

Abstract
Bioavailability of contaminants is a prerequisite for their effective biodegradation in soil. The average bulk concentration of a contaminant, however, is not an appropriate measure for its availability; bioavailability rather depends on the dynamic interplay of potential mass transfer (flux) of a compound to a microbial cell and the capacity of the latter to degrade the compound. In water-unsaturated parts of the soil, mycelia have been shown to overcome bioavailability limitations by actively transporting and mobilizing organic compounds over the range of centimeters. Whereas the extent of mycelia-based transport can be quantified easily by chemical means, verification of the contaminant-bioavailability to bacterial cells requires a biological method. Addressing this constraint, we chose the PAH fluorene (FLU) as a model compound and developed a water unsaturated model microcosm linking a spatially separated FLU point source and the FLU degrading bioreporter bacterium Burkholderia sartisoli RP037-mChe by a mycelial network of Pythium ultimum. Since the bioreporter expresses eGFP in response of the PAH flux to the cell, bacterial FLU exposure and degradation could be monitored directly in the microcosms via confocal laser scanning microscopy (CLSM). CLSM and image analyses revealed a significant increase of the eGFP expression in the presence of P. ultimum compared to controls without mycelia or FLU thus indicating FLU bioavailability to bacteria after mycelia-mediated transport. CLSM results were supported by chemical analyses in identical microcosms. The developed microcosm proved suitable to investigate contaminant bioavailability and to concomitantly visualize the involved bacteria-mycelial interactions.
Keywords: Environmental Sciences, Issue 94, PAH, bioavailability, mycelia, translocation, volatility, bioreporter, CLSM, biodegradation, fluorene
Introduction
Soil is densely populated by a wide range of microorganisms1,2 such as bacteria. However, conditions in this habitat are challenging, especially in terms of water availability3. Bacteria permanently need to search for optimal conditions in heterogeneous environments4, but the absence of continuous water films is resulting in restricted mobility5 hindering them to spread freely. Also, diffusion rates of solutes (e.g., nutrients) are lowered under unsaturated conditions6. Thus, bacteria and nutrients are often physically separated and nutrient accessibility is limited3. As a consequence, a transport vector for chemical compounds which does not require a continuous water-phase could help to overcome these limitations. In fact, many microorganisms such as fungi and oomycetes have developed a filamentous growth form enabling them to grow through air-filled pore spaces thereby reaching and mobilizing also physical separated nutrients7 and carbonaceous8 substances over long distances. They may even act as biological transport vectors which deliver sugars and other energy sources to bacteria9. Uptake and transport in mycelial organisms has also been shown for hydrophobic organic pollutants such as polycyclic aromatic hydrocarbons (PAH) in Pythium ultimum10 or in arbuscular mycorrhizal fungi11. Since PAH are ubiquitous and poorly water soluble contaminants12 in soil, mycelia-mediated transport might help to increase contaminant bioavailability for potential bacterial degraders. Whereas the total amount of contaminant transport can be quantified directly by chemical means10, bioavailability of contaminants transported by mycelia to degrading bacteria and other organisms cannot be assessed easily.
The following protocol presents a method to evaluate the impact of mycelia on contaminant bioavailability to bacterial degraders in a direct manner; it allows gathering information about the spatiotemporal impact of contaminants on microbial ecosystems. We describe how to set up an elaborate unsaturated microcosm system mimicking air-water interfaces in soil by linking a physically separated PAH point source with PAH-degrading bioreporter bacteria via mycelial transport vectors. Because airborne transport is excluded, the effect of mycelial-based transport on PAH bioavailability for bacteria can be studied in an isolated way. In more detail, three-ring PAH fluorene, the mycelial organism Pythium ultimum and the bioreporter bacterium Burkholderia sartisoli RP037-mChe13 were applied in the described microcosm setups. The bacterium B. sartisoli RP037-mChe was originally constructed to study phenanthrene fluxes to the cell14 and expresses enhanced green fluorescent protein (eGFP) as a result of the PAH flux to the cell, whereas the red fluorescing mCherry is expressed constitutively. Detailed information on the reporter construction is given by Tecon et al.13 In preliminary tests, the bacterium revealed no swimming and only very slow swarming ability. It was able to migrate slowly on hyphae of Pythium ultimum when applied as a dense suspension on top of the hyphae. Since bacteria were embedded in agarose in the following protocol, migration on hyphae did not occur.
Using confocal laser scanning microscopy (CLSM), the bioreporter bacteria can be visualized directly in the microcosms and expression of eGFP can be quantified in relation to the amount of cells (proportional to the mCherry signal) with the help of the software ImageJ. This allows comparing bioavailability qualitatively in different scenarios (i.e., higher or lower). FLU was found to be bioavailable after mycelial transport by P. ultimum (i.e., it was higher than in a negative control). Furthermore, the protocol describes how to quantify the total amount of mycelia-mediated transport via chemical means and to verify contaminant bioavailability using silicon-coated glass fibers (SPME fibers) in identical microcosms. Results using this microcosm setup have been published and discussed for the combination of P. ultimum, fluorene and B. sartisoli RP037-mChe15. Here, the focus lies on a detailed method description and the identification of potential pitfalls of the protocol to provide this knowledge for potential further applications. Further applications may involve various fungal, bacterial species (e.g., from contaminated sites), and other contaminants (e.g., pesticides) or contaminant-supply (e.g., aged soils).
Protocol
1. Preparation of Dishes, Slides and Incubation Chambers
Prepare the following material for each microcosm: one big plastic Petri dish bottom (d = 10 cm), one modified (see step 1.2) small plastic Petri dish bottom (d = 5 cm) with lids and one counting chamber slide with three cavities.
Take the desired number of Petri dish bottom parts (d = 5 cm). Remove part of the brim with a saw to exactly fit a slide (26 mm edge length). To sterilize the system, soak Petri dish bottoms and lids in 70% ethanol O/N and dry them for at least 2 hr in a flow cabinet under UV-light. Store the Petri dishes in a tightly sealed plastic container, which had also been exposed to UV-light for 2 h.
Wipe the desired number of slides with 70% ethanol and air-dry them. Wrap slides in aluminum foil and put them in a muffle furnace for 5 hr at 450 °C to remove possible contaminations.
Prepare glass incubation chambers with lids. Clean with 70% ethanol and expose open chambers to UV-light for at least 2 hr in a flow cabinet. NOTE: Large desiccators (diameter around 30 cm) with lids may be used for this purpose; each desiccator may house up to ten microcosm setups.
2. Preparation of Media and Cultures
To run 20 parallel microcosms, obtain 100 ml of modified tryptone-yeast medium (mTY)14, about 150 ml of 21C minimal medium16 (MM), 5 ml of minimal medium agar (MMA; 0.5%, w/v), three potato dextrose agar plates (PDA; 1.5%, w/v) for P. ultimum, four blank plates, one PDA plate containing 2 mg L-1 cycloheximide, four agarose plates (3%, w/v) containing 0.3 g ml-1 activated carbon and 40 mg of FLU in 2 mg portions.
Prepare mTY containing 50 mg L-1 of kanamycin (kanamycin is required to maintain the eGFP reporter plasmid) and MM containing 10 mM acetate for bacterial cultivation. Use MM without acetate to prepare 0.5% MMA for the immobilization of bacteria in the microcosms and pour plates (each 20 ml) of 1.5% PDA for cultivation of Pythium ultimum and microcosm setups.
Inoculate some hyphae of P. ultimum on three fresh PDA plates and incubate in the dark at 25 °C for about 72 hr. Then ensure that the plates are overgrown completely by fresh and fluffy mycelium.
Start two cultures of B. sartisoli RP037-mChe in 50 ml of mTY from a glycerol stock. Incubate at 30 °C with 230 rpm rotary flask movement O/N.
Measure culture optical density (OD) at 578 nm and centrifuge at 1,000 x g for 10 min (for B. sartisoli RP037-mChe, the expected OD578 is about 1.0 and cells are in exponential phase). Discard supernatant and re-suspend the cells in an appropriate amount of MM to reach an OD578 of 0.4. Inoculate 20 ml of MM containing 50 mg L-1 kanamycin with 400 µl of the resuspended cells. Incubate at 30 °C with 230 rpm rotary flask movement O/N.
Measure OD578 of the culture in MM (for B. sartisoli RP037-mChe, the expected OD578 is about 0.2 and cells are in early exponential phase). Centrifuge culture at 2,000 x g for 10 min and re-suspend the cells in an appropriate amount of MM to reach an OD of 0.4. Mix 50 µl of the cell suspension with 500 µl of warm, liquid MMA in 2 ml tubes and store at 50 °C until usage. Use 150 µl of cells in MMA for each microcosm.
Pour plates of agarose gel containing activated carbon. Melt agarose (3%; w/v) in double distilled water. Mix thoroughly with activated carbon (0.3 g ml-1) and pour mixture into plastic Petri dishes (d = 10 cm).
3. Microcosm Mounting
NOTE: The complete microcosm setup is depicted schematically in Figure 1. Please refer to Figure 1 for all following individual steps. The following steps are describing the preparation of a sample (SAM) with mycelial transport vectors and three different control setups (CONAIR, CONNEG, CONPOS). A summary of all different setups can be found in Table 1.
Take desired number of Petri dishes and slides and expose them to UV light under a flow cabinet for 30 min.
Place the small Petri dish inside the big one and fit the slide through the gap of the small Petri dish. Take one sample of the warm cell suspension in MMA and add 150 µl into the middle cavity of one slide. Repeat this step until the middle cavities of all slides are filled with MMA. Wait 5 min for the MMA to solidify.
Use a 1 cm cork borer to punch out circular patches from a blank PDA plate. Place one patch at a distance of 2 mm next to the MMA inside the small Petri dish. Add this to promote mycelial growth in the setup (PDA 3 in Figure 1).
- Cut curved PDA patches as mechanical barriers. Use a clean and sterile small Petri dish bottom part and press it into a PDA plate. Use a spatula to cut another smaller ring at a distance of 0.5 cm into the agar which results in a PDA-ring.
- Cut out a piece, which exactly fits into the gap of the small Petri dish in the microcosm setup and place it inside the gap on top of the slide. Ensure that the distance between the MMA and the circular PDA barrier is about 2 mm. Close the lid of the small Petri dish gently pressing it into the PDA barrier.
- Repeat steps 3.4 and 3.4.1 with a PDA plate containing 2 g L-1 of cycloheximide. NOTE: This is the control setup for airborne transport (CONAIR) towards the bioreporter cells, since mycelia are inhibited by cycloheximide and do not overgrow MMA.
Use a 1 cm cork borer to punch out circular patches of a blank PDA plate. Use another 0.5 cm cork borer to cut out the middle of the first patch. Place the resulting small PDA-ring at a distance of 1 mm next to the PDA barrier (PDA 1 in Figure 1).
- Add 2 mg of FLU into the hole of PDA 1. Use a 1 cm cork borer to cut out circular patches from PDA plates overgrown with P. ultimum and place one patch (PDA 2 in Figure 1) bottom down onto the PDA ring so that the mycelial mat is facing the FLU crystals.
- Repeat the previous step without adding FLU crystals. NOTE: This is the negative control setup (CONNEG) to determine background fluorescence of bioreporter cells.
Use a 1 cm cork borer to punch out circular patches from the agar containing activated carbon. Place four patches in each microcosm setup to further decrease the gaseous FLU concentration (Figure 1).
Prepare a positive control (CONPOS). Add some FLU crystals to 200 µl of MMA cell suspension and put it in one cavity of an empty slide.
- Place a Petri dish (d = 10 cm) with some powder of activated carbon on the bottom of the incubation chamber to minimize gaseous FLU concentration in the chamber. Also, place four to five 50 ml beakers with sterile water on the bottom to maintain high humidity in the chamber.
- Transfer microcosm setups into incubation chambers and incubate at 25 °C for 96 hr. Place samples randomly in the different growth chambers to exclude location effects. NOTE: This incubation time applies to the oomycete P. ultimum. It might be different for other organisms.
4. Confocal Laser Scanning Microscopy
Ideally use a CLSM setup with upright microscope. NOTE: It may be equipped with conventional lasers offering e.g., 488, 561 and 633 nm lines for excitation or with a tunable white laser source.
Use the following settings for collection of images: Excitation: 490 nm (eGFP) and 585 nm (mCherry) at the appropriate laser intensity (simultaneous excitation); emission ranges: 500 to 550 nm (eGFP) and 605 to 650 nm (mCherry); objective lens: 63X NA 0.9 water immersible (due to its long working distance); step size for z-stacks: 0.5 µm.
Open microcosm setup and use a scalpel to remove PDA 1, 2, 3 and the PDA barrier. Put a glass cover slip on top of the MMA and press it gently to remove air bubbles. Mount slide on microscope stage. Add a droplet of water on top of the cover slip and use mCherry settings to find bioreporter cells in the sample.
Get a quick overview of the sample and optimize signal to noise ratio using a glow-over-under lookup table for red (mCherry) and green (eGFP) channel. Choose a random position on the sample to analyze bioreporter fluorescence. NOTE: hyphae may show autofluorescence in the range of bioreporter emission (e.g., green autofluorescence)17. Be sure to select areas without hyphae if this is the case.
- Define and record z-stacks for each position in the green (eGFP) and the red (mCherry) channel. Avoid bleaching of the sample by long exposure to epifluorescence light or laser light.
- Repeat previous step for ten random, evenly distributed positions.
Repeat all steps using the same settings for all samples of the test track (SAM), CONAIR , CONNEG and CONPOS.
5. Image Analysis
NOTE: To analyze red (mCherry) and green (eGFP) fluorescence in the z-stacks recorded, amongst other options the free software ImageJ18 (http://rsb.info.nih.gov/ij/download.html) can be used.
Make sure to install the logi_tool plugin (http://downloads.openmicroscopy.org/bio-formats/4.4.10/).
- Open file in ImageJ. Select “split channels” and close green channel. Use the macro ‘mCherry area’ provided as supplementary code file to quantify area of red fluorescence in z-stack. Adjust preferred settings in the macro (indicated in bold).
- Output is a table with the measured pixels above threshold (and above the defined minimum size) in z- stack. NOTE: The sum of all values is the total number of red pixels (area) in the stack.
- Open file in ImageJ. Select “split channels” and close red channel. Use the macro ‘eGFP int’ provided as supplementary code file to quantify intensity of green fluorescence in z- stack. Adjust preferred settings in the macro (indicated in bold).
- Output is a table with the mean intensities and area of all green objects above threshold (and above the defined minimum size) in z-stack. To calculate the total intensity of green pixels in the stack, multiply each mean intensity by the corresponding area.
Calculate the relative eGFP induction (eGFPrel):
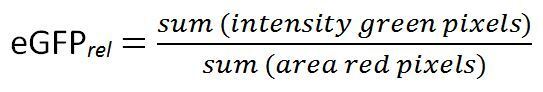 (1) NOTE: Since mCherry is expressed constitutively, the resultant value of eGFPrel is a relative measure for the amount of eGFP expressed by a certain amount of cells. Please also refer to Table 3 for further information on calculation of eGFPrel.
(1) NOTE: Since mCherry is expressed constitutively, the resultant value of eGFPrel is a relative measure for the amount of eGFP expressed by a certain amount of cells. Please also refer to Table 3 for further information on calculation of eGFPrel.
6. Chemical Quantification
NOTE: Microcosm setups may also be used to perform chemical quantification of translocated FLU amounts with or without an abiotic contaminant sink.
Wrap 15 ml glass vials, 1 ml GC/MS vials and inserts in aluminum foil. Fill evaporating dishes with Na2SO4; place all in muffle furnace for 5 hr at 450 °C to remove possible contaminations. Store the Na2SO4 inside of an activated desiccator.
Cut PDMS coated glass fibers into 0.5 cm length. Wash fibers by shaking four times 2 hr in fresh methanol. Repeat washing steps another four times with ultrapure water and store clean fibers in ultrapure water in a sealed glass vial.
Prepare 1.5 L of toluene/acetone (3:1, v/v) containing 10 µg L-1 phenanthrene-d10.
- Perform steps 1, 2 and 3 of the protocol. Prepare microcosms without bioreporter cells to study FLU transport (SAM(-) and CONAIR(-)) and with bioreporter cells (SAM) to verify FLU degradation.
- Optional (without bioreporter cells): Place three PDMS coated glass fibers (usually used for SPME experiments) as artificial and quantifiable contaminant sink inside MMA of SAM(-) using fine forceps.
- Remove glass fibers using fine forceps and transfer each one into a GC/MS vial with insert.
- Add 200 µl of toluene and shake vertically O/N and analyze via GC/MS. Use the settings provided in Table 2.
- Transfer MMA from the middle cavity into a 15 ml glass vial and add about three spatulas of Na2SO4. Mix thoroughly using a spatula and add 10 ml of solvent. Vortex the mixture and shake horizontally O/N in the dark.
- Transfer 100 µl of sample into GC/MS vial with insert. Add 10 µl of acenaphthylene-d08 (10 mg L-1) and analyze via GC/MS using the same program as described in 6.4.3.
Representative Results
The results presented here have already been published earlier15. Please refer to the article for detailed mechanistic and environmental discussion.
After image recording via CLSM, a maximum intensity projection can be conducted using the respective microscope software or ImageJ to gain a first visual impression of the sample and the controls (Figure 2). Later, the data sets may be projected differently in order to show meaningful features by specific visualization software. The positive control (CONPOS) shows distinct eGFP induction (Figure 2A), whereas the airborne control (CONAIR) exhibits only background eGFP fluorescence (Figure 2B). eGFP fluorescence in the test sample (SAM) appears to be elevated compared to CONAIR, yet not as marked as CONPOS (Figure 2C). The mCherry fluorescence is independent from PAH degradation and therefore similar for all samples. However, slightly elevated eGFP signals will not be detected by this method and subsequent image analysis is essential.
For the tested microcosms, visual inspection was complemented by image analysis and calculation of relative eGFP induction (cf. step 5 ff of the protocol; Figure 3). The green background fluorescence for non-induced B. sartisoli RP037-mChe is calculated with CONNEG. Under the given conditions, the relative eGFP induction was found to be eGFPrel=0.53. No significant difference could be detected between CONNEG and CONAIR (eGFPrel=0.54; p>0.95) thus excluding any vapor-phase FLU transport towards the bioreporter cells. For CONPOS, highly elevated eGFP induction (eGFPrel=53.7) was found confirming the vitality of the cells in MMA after 96hr and representing the maximum eGFP induction. In the presence of mycelia and FLU (SAM), eGFP was induced significantly in the cells compared to the negative controls (eGFPrel=1.15; p<0.001). However, the eGFP induction is relatively low compared to the positive control (cf. Table 3, point 8).
Transport of FLU by mycelia of P. ultimum in the microcosm setup was quantified chemically (cf. step 6 ff of the protocol; Figure 4). In the presence of FLU and P. ultimum (SAM(-)), mycelia translocated about 25ng of FLU within 96hr which equals a transport rate of 37.5pmold-1cm-1. Controls in the absence of mycelia (CONAIR(-)) revealed gaseous FLU transport into the MMA in the range of only 2ng within 96hr, i.e., induction of the bioreporter by vapor-phase-concentrations could be excluded. When B.sartisoli RP037-mChe was present (SAM), < 1ng of FLU was detected in the MMA patch. This verified effective bacterial FLU degradation subsequent to the mycelia-mediated translocation.
It is possible to investigate the influence of bacterial degradation, i.e., of a FLU sink, on FLU translocation by adding an abiotic contaminant sink to the system (cf. step 6.4.1 ff of the protocol). Thus, one is able to quantify the amount of FLU absorbed by the fibers and the amount left inside of the MMA and overlying hyphae. For SAM(-), the total translocated amount is independent from the presence of a contaminant sink (p>0.9; Figure 5A). However, a microcosm with increased FLU uptake area was also tested as described in detail earlier15. There, a significant increase of the translocated FLU amount could be detected (Figure 5B).
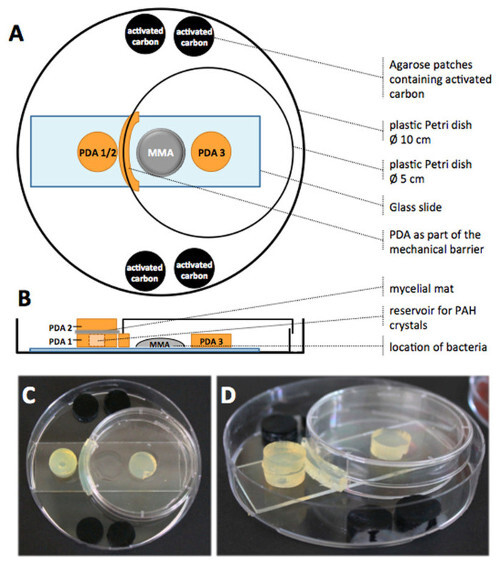 Figure 1. Schematic drawing and photos of the complete microcosm setup. View from top (A) and from the side (B). All agar patches were placed on top of a slide with three small cavities. Two mg of FLU crystals were placed inside a cavity (d = 5 mm) in the middle of PDA 1. PDA 2 was freshly overgrown with hyphae of P. ultimum facing PDA 1. A curved PDA piece was placed between PDA 2 and the MMA patch to create a mechanical FLU barrier together with the lid of a Petri dish (d = 5 cm). Four agar patches containing activated carbon were placed in the setup to further decrease the gaseous FLU concentration. Photos of the complete microcosm from top (C) and diagonal view (D) without microorganisms and FLU. Figures 1A and 1B have been modified with permission from Schamfuss et al.15 Adapted with permission from Schamfuss, S. et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 47, 6908-6915. Copyright (2013) American Chemical Society. Please click here to view a larger version of this figure.
Figure 1. Schematic drawing and photos of the complete microcosm setup. View from top (A) and from the side (B). All agar patches were placed on top of a slide with three small cavities. Two mg of FLU crystals were placed inside a cavity (d = 5 mm) in the middle of PDA 1. PDA 2 was freshly overgrown with hyphae of P. ultimum facing PDA 1. A curved PDA piece was placed between PDA 2 and the MMA patch to create a mechanical FLU barrier together with the lid of a Petri dish (d = 5 cm). Four agar patches containing activated carbon were placed in the setup to further decrease the gaseous FLU concentration. Photos of the complete microcosm from top (C) and diagonal view (D) without microorganisms and FLU. Figures 1A and 1B have been modified with permission from Schamfuss et al.15 Adapted with permission from Schamfuss, S. et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 47, 6908-6915. Copyright (2013) American Chemical Society. Please click here to view a larger version of this figure.
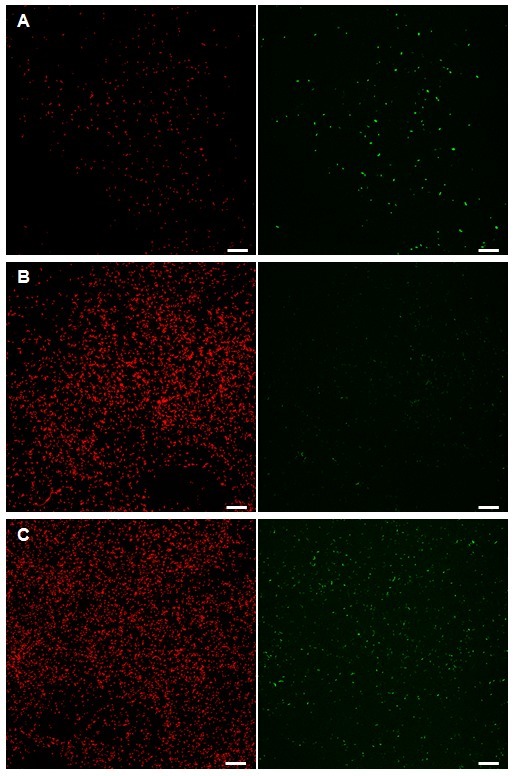 Figure 2. Maximum intensity projections of confocal laser scanning micrographs. Micrographs are visualizing the mCherry and eGFP induction of the bioreporter bacterium B. sartisoli RP037-mChe in MMA. When the bacteria had contact to a crystalline FLU source in MMA (CONPOS; A), the eGFP signal is elevated compared to controls (CONAIR; B). Samples with FLU and P. ultimum (SAM; C) also show an elevated eGFP signal, however less marked than the positive control. mCherry is expressed constitutively and therefore serves as a visual control to detect the cells. (Magnification 630X, bar 20 µm). Please click here to view a larger version of this figure.
Figure 2. Maximum intensity projections of confocal laser scanning micrographs. Micrographs are visualizing the mCherry and eGFP induction of the bioreporter bacterium B. sartisoli RP037-mChe in MMA. When the bacteria had contact to a crystalline FLU source in MMA (CONPOS; A), the eGFP signal is elevated compared to controls (CONAIR; B). Samples with FLU and P. ultimum (SAM; C) also show an elevated eGFP signal, however less marked than the positive control. mCherry is expressed constitutively and therefore serves as a visual control to detect the cells. (Magnification 630X, bar 20 µm). Please click here to view a larger version of this figure.
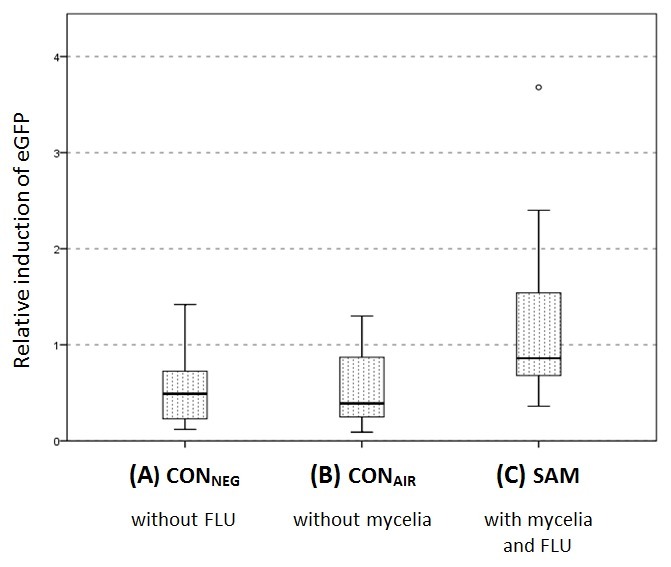 Figure 3. Relative eGFP induction in B.sartisoli RP037-Che for CONNEG, CONAIR and SAM with mycelia and FLU. Comparison of the relative eGFP induction (eGFPrel) by B.sartisoli RP037-Che after 96hr in the absence of FLU (CONNEG), in the presence of FLU but absence of P.ultimum (CONAIR) and in the presence of both FLU and P.ultimum (SAM). Statistically significant differences to CONNEG are marked by an asterisk. Experiments were performed in independent triplicates for CONAIR and SAM and duplicates for CONNEG, respectively. One sample was tested for CONPOS and eGFPrel was calculated with 53.7. This figure has been modified with permission from Schamfuss et al.15 Adapted with permission from Schamfuss, S. et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 47, 6908-6915. Copyright (2013) American Chemical Society.
Figure 3. Relative eGFP induction in B.sartisoli RP037-Che for CONNEG, CONAIR and SAM with mycelia and FLU. Comparison of the relative eGFP induction (eGFPrel) by B.sartisoli RP037-Che after 96hr in the absence of FLU (CONNEG), in the presence of FLU but absence of P.ultimum (CONAIR) and in the presence of both FLU and P.ultimum (SAM). Statistically significant differences to CONNEG are marked by an asterisk. Experiments were performed in independent triplicates for CONAIR and SAM and duplicates for CONNEG, respectively. One sample was tested for CONPOS and eGFPrel was calculated with 53.7. This figure has been modified with permission from Schamfuss et al.15 Adapted with permission from Schamfuss, S. et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 47, 6908-6915. Copyright (2013) American Chemical Society.
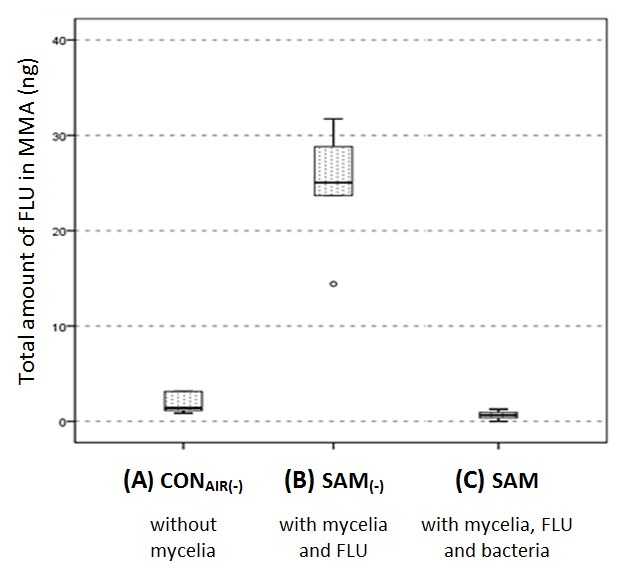 Figure 4. Chemical FLU quantification. Total amounts of FLU in ng extracted from the MMA patch (cf. Figure 1) after 96hr for vapor-phase transport without cells (CONAIR(-)), mycelial transport without cells (SAM(-)) and mycelial transport with cells (SAM). This figure has been modified with permission from Schamfuss et al.15 Adapted with permission from Schamfuss, S. et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 47, 6908-6915. Copyright (2013) American Chemical Society.
Figure 4. Chemical FLU quantification. Total amounts of FLU in ng extracted from the MMA patch (cf. Figure 1) after 96hr for vapor-phase transport without cells (CONAIR(-)), mycelial transport without cells (SAM(-)) and mycelial transport with cells (SAM). This figure has been modified with permission from Schamfuss et al.15 Adapted with permission from Schamfuss, S. et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 47, 6908-6915. Copyright (2013) American Chemical Society.
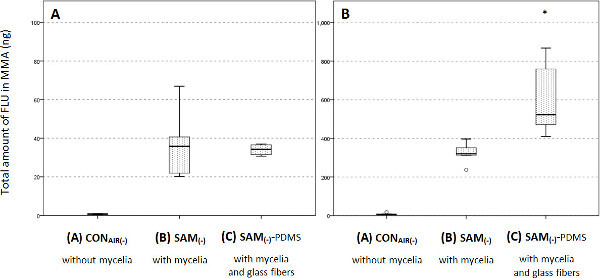 Figure 5. Chemical FLU quantification in the presence of an abiotic contaminant sink. (A) Total amounts of FLU in ng extracted from the MMA patch (cf. Figure 1) after 96hr for mycelial transport with or without artificial contaminant sink (SAM(-) with or without PDMS coated fibers). (B) Same results for a varied test track with increased mycelial FLU uptake area. This figure has been modified with permission from Schamfuss et al.15 Adapted with permission from Schamfuss, S. et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 47, 6908-6915. Copyright (2013) American Chemical Society.
Figure 5. Chemical FLU quantification in the presence of an abiotic contaminant sink. (A) Total amounts of FLU in ng extracted from the MMA patch (cf. Figure 1) after 96hr for mycelial transport with or without artificial contaminant sink (SAM(-) with or without PDMS coated fibers). (B) Same results for a varied test track with increased mycelial FLU uptake area. This figure has been modified with permission from Schamfuss et al.15 Adapted with permission from Schamfuss, S. et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 47, 6908-6915. Copyright (2013) American Chemical Society.
| Name | FLU | Mycelia | Cells | Application | Comments |
| SAM | + | + | + | CLSM | |
| GC/MS | |||||
| SAM(-) | + | + | - | GC/MS | also possible with PDMS fibers as artificial contaminant sink |
| CONAIR | + | (+) | + | CLSM | mycelia only growing up to PDA barrier; MMA not covered |
| CONAIR(-) | + | (+) | - | GC/MS | mycelia only growing up to PDA barrier; MMA not covered |
| CONNEG | - | + | + | CLSM | |
| CONPOS | + | - | + | CLSM | cells are directly exposed to FLU crystals |
Table 1. Overview of all sample types. Summary of all control and test samples including application (bioreporter assay – CLSM or chemical quantification – GC/MS) and composition (FLU, mycelia and bioreporter cells).
| Oven | |
| Initial temp | 50 °C |
| Initial time | 2 min |
| Rate | 15 °C min-1 |
| Final temp | 300 °C |
| Final time | 6.33 min |
| Injector (PTV) | |
| Injection Volume | 0.5 µl |
| Mode | splitless |
| Purge | 2.00 min |
| Purge flow | 50.0 ml min-1 |
| Initial temp | 80 °C |
| Initial time | 0.02 min |
| Ramps: Rate | 600 °C min-1 |
| Final temp | 300 °C |
| Final time | 10 min |
| Column | |
| Model | J&W DB-5MS |
| Nominal length | 20 m |
| Nominal diameter | 180 µm |
| Nominal film thickness | 0.18 µm |
| Carrier gas | helium 0.8 ml min-1 |
| MSD Transferline | 280 °C |
| MSD | |
| Sim mode | |
| MS Source | 230 °C |
| MS Quad | 150 °C |
Table 2. Settings for GC/MS. Summary of all settings for GC and MS to quantify fluorene in the microcosm setups.
| PITFALLS | COMMENT | |
| 1 | Preliminary growth and vitality tests | Study the growth of mycelial organism of choice. How long does it take to reach the MMA patch? Is it inhibited by the cycloheximide barrier? Also check whether bioreporter cells stay vital in MMA the whole time via plaiting assays. If not, one also may add a carbon source to the MMA which does not result in bioreporter induction. Exclude possible mutual inhibition of bacteria and mycelia. |
| 2 | Mycelial growth rate | The growth rate of the mycelial organism should not be too slow, because otherwise gas-phase transport of PAH increases with prolonged incubation time. The protocol may be adapted to add PAH at a later time point, but then again, mycelia at the inoculation point may not be active anymore. |
| 3 | Bacterial mobility | If a point scanning laser microscope is used and the bacterial strain is motile, the recording and quantification of z-stacks may be impossible due to movement inside of MMA. Thus, bacterial movement should be prevented, e.g., by increasing the solidity of MMA, by using CLSM with a fast scanner or a spinning disk laser microscope. |
| 4 | Vapor-phase transport | Vapor-phase transport of the chemical (e.g., PAH) towards the bioreporter cells must be excluded sufficiently since bioreporter cells are extremely sensitive to chemicals in the vapor-phase. This is the crucial point of the whole protocol in order to prevent false-positive results caused by gaseous transport. The gas phase concentration can be estimated via chemical analysis of CONAIR(-) and vapor-phase concentrations may be adjusted for example by adding more or less agarose patches with activated carbon. |
| 5 | Toxicity | Keep in mind, that high concentrations of the tested chemical might have toxic effects on the mycelial organism and/or bacteria. |
| 6 | Autofluorescence | Check if the chosen mycelial organism shows some autofluorescence in the range of the expected bioreporter signal. Be sure to check at different growth stages since autofluorescence may vary17. If autofluorescence is detected, be sure to record z-stacks in mycelia-free areas. |
| 7 | Bleaching | mCherry fluorescence is usually stable and not sensitive to bleaching in the applied bioreporter cells. In contrast, eGFP bleaches rather quickly. Therefore, don’t use the eGFP channel to visualize the sample prior to z-stack recording. |
| 8 | eGFP induction | We noticed a low eGFP induction in SAM compared to CONPOS. This is accounted for by the strong spatial restriction and low accessibility of the FLU source to maintain very low vapor-phase concentration of FLU (cf. last results paragraph). Depending on the chosen bioreporter strain and chemical, the uptake area at PDA 1 may be varied to increase the transport rate (Figure 5B). However, it has to be considered that this also increases vapor-phase transport towards the bioreporter cells. |
| 9 | Calculation of eGFPrel | The presented calculation method for eGFPrel where the intensity of green pixels is compared to the area of red pixels is only one possibility. Please refer to the discussion for further information. |
| 10 | Pixel size | Please be aware that the magnification of the chosen lens and a potentially applied zoom factor affect the pixel size of the image. This must be taken into consideration prior to image analysis. |
| 11 | Bioavailable fractions | Keep in mind that the bioavailability of the transported compound is assessed qualitatively (not quantitatively) via eGFP induction. No correlation of the calculated relative eGFP-induction and the bioavailable fraction was attempted. However, this may be addressed in future applications. |
| 12 | Detection limits | Chemical detection. For chemical quantification of organic compounds, a wide range of concentrations can be detected reliably. We detected amounts in the range 0ng and 1,000ng. eGFP quantification. We compared relative eGFP induction in samples without contaminant transport (i.e., 0ng transported), with mycelia-mediated transport (i.e., between 20 and 40ng transported; cf. Figures 4 and 5) and with direct contact to the contaminant source (i.e., maximum available amount). These three cases proved to be well distinguishable with the described method. However, we cannot make a more detailed statement on the resolution of the relative eGFP induction. This issue may be addressed in the future by linking different contaminant amounts with the correlating eGFP induction in the microcosm setups. |
| 13 | Petri dish material | Plastic Petri dishes may result in an underestimation of mycelial PAH translocation and/or bioavailability. This should not be problematic, if qualitative statements are aspired. In case precise quantitative measurements are required, application of glass Petri dishes should be considered despite preparation and handling will be more inconvenient. |
Table 3. Discussion of possible pitfalls. Possible pitfalls prior to and during the experiment and proposed comments and options.
Discussion
The presented microcosm setup proved suitable to study bioavailability of spatially separated chemicals to degrading organisms after uptake and transport by mycelia. Potential gas-phase transport of partially volatile compounds is prevented and bacterial bioreporter cells can be visualized without elaborate sample preparation and thus with minimal disturbance of the sensitive system. At the same time, chemical analysis of the sample can be easily conducted allowing for a good control of the gained results and for quantification of the total transport. However, some points have to be carefully considered prior to and while performing the experiment. An important point is to keep the microcosms free of any bacterial or chemical cross contaminations. This can be challenging, since many microcosms are run in parallel and thus, big incubation chambers (such as desiccators) are required. Hence, it is crucial to sterilize all parts carefully prior to the setup construction. Another critical point may be the application of plastic Petri dishes for the microcosms. Although PAH and bioreporter do not get in direct contact, some amount of PAH may be absorbed by the plastic material. However, we consider this not problematic for the described protocol, since this would result, if any, in an underestimation of mycelial PAH-translocation and/or bioavailability. In any case, if precise quantification is required, it should be considered exchanging the plastic parts with glass parts, although handling will be more inconvenient. Other points that should be considered involve the chosen bacterial and fungal strains (possible mutual inhibition, growth rate, mobility and possible autofluorescence), the chosen chemicals (toxic effects on microorganisms, volatility, detection limits) and practical aspects (bleaching, limitation of eGFP induction and calculation of eGFPrel). A summary of potential pitfalls and respective comments can be found in Table 3.
We used this model microcosm setup to demonstrate mycelial FLU transport and subsequent bacterial degradation in an unsaturated system directly on a cell-mycelia interaction level. Our model set-up can be varied and adjusted to meet different needs with various bacterial and fungal strains, chemical compounds, etc. Thus, the system could be used for example (i) to evaluate the influence of mycelia on bacteria in the presence of an otherwise restricted chemical (e.g., toxic) source, (ii) to predict the influence of mycelia on the bioavailability of compounds for bacterial degradation, (iii) to study contaminant bioavailability depending on the distance of bacteria to the transport vector or (iv) to investigate the effect of different chemical sources (e.g., crystalline, in soil or dissolved). However, the adjustment of the system to alternate conditions will be delicate and should be conducted step by step in order to achieve clear results. So far, we have not tested the system for other bacterial or fungal strains and other compounds than PAH. However, since mycelial networks are known to transport also other contaminants than PAH like pesticides19 or soluble substances like salicylate20, we think that the system can be expanded to different chemical compounds given a compound-specific bacterial biosensor.
Please note that depending on the applied bioreporter strain a different calculation of eGFPrel might be preferable. Instead of equation (1) the two following options could be applied:
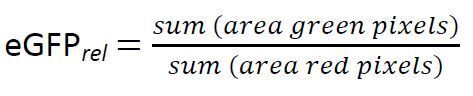 (2)
(2)
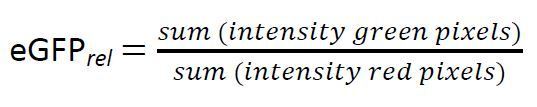 (3)
(3)
However, in the presented protocol equation (1) was chosen due to the following reasons: (i) eGFP intensity was reported to correlate to the PAH flux to the cell14 whereas (ii) mCherry was reported to show considerable variations for different cells13. Equation (2) and (3), in contrast, may be chosen to prevent false information from potential light artefacts in the sample. In any case, we compared all three calculation methods with each other, whereby no statistically significant differences could be found. Still, due to the reported variations in the fluorescence intensity of mCherry, we recommend using equation (1) or (2).
In the end, some limitations of the technique should be considered. Since the microcosms were designed to prevent gas-phase transport of the tested compound, mycelial uptake of the compound is reduced. This results in lower transport rates than would be expected with unhindered uptake. Hence, the sensitivity of the bioreporter strain must be high enough to detect small changes in bioavailable fractions. Further, the setup will probably not be suitable for fungal strains with a low growth rate. In this case, long term incubation might favor gas-phase transport of the contaminant thus leading to false-positive results. It might be possible to modify the setup in a way that the contaminant can be applied at a later time point. However, hyphae may then be already inactive or dead at the inoculation point. Finally, as stated above, the range of chemical compounds which are transported by mycelial networks is big. However, the availability of an appropriate bioreporter strain might be a limiting key factor.
Disclosures
The authors declare that they have no competing financial interest.
Acknowledgments
Funding by the German Environmental Foundation (DBU) is acknowledged. The authors thank Ute Kuhlicke for technical help with CLSM analysis and Birgit Würz, Rita Remer, and Jana Reichenbach for skilled experimental help. The authors would particularly like to thank Prof. Jan Roelof van der Meer and Dr. Robin Tecon for fruitful discussion and providing the bioreporter strain. It contributes to the ‘Chemicals in the Environment’ (CITE) research program of the Helmholtz Association.
References
- Holden PA, Fierer N. Microbial processes in the vadose zone. Vadose Zone Journal. 2005;4:1–21. [Google Scholar]
- Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: The unseen majority. Proceedings of the National Academy of Sciences. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft TL, et al. Microbial abundance and activities in relation to water potential in the vadose zones of arid and semiarid sites. Microbial ecology. 1993;26:59–78. doi: 10.1007/BF00166030. [DOI] [PubMed] [Google Scholar]
- Wang G, Or D. Aqueous films limit bacterial cell motility and colony expansion on partially saturated rough surfaces. Environ. Microbiol. 2010;12:1363–1373. doi: 10.1111/j.1462-2920.2010.02180.x. [DOI] [PubMed] [Google Scholar]
- Griffin DM. In: Water Potential Relations in Soil Microbiology SSSA Special Publication. Parr JF, Gardner WR, Elliott LF, editors. Soil Science Society of America; 1981. pp. 141–151. [Google Scholar]
- Papendick RI, Camprell GS. In: Water Potential Relations in Soil Microbiology SSSA Special Publication. Parr JF, Gardner WR, Elliott LF, editors. Soil Science Society of America; 1981. pp. 1–22. [Google Scholar]
- Boswell GP, Jacobs H, Davidson FA, Gadd GM, Ritz K. Functional consequences of nutrient translocation in mycelial fungi. J. Theor. Biol. 2002;217:459–477. doi: 10.1006/jtbi.2002.3048. [DOI] [PubMed] [Google Scholar]
- Jennings DH. Translocation of solutes in fungi. Biol. Rev. Camb. Philos. Soc. 1987;62:215–243. [Google Scholar]
- Bais HP, Weir TL, Perry LG, Gilroy S, Vivanco JM. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 2006;57:233–266. doi: 10.1146/annurev.arplant.57.032905.105159. [DOI] [PubMed] [Google Scholar]
- Furuno S, et al. Mycelia promote active transport and spatial dispersion of polycyclic aromatic hydrocarbons. Environ. Sci. Technol. 2012;46:5463–5470. doi: 10.1021/es300810b. [DOI] [PubMed] [Google Scholar]
- Gao Y, Cheng Z, Ling W, Huang J. Arbuscular mycorrhizal fungal hyphae contribute to the uptake of polycyclic aromatic hydrocarbons by plant roots. Bioresour. Technol. 2010;101:6895–6901. doi: 10.1016/j.biortech.2010.03.122. [DOI] [PubMed] [Google Scholar]
- Semple KT, Morriss AWJ, Paton GI. Bioavailability of hydrophobic organic contaminants in soils: fundamental concepts and techniques for analysis. Eur. J. Soil. Sci. 2003;54:809–818. [Google Scholar]
- Tecon R, Binggeli O, vander Meer JR. Double-tagged fluorescent bacterial bioreporter for the study of polycyclic aromatic hydrocarbon diffusion and bioavailability. Environ. Microbiol. 2009;11:2271–2283. doi: 10.1111/j.1462-2920.2009.01952.x. [DOI] [PubMed] [Google Scholar]
- Tecon R, Wells M, vander Meer JR. A new green fluorescent protein-based bacterial biosensor for analysing phenanthrene fluxes. Environ. Microbiol. 2006;8:697–708. doi: 10.1111/j.1462-2920.2005.00948.x. [DOI] [PubMed] [Google Scholar]
- Schamfuss S, et al. Impact of mycelia on the accessibility of fluorene to PAH-degrading bacteria. Environ. Sci. Technol. 2013;47:6908–6915. doi: 10.1021/es304378d. [DOI] [PubMed] [Google Scholar]
- Smibert RM, Krieg RM, et al. In: Manual of methods for general bacteriology. Gerhardt P, Murray RGE, Costilow RN, Nester EW, Wood WA, Krieg NR, Phillips GB, editors. Vol. 19. Washington, D.C: American Society for Microbiology; 1981. pp. 409–443. [Google Scholar]
- Wu CH, Warren HL. Natural autofluorescence in fungi and its correlation with viability. Mycologia. 1984;76:1049–1058. [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen C, Sylvia D. Ch. 8 Concepts in Mycorrhizal Research. In: Mukerji KG, editor. Handbook of Vegetation Science. Vol. 19. Netherlands: Springer; 1996. pp. 195–222. [Google Scholar]
- Furuno S, et al. Fungal mycelia allow chemotactic dispersal of polycyclic aromatic hydrocarbon-degrading bacteria in water-unsaturated systems. Environ. Microbiol. 2010;12:1391–1398. doi: 10.1111/j.1462-2920.2009.02022.x. [DOI] [PubMed] [Google Scholar]