °C for long-term storage.
- Prepare LacZ fixative using 80 µl of 25% glutaraldehyde, 5 ml of 4% PFA, 20 µl of 1 M MgCl2, 100 µl of 500 mM EGTA, 1 ml of 1 M phosphate buffer pH 7.4 and ddH2O up to a final volume of 10 ml.
- Prepare LacZ buffer A using 1 ml of 1 M MgCl2, 5 ml of 500 mM EGTA, 50 ml of 1 M phosphate buffer pH 7.4 and ddH2O up to a final volume of 500 ml.
- Prepare LacZ buffer B using 1 ml of 1 M MgCl2, 5 ml of 500 mM EGTA, 5 ml of 1% sodium deoxycholate, 100 ml of 10% NP-40, 50 ml of 1 M phosphate buffer pH 7.4 and ddH2O up to a final volume of 500 ml.
- Prepare LacZ buffer C by adding 10 µl of NBT stock solution and 12.5 µl of X-gal stock solution to 1 ml of LacZ buffer B. Make fresh just before use and cover with aluminium foil.
Dry the slides at RT at least for 10-15 min (meanwhile prepare the slides with a PAP pen).
Fix the section with LacZ fixative for 2 min at room temperature in a humidified chamber inside a chemical fume-hood. Wash 3 times with PBS supplemented with 2 mM MgCl2.
Rinse the slides with LacZ buffer A. Wash the slides with LacZ buffer A 3 times for 10 min each at RT. Wash 2 times with LacZ buffer B for 5 min each. Incubate the slides in LacZ buffer C at 30-37 °C for 6 hr or overnight in a humidified chamber. NOTE: Add sufficient volume of LacZ buffer C; slides should not dry out during incubation.
After appropriate incubation wash the slides with PBS supplemented with 2 mM MgCl2. Rinse sections briefly with ddH2O. Coverslip with Mayer’s glycine gelatin. Suitable for a light microscope equipped with a differential interference contrast (DIC) imaging set-up.
6. Gender and Tracer Genotyping
Prepare tail lysis buffer using 1 ml of 1 M Tris-HCl pH 8.5, 100 µl of 500 mM EGTA, 200 µl of 10% SDS, 400 µl of 5 M NaCl and ddH2O to make up a final volume of 10 ml.
Add 500 µl of tail lysis buffer to each Eppendorf tube containing a tail biopsy. Add proteinase K to a final concentration of 100 mg/ml. Incubate overnight on a shaker at 55 °C.
Centrifuge samples at 17,000 x g for 10 min at RT. Transfer the supernatant to a new centrifuge tube. Add 500 µl of isopropanol to the supernatant. Mix for at least 5 min by inverting the tubes up and down.
Spin in centrifuge at 17,000 x g for 5 min at RT. Pour off the supernatant. Add 200 µl of 70% ethanol. Shake for at least 5 min.
Centrifuge at 17,000 x g for 5 min. Remove supernatant with a Pipetman (do not pour off). Dry the pellet in a warm chamber (37 °C) for 15-30 min.
Resuspend pellet in 100 µl of low TE pH 8.0 or water using wide-bore filtered tips. Put in 55 °C for 1 hr and at 37 °C overnight for proper dissolving. Store at 4 °C.
Use 1 µl of DNA for PCR reaction (50 μl reaction mixture containing 1 μl of tail DNA, 2.5 μl DMSO, 10 pM of each primer, 25 μM of each dNTP, and 1M betaine). PCR primers for genotyping and gender identification are listed in the Materials table.
Representative Results
This section shows representative results that can be obtained working with the R26-BIZ (BL-IRES-τlacZ) and the R26-GTT (GFP-TTC) alleles. Here we use the R26-BIZ and the R26-GTT alleles to analyze the maturation of the neural circuits regulating the reproductive axis. Reproduction in vertebrates is centrally controlled by a small subset of neurons in the hypothalamus, which secrete gonadotropin-releasing hormone (GnRH). Kisspeptin, a potent activator of GnRH neurons, has been implicated in regulating the activity of GnRH neurons, however when the neural circuits between GnRH and kisspeptin neurons are established in the developing female mouse brain was not known5. First we show that expression of the BIZ and GTT transgenes are specifically activated by Cre-dependent recombination in embryonic kisspeptin neurons in the arcuate nucleus (ARC) (Figure 2) using a kisspeptin-specific Cre driver line10. We then demonstrate that kisspeptin neurons in the ARC already communicate with a specific subset of GnRH neurons in utero (Figure 3A, B). Furthermore, we show that ARC kisspeptin neurons are upstream of GnRH neurons (Figure 3A, C). Taken together our findings indicate that the neural circuits between kisspeptin and GnRH neurons are fully established and operative in the female mouse brain before birth.
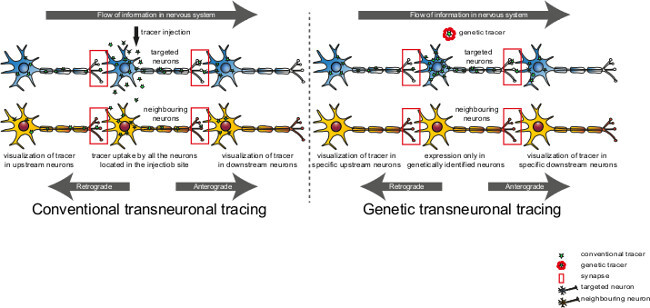 Figure 1: Transsynaptic transfer of tracers using conventional or genetic approaches. In the conventional approach, a transsynaptic tracer is taken up by all the neurons at the injection site thus potentially labeling non-specific unrelated pathways. In the genetic approach, the tracer is selectively expressed by genetically defined neurons thus specifically visualizing neurons connected to the tracer-expressing cells. Please click here to view a larger version of this figure.
Figure 1: Transsynaptic transfer of tracers using conventional or genetic approaches. In the conventional approach, a transsynaptic tracer is taken up by all the neurons at the injection site thus potentially labeling non-specific unrelated pathways. In the genetic approach, the tracer is selectively expressed by genetically defined neurons thus specifically visualizing neurons connected to the tracer-expressing cells. Please click here to view a larger version of this figure.
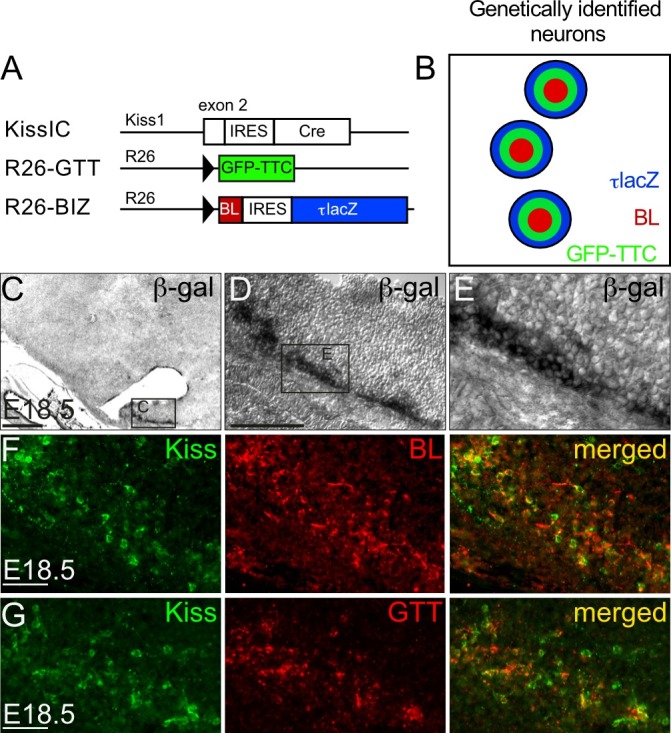 Figure 2: Faithful activation of the BIZ and GTT transgene in embryonic ARC kisspeptin neurons. (A) Breeding strategy to activate BIZ and GTT expression in kisspeptin neurons. We bred R26-BIZ and R26-GTT mice with Kisspeptin-IRES-Cre (KissIC) mice10, in which Cre-recombinase is expressed under the control of the Kiss1 promoter. (C-E) β-gal enzymatic activity identifies the tracer-producing cells and is restricted to the ARC in the brain of a female KissIC/R26-BIZ embryo at embryonic day (E) 18.5. (F-G) Double immunofluorescence for kisspeptin (green) and BL (red) (F) or GTT (red) (G) demonstrates faithful activation of BL or GTT expression by Cre-mediated recombination in kisspeptin neurons in KissIC/R26-BIZ or KissIC/R26-GTT female mouse embryos, respectively. Scale bars (C) 500 µm, (D) 200 µm, (F, G) 50 µm. Please click here to view a larger version of this figure.
Figure 2: Faithful activation of the BIZ and GTT transgene in embryonic ARC kisspeptin neurons. (A) Breeding strategy to activate BIZ and GTT expression in kisspeptin neurons. We bred R26-BIZ and R26-GTT mice with Kisspeptin-IRES-Cre (KissIC) mice10, in which Cre-recombinase is expressed under the control of the Kiss1 promoter. (C-E) β-gal enzymatic activity identifies the tracer-producing cells and is restricted to the ARC in the brain of a female KissIC/R26-BIZ embryo at embryonic day (E) 18.5. (F-G) Double immunofluorescence for kisspeptin (green) and BL (red) (F) or GTT (red) (G) demonstrates faithful activation of BL or GTT expression by Cre-mediated recombination in kisspeptin neurons in KissIC/R26-BIZ or KissIC/R26-GTT female mouse embryos, respectively. Scale bars (C) 500 µm, (D) 200 µm, (F, G) 50 µm. Please click here to view a larger version of this figure.
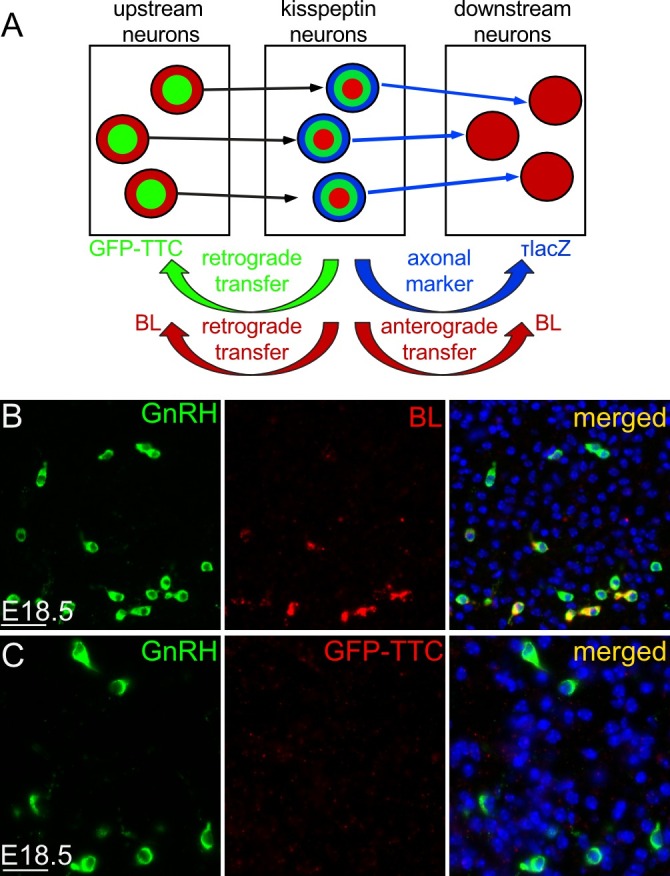 Figure 3: GnRH neurons are synaptically connected to and downstream of kisspeptin neurons. (A) Combinatorial genetic bidirectional transsynaptic tracing. While τlacZ is confined to the Cre-expressing kisspeptin neurons, BL is transsynaptically transferred to upstream (presynaptic) and downstream (postsynaptic) neurons in KissIC/R26-BIZ mice. In contrast, GFP-TTC is transsynaptically transferred to presynaptic, but not postsynaptic neurons in KissIC/R26-GTT mice. Postsynaptic neurons have τlacZ-positive axon fibers in their close vicinity. (B, C) Double immunofluorescence for BL (red; B) or GTT (red; C) and GnRH (green) on a sagittal section through the whole head of a female KissIC/R26-BIZ or KissIC/R26-GTT embryo at E18.5. (B) Note that some, but not all GnRH neurons contain BL. These data demonstrate that a subset of embryonic GnRH neurons is synaptically connected to ARC kisspeptin neurons. (C) Note that none of the GnRH neurons contain GTT. Exclusive transsynaptic transfer of BL but not GTT to GnRH neurons demonstrates that GnRH neurons are downstream of kisspeptin neurons. Scale bars (B, C) 50 µm. Please click here to view a larger version of this figure.
Figure 3: GnRH neurons are synaptically connected to and downstream of kisspeptin neurons. (A) Combinatorial genetic bidirectional transsynaptic tracing. While τlacZ is confined to the Cre-expressing kisspeptin neurons, BL is transsynaptically transferred to upstream (presynaptic) and downstream (postsynaptic) neurons in KissIC/R26-BIZ mice. In contrast, GFP-TTC is transsynaptically transferred to presynaptic, but not postsynaptic neurons in KissIC/R26-GTT mice. Postsynaptic neurons have τlacZ-positive axon fibers in their close vicinity. (B, C) Double immunofluorescence for BL (red; B) or GTT (red; C) and GnRH (green) on a sagittal section through the whole head of a female KissIC/R26-BIZ or KissIC/R26-GTT embryo at E18.5. (B) Note that some, but not all GnRH neurons contain BL. These data demonstrate that a subset of embryonic GnRH neurons is synaptically connected to ARC kisspeptin neurons. (C) Note that none of the GnRH neurons contain GTT. Exclusive transsynaptic transfer of BL but not GTT to GnRH neurons demonstrates that GnRH neurons are downstream of kisspeptin neurons. Scale bars (B, C) 50 µm. Please click here to view a larger version of this figure.
| Features | Transsynaptic tracers | Neurotropic viruses |
| Mode of application | Expressed as a transgene | Mechanically injected |
| Direction of spread | Anterograde, retrograde & bidirectional | Retrograde & anterograde |
| Synaptic efficacy | Multisynaptic | Multisynaptic or monosynaptic (retrograde only) |
| Signal strength | Weak, decreases at every synapse | Strong, signal amplification at every synapse |
| Immune response | None; expressed as endogenous protein | Strong immune response, lethal |
| Spatial and temporal resolution | Depends on promoter of choice | Highly selective due to mechanical injections at individual sites |
Table 1: Comparison of features of genetic transsynaptic tracers and genetically modified neurotropic viruses
Discussion
Expressing transsynaptic tracers as transgenes to trace the neural circuits of genetically defined neuronal populations has several advantages compared to the stereotaxic injection of tracers or neurotopic viruses. First, the tracer is produced as an endogenous protein and therefore does not elicit any immune response and a selective neural pathway can be analyzed in different animals with high reproducibility. Second, because this is a non-invasive method it can be utilized to trace the circuits from neurons not easily accessible for stereotaxic injections, for example in utero. Limitations include a generally low efficacy in transsynaptic transfer, which may result in difficulties in detecting the tracer. Furthermore, the tracer molecule gets diluted at every synapse. In contrast, neurotropic viruses replicate and viral replication in turn then leads to signal amplification after crossing the synapse. However, viral replication is also a major limitation of the use of neurotropic viruses due to intrinsic viral cytotoxicity. Some features of genetic transsynaptic tracers and genetically modified neurotropic viruses are compared in Table 1.
The R26-BIZ and R26-GTT alleles complement each other and facilitate the visualization of the neural circuits of a genetically identified neuronal population upon Cre-mediated recombination. The R26 promoter is well characterized and mediates ubiquitous expression with modest strength11,12. Using highly sensitive methods for tracer detection such as the tyramide signal amplification (TSA) system allows the identification of even minute amounts of tracer molecules transferred across synapses. Another major advantage of using the R26 promoter to drive tracer expression is that the expressing neurons continuously synthesize the tracers throughout their life. This makes it possible to analyze circuit maturation for prolonged time periods5. In contrast, this is not possible when using neurotropic viruses due to their cytotoxicity. Importantly, our binary genetic system and the use of the R26 promoter uncouples tracer production from potentially heavily regulated endogenous promoters driving Cre expression (in this case, the Kiss1 promoter), which may be regulated by a variety of genetic and epigenetic factors13. Hence transsynaptic transfer of BL and GTT reflects actual synaptic communication between two neuronal populations.
Cre-mediated recombination is an irreversible event and therefore it converts developmentally transient gene expression into stable transgene expression. The use of an inducible Cre system14 can potentially unmask a developmental effect due to transient Cre expression.
In conclusion, the two novel R26-BIZ and R26-GTT mouse strains can be used to analyze local and long range neural circuits that might be composed of different classes and types of neurons originating from any brain region or even from the spinal cord in a Cre-dependent manner.
Disclosures
No conflict of interest declared.
Acknowledgments
We thank Michael Candlish for critical comments on the manuscript. This project was supported by the Deutsche Forschungsgemeinschaft grants BO1743/6 and SFB/TRR 152 P11 and Z02 to Ulrich Boehm.
References
- Vercelli A, Repici M, Garbossa D, Grimaldi A. Recent techniques for tracing pathways in the central nervous system of developing and adult mammals. Brain. Res. Bull. 2000;51:11–28. doi: 10.1016/s0361-9230(99)00229-4. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Zeng H. Genetic approaches to neural circuits in the mouse. Annu. Rev. Neurosci. 2013;36:183–215. doi: 10.1146/annurev-neuro-062012-170307. [DOI] [PubMed] [Google Scholar]
- Lanciego JL, Wouterlood FG. A half century of experimental neuroanatomical tracing. J. Chem. Neuroanat. 2011;42:157–183. doi: 10.1016/j.jchemneu.2011.07.001. [DOI] [PubMed] [Google Scholar]
- DeFalco J, et al. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 2001;291:2608–2613. doi: 10.1126/science.1056602. [DOI] [PubMed] [Google Scholar]
- Kumar D, et al. Murine arcuate nucleus kisspeptin neurons communicate with GnRH neurons in utero. J. Neurosci. 2014;34:3756–3766. doi: 10.1523/JNEUROSCI.5123-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz LF, Montmayeur JP, Echelard Y, Buck LB. A genetic approach to trace neural circuits. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3194–3199. doi: 10.1073/pnas.96.6.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos U, Kissa K, ST Cloment C, Brulet P. Retrograde trans-synaptic transfer of green fluorescent protein allows the genetic mapping of neuronal circuits in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 2002;99:10120–10125. doi: 10.1073/pnas.152266799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roux N, et al. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. U.S.A. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminara SB, et al. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- Mayer C, et al. Timing and completion of puberty in female mice depend on estrogen receptor alpha-signaling in kisspeptin neurons. Proc. Natl. Acad. Sci. U.S.A. 2010;107:22693–22698. doi: 10.1073/pnas.1012406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Seibler J, et al. Single copy shRNA configuration for ubiquitous gene knockdown in mice. Nucleic Acids Res. 2005;33:e67. doi: 10.1093/nar/gni065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semaan SJ, Kauffman AS. Emerging concepts on the epigenetic and transcriptional regulation of the Kiss1 gene. Int. J. Dev. Neurosci. 2013;31:452–462. doi: 10.1016/j.ijdevneu.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, et al. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]