

Abstract
The accessibility of high-throughput sequencing has revolutionized many fields of biology. In order to better understand host-associated viral and microbial communities, a comprehensive workflow for DNA and RNA extraction was developed. The workflow concurrently generates viral and microbial metagenomes, as well as metatranscriptomes, from a single sample for next-generation sequencing. The coupling of these approaches provides an overview of both the taxonomical characteristics and the community encoded functions. The presented methods use Cystic Fibrosis (CF) sputum, a problematic sample type, because it is exceptionally viscous and contains high amount of mucins, free neutrophil DNA, and other unknown contaminants. The protocols described here target these problems and successfully recover viral and microbial DNA with minimal human DNA contamination. To complement the metagenomics studies, a metatranscriptomics protocol was optimized to recover both microbial and host mRNA that contains relatively few ribosomal RNA (rRNA) sequences. An overview of the data characteristics is presented to serve as a reference for assessing the success of the methods. Additional CF sputum samples were also collected to (i) evaluate the consistency of the microbiome profiles across seven consecutive days within a single patient, and (ii) compare the consistency of metagenomic approach to a 16S ribosomal RNA gene-based sequencing. The results showed that daily fluctuation of microbial profiles without antibiotic perturbation was minimal and the taxonomy profiles of the common CF-associated bacteria were highly similar between the 16S rDNA libraries and metagenomes generated from the hypotonic lysis (HL)-derived DNA. However, the differences between 16S rDNA taxonomical profiles generated from total DNA and HL-derived DNA suggest that hypotonic lysis and the washing steps benefit in not only removing the human-derived DNA, but also microbial-derived extracellular DNA that may misrepresent the actual microbial profiles.
Keywords: Molecular Biology, Issue 94, virome, microbiome, metagenomics, metatranscriptomics, cystic fibrosis, mucosal-surface
Introduction
Viral and microbial communities associated with the human body have been investigated extensively in the past decade through the application of sequencing technologies1,2. The outcomes have led to the recognition of the importance microbes in human health and disease. The major initiative came from the human microbiome project that describes the bacteria (and some archaea) residing on human skin, and within oral cavities, airways, urogenital tract, and gastrointestinal tract3. Further microbiome studies of healthy human airways through bronchoalveolar lavage (BAL)4,5 and nasopharyngeal swabs4 have shown that the lung can serve as an environmental sampling device, results in transient microbial colonization in the airways. However, the impact of microbial colonization in impaired airway surfaces can lead to severe and chronic lung infections, such as those seen in Cystic Fibrosis (CF) patients.
CF is a lethal genetic disease caused by the mutation in Cystic Fibrosis Transmembrane Regulator (CFTR) gene6. These mutations give rise to defective CFTR proteins that in turn affect transepithelial ion transport across the apical surface of the epithelium. The disease affects multiple organ systems, but the majority of mortality and morbidity is attributable to CF lung disease7. The CF lung provides a unique ecosystem for microbial colonization. The defect in ion transport causes mucus to build up in the CF airways, creating microenvironments consisting of aerobic, microaerophilic, and anaerobic compartments anchored by a static nutrient-rich mucosal surface. This environment facilitates the colonization and proliferation of microbes, including viral, bacteria, and fungi. Acute and chronic pulmonary microbial infections lead to constant but ineffective immune responses, resulting in extensive airway remodeling, loss of pulmonary capacity, and ultimately pulmonary failure.
Bacterial communities associated with the CF lung have been well described using both culture-dependent and culture-independent approaches, which include using 16S ribosomal RNA (rRNA) gene sequencing8 and shotgun metagenomics9,10. The 16S rRNA-based approach is able to characterize a wide range of microbial species and capture broad shifts in community diversity. However, it is limited in its resolution in defining the communities (summarized in Claesson et al. 201011) and the predictions of metabolic potentials are limited to those general functions known for the taxa identified. Therefore, 16S rRNA gene sequencing methods are insufficient for the necessary taxonomic and functional analytic accuracy of the diverse microbial communities present in CF lungs. The metagenomic approach described here complements the 16S rRNA-based approach, overcomes its limitations, and enables a relatively effective way to analyze both the microbial community taxonomy and genetic contents in CF lungs.
Microbial DNA isolated from animal-associated samples often contains a large amount of host DNA. CF sputum or lung tissue samples usually contain a large amount of human DNA released by neutrophils in the immune response, often greater than 99% of the total DNA12–14. Although some intact human cells may be present, most of this DNA is free in solution or adsorbed to the surface of microbes. In addition, the presence of exceptionally viscous mucus plugs, cellular debris, and other unknown contaminants further complicate isolation of microbial cells. Several methods were tested for depleting these samples of human DNA, including Percoll gradients to separate human from microbial cells15, treatment with DNase I, ethidium bromide monoazide to selectively degrade human DNA16, and the MolYsis kit, all with limited success. To date the most effective microbial DNA purification procedure for CF sputum has been a modification of the process described by Breitenstein et al. (1995)17. This approach, herein known as hypotonic lysis (HL) method, uses a combination of β-mercaptoethanol to reduce mucin disulfide bonds, hypotonic lysis of eukaryotic cells, and DNase I treatment of soluble DNA9. Despite the lack of alternatives the HL method raised some concerns due to (i) possible biases resulting from unwanted lysis of microbes and (ii) whether the observed fluctuations in community composition9,10 are an artifact of variations associated with the sample processing. In addition to the generation of shotgun metagenomes, we address these issues by comparing the 16S rRNA gene profiles of the total DNA and microbial DNA extracted from the HL method using the same set of sputum samples collected from a single patient across seven consecutive days.
Compared to microbial communities, the characterization of viral communities associated with animals is limited18,19. The viral communities in CF airways have only been characterized minimally20–22. The first metagenomic study characterizing the DNA of viral communities in CF airways showed that most viruses associated with CF lungs are phages20. The metabolic potential of phage in CF and non-CF individuals was significantly different. Specifically, the phage communities in CF individuals carried genes reflective of bacterial host adaptations to the physiology of CF airways, and bacterial virulence20. Subsequent metagenomic studies of viruses in CF lung tissue demonstrated distinct spatial heterogeneity of viral communities between anatomical regions22. In addition, CF lung tissue harbored the lowest viral diversity observed to date in any ecosystem22. Most viruses identified were phages with the potential to infect CF pathogens. However, eukaryotic viruses such as herpesviruses, adenoviruses, and human papilloma viruses (HPV) were also detected. In one event, where cysts in the lung tissue were observed during dissection, more than 99% of a human papillomavirus genome was recovered, even though the patient was never diagnosed with a pulmonary papilloma or carcinoma. This indicates that the viral diversity present not only reflects the severity of tissue damage, but may also expose and explain an underlying uncharacterized disease. The protocols described here provide a simple, yet powerful way to isolate viral-like particles (VLPs) from samples that consist of large amounts of thick mucus, host and microbial cells, free DNA, as well as cell debris.
Complementing metagenomics, metatranscriptomics is used to monitor the dynamics in gene expression across the microbial community and the host9,23. In this case, both microbial and host mRNA need to be preferentially selected. Since bacterial mRNAs are not polyadenylated, an oligo-dT-based mRNA pull-down method cannot be exploited. Polyadenylation-dependent RNA amplification cannot be used in host-associated samples if the samples are known to contain large amounts of eukaryotic mRNA. Many animal-associated samples, including CF sputum, contain a high density of cells in addition to high amounts of cellular debris and nucleases that include RNases. Therefore, another challenging task is to prevent extensive RNA degradation during metatranscriptome processing. In most cases, total RNA extracted from CF sputum is partially degraded, limiting the downstream applications and utility of the derived RNA. In recent years, several approaches for rRNA depletion have been developed and adapted in commercially available kits. The efficacy of these approaches is however limited, especially when working with partially degraded rRNA9,24. The methods employed here allowed for the retrieval of partially degraded total RNA suitable for efficient downstream total rRNA removal. Direct comparison of the efficiency in rRNA removal from partially degraded total RNA comparing two different kits was illustrated by Lim et al. (2012)9.
Overall, the goal of this manuscript is to provide a complete set of protocols (Figure 1) to generate viral and microbial shotgun metagenomes, and a metatranscriptome, from a single animal-associated sample, using induced sputum sample as an example. Molecular laboratory workflow should include separate pre- and post-amplification areas to minimize cross-contamination. The methods are easily adaptable to other sample types such as tissue22, nasopharyngeal and oropharyngeal swabs25, bronchoalveolar lavage (BAL) and coral (unpublished data). Each sample should be processed immediately upon collection especially when microbial metagenomics and metatranscriptomics studies are desired. If the samples were frozen, it limits the isolation of intact microbial cells for microbial metagenomes as freezing potentially disrupt the cell integrity. However, freezing does not preclude metatranscriptomics and viral isolation, but the quality of RNA and amount of viral particles recovered may be affected through the freeze-thaw process. It is important to note that induced sputum has served as the primary source of samples in many studies associated with adult CF patients and other chronic pulmonary diseases26,27 as BAL can be too invasive. In our studies, sputum samples were collected with a careful and consistent sampling method, i.e., following mouthwash and rinsing of the oral cavity using sterile saline solution to keep oral microbes contamination within the sputum samples to a minimum.
Protocol
NOTE: Induced sputum samples were collected in accordance with the University of California Institutional Review Board (HRPP 081500) and San Diego State University Institutional Review Board (SDSU IRB#2121), by the research coordinator of the University of California, San Diego (UCSD) adult CF clinic.
1. Sample Collection and Pre-treatment (Pre-treat Samples within 30 Min After Collection)
Prior to sample collection, label four 15 ml tubes as: (i) viral metagenome, (ii) microbial metagenome, (iii) metatranscriptome, and (iv) extra sputum. Repeat for each sample. Add 2 ml of 0.1 mm silica beads into the tube labeled “Metatranscriptome”, followed by 6 ml of guanidine isothiocyanate-based RNA lysis buffer (GITC-lysis buffer).
During sample collection, use sterile saline solution (60 ml) as a mouth rinse to minimize contamination by oral microbes. Collect sputum samples over a 30 min time period after the inhalation of 4 ml of 7% hypertonic saline via a nebulizer. Process samples immediately, as described below.
- Dilute the sample to a total volume of 8 ml.
- Estimate sample volume by weighing empty sputum cup before and after sample collection.
- If the volume of sample is less than 8 ml, add appropriate amount of 0.02 μm-filtered 1x PBS to generate a total sample volume of at least 8 ml.
- Immediately homogenize the sample with a 3 ml syringe until no visible clumps remain within the sputum
- Using the same syringe, draw up 2 ml of sputum and proceed immediately to step 1.4.
- Preserving Total RNA from Sputum Sample
- Inject 2 ml sputum sample from step 1.3.4 into the “metatranscriptome” tube containing silica beads and GITC-lysis buffer.
- Close the lid and seal the tube securely with Parafilm to avoid leakage.
- Homogenize the sputum immediately at medium speed for 10 min. Depending on the vortexer available, place the tube horizontally and secure with tape if necessary.
- Keep the tube at 4 °C or in an ice box and transport to the laboratory if necessary.
Using the same syringe, aliquot 2 ml of sputum each into the tubes labeled “viral metagenome” and “microbial metagenome” and transfer the remaining sputum from the sputum cup into the tube labeled “extra sputum”.
Store all tubes at 4 °C or ice box and transport to the laboratory if necessary.
2. Generating Viral Metagenome
- Preparation of Buffers and Solutions
- Prepare 50 mM dithiothreitol (DTT) in advance and store at 4 °C. This is stable for 2 weeks.
- Prepare Saline Magnesium (SM) buffer (250 ml): 1 M NaCl, 10 mM MgSO4, 50 mM Tris-HCl; adjust pH to 7.4. Filter sterilize (0.02 µm pore size) and store at room temperature.
- Prepare DNase I enzyme to 100 U/µl (in molecular grade water) from lyophilized bovine pancreas DNase I according to the activity defined by Dornase unit/mg dry weight.
- Prepare 10x DNase I buffer (50 ml): 100 mM MgCl2, 20 mM CaCl2; adjust pH to 6.5. Filter sterilize (0.02 µm pore size) and store at room temperature.
- Prepare 4% paraformaldehyde.
- Prepare 200x TE Buffer: 2 M Tris-HCl (pH 8.5), 0.2 M EDTA. Filter sterilize (0.02 µm pore size) and store at room temperature.
- Prepare 10 ml of 10% Sodium Dodecyl Sulfate (SDS) using molecular grade water.
- Prepare 50 ml CTAB/NaCl (10% CTAB. 700 mM NaCl) using molecular grade water. Dissolve CTAB overnight. If precipitates persist, heat up the solution at 65 °C. Solution is highly viscous in room temperature. NOTE: The filtration of buffers using 0.02 μm filter allows the removal of viral-like particles within the solution, but not free nucleic acid contamination.
- Sample Pre-treatment
- Prepare appropriate amount of fresh 6.5 mM dithiothreitol (DTT).
- Dilute the homogenate by adding 0.02 µm-filtered SM buffer to generate a total volume of 6 ml.
- To aid in mucus dissolution, add equal volume (6 ml) of 6.5 mM dithiothreitol (DTT) to the sample, vortex vigorously to mix and incubate for 1 hr at 37 °C.
- Vortex the treated sample vigorously and spin at 10 °C, 3,056 x g for 15-20 min.
- Collect the supernatant into a new 15 ml tube.
- Repeat step 2.2.3 and 2.2.5 for the next sample.
- Transfer and filter the supernatant with a 0.45 µm filter mounted on a syringe into a new 15 ml tube. NOTE: If the filter clogs, retrieve the samples from the syringe and omit the filtration step.
- Take a 100 µl subsample of the 0.45 µm-filtered sample, perform chloroform and DNase I treatment (see section 2.3.12-2.3.15) and add equal volume of 4% paraformaldehyde to fix the sample for epifluorescence microscopy (Figure 2A).
- For a “catch-all” viral particles enrichment approach (see Discussion), go to step 2.3.12 to omit viral particles selection based on cesium chloride gradient ultracentrifugation. However, this may result in chloroform-resistant bacterial contamination and higher amount of host-DNA in the viral lysate.
- Viral-like Particles (VLPs) Enrichment and Purification
- Prepare individual cesium chloride (CsCl) solutions by dissolving the appropriate amount of CsCl with non-filtered SM buffer to the desired density (1.7 g/ml, 1.5 g/ml, 1.35 g/ml, and 1.2 g/ml). Filter each solution through a 0.02 µm filter prior to use.
- Set up CsCl gradient as shown in Figure 2B.
- Load 1 ml of 1.7 g/ml into each tube, load 1 ml of 1.5 g/ml into each tube, load 1 ml of 1.35 g/ml into each tube, load 1.2 g/ml into each tube (optional), and finally load 6-8 ml sample into the respective tube. Mark individual layers to denote the location of each fraction.
- Balance each opposing pair of tubes to within 1 mg.
- Carefully load each tube into the spin bucket. Spin all buckets even if they are empty. Load the spin bucket onto the rotor.
- Centrifuge at 82,844 x g at 4 °C for 2 hr.
- Following centrifugation, carefully remove the tubes from the holder without disrupting the density gradients.
- Using a 3 ml syringe with an 18 G needle, pierce the tube just below the 1.5 g/ml density layer (Figure 2, red arrow) and pull ~1.5 ml into the syringe.
- Collect the upper fraction by slowly removing the needle and allowing the remaining fraction in the tube to drip into a new 15 ml tube through the puncture. Label this as “upper fraction waste”.
- Collect the 1.5 g/ml fraction (containing VLPs) from the syringe by ejecting the contents into two new microfuge tubes.
- Repeat step 2.3.8-2.3.10 for all samples.
- Add 0.2 volume of chloroform into the viral concentrate, shake vigorously, incubate at RT for 10 min, spin at max speed for 5 min, and collect the aqueous phase.
- Add 10x DNase buffer and DNase I (final concentration = 2.5 U/µl) into the chloroform-treated viral concentrate, and incubate at 37 °C for 1.5-2 hr.
- Inactivate the DNase activity at 65 °C for 15 min.
- Remove 15 µl of the chloroform- and DNase I-treated viral fraction into a new tube, and add 15 µl 4% paraformaldehyde to fix the sample for epifluorescence microscopy.
- DNA Extraction
- Pool viral concentrates from each sample into a cleaned and autoclaved 50 ml Oak Ridge high-speed centrifuge tube.
- Add the following: 0.1 volume 200x TE buffer, 10 μl 0.5 M EDTA per ml of sample, 1 volume of formamide, and 10 μl glycogen. Mix well and incubate at room temperature for 30 min.
- Using the new volumes, add 2 volumes of room temperature 100% ethanol. Mix well and incubate at 4 °C for at least 30 min.
- Pellet DNA by spinning the tube at 17,226 x g for 20 min, at 4 °C using a SS-34 rotor.
- Discard the supernatant carefully by using a serological pipette. Wash the pellet twice with ice-cold 70% ethanol.
- Remove as much liquid as possible and allow the pellet to air-dry at room temperature for 15 min.
- Resuspend the DNA pellet in 567 μl of 1x TE buffer (pH 8.0). NOTE: Allow at least 15 min for complete resuspension at room temperature. Store the resuspended DNA overnight at 4 °C until further processing.
- Transfer the entire 567 μl of resuspended DNA solution into a new 1.5 ml microfuge tube. Add 30 μl of pre-warmed 10% SDS and 3 μl of proteinase K (20 μg/ml), mix thoroughly and incubate for 1 hr at 56 °C. Pre-warm CTAB/NaCl at 65 °C.
- Add 100 μl of 5 M NaCl and mix thoroughly. Add 80 μl of pre-warmed CTAB/NaCl solution, vortex, and incubate for 10 min at 65 °C.
- Add equal volume of chloroform, vortex to mix, and spin at 16,100 x g for 5 min.
- Transfer the supernatant to a new 1.5 ml microfuge tube. Add an equal volume of phenol/chloroform, vortex to mix, and spin at 16,100 x g for 5 min.
- Transfer the supernatant to a new 1.5 ml microfuge tube. Add an equal volume of chloroform, vortex to mix, and spin at 16,100 x g for 5 min.
- Transfer the supernatant to a new 1.5 ml microfuge tube. Add equal volume of isopropanol to the supernatant fraction, mix, and incubate at -20 °C for at least 30 min.
- Pellet the DNA, spin at 16,100 x g for 15 min at 4 °C. Pipette off the supernatant carefully and wash the pellet twice with ice-cold 70% ethanol.
- Perform a short spin and remove the remaining ethanol from the tube. Air dry the pellet for 15 min.
- Resuspend the DNA pellet with 50 μl of elution buffer (5 mM Tris, pH 8.5). Allow the pellet to rehydrate for at least 5 min in room temperature.
- Quantify the DNA using a high-sensitivity fluorescence-based assay.
- Amplification using Phi29 Polymerase (Optional)
- Prepare 2x annealing buffer: 80 mM Tris-HCl (pH 8.0), 20 mM MgCl2.
- Dilute Phi29 DNA polymerase to 5 U/μl.
- Pre-mix sample buffer, comprised of 50 μl random hexamer primer (100 μM), 125 μl 2x annealing buffer, and 25 μl water. Aliquot and store at -20 °C.
- Pre-mix reaction buffer, comprised of 100 μl Phi29 10x buffer, 40 μl 10 mM dNTPs, and 560 μl water. Aliquot and store at -20 °C.
- Add 1 μl template DNA into 4 μl sample buffer.
- Incubate the mixture at 95 °C for 3 min and cool on ice.
- Add 14 μl of reaction buffer into the mixture from 2.4.4, mix by pipetting up and down.
- Add 1 μl of Phi29 DNA polymerase, mix by pipetting up and down, and incubate at 30 °C for 18 hr followed by 65 °C for 10 min.
- Clean up the reactions using genomic DNA columns or phenol/chloroform and ethanol precipitations.
- Epifluorescence Microscopy (Refer to Haas et al. 201428 for filtration system setup) NOTE: Following isolation and purification, epifluorescence microscopy with nucleic acid dyes can be used to verify the presence and purity of viral particles in samples (Figures 2A and 2C). Free DNA in the sample can give rise to high background fluorescence. Therefore, the sample should be DNase I-treated prior to fixation and staining for micrographs.
- Prepare mount solution (0.1% ascorbic acid, 50% glycerol). Add 100 μl of 10% ascorbic acid to 4.9 ml of 1x phosphate buffered saline (PBS), mix thoroughly. Add 5 ml of 100% glycerol to the mixture, mix thoroughly and label the tube as “mount”.
- Filter mount using a 0.02 μm alumina matrix disposable syringe filter, aliquot into microfuge tubes, and store at -20 °C.
- Aliquot 100 μl of sample into a new microfuge tube and add equal volume of 4% paraformaldehyde to fix the VLPs. Incubate the mixture at room temperature for at least 10 min.
- Make up the volume to 1 ml by adding 800 μl of 0.02 μm-filtered water. Add 1 μl of SYBR Gold stain into the tube and incubate at room temperature for 10 min.
- Set up the filtration system by turning on the vacuum pump between -9 and -10 psi (-62.1 and -68.9 kPa).
- Wash the pedestal with water and place a 0.02 µm alumina matrix filter with annular polypropylene support ring into the filter pedestal.
- Place a filter tower on top of the filter pedestal with the filter and secure with a clamp.
- Pipette the contents from the 1.5 ml microfuge tube into the filter tower, and allow a few minutes for sample to filter through.
- Label and pipette 10 µl of mount reagent into a microscopic slide.
- Leave the vacuum on while removing the filter tower and clamp.
- Carefully remove the filter from the filter pedestal and blot the bottom of the filter with a Kimwipe, then place the filter directly on top of the mount on the microscopic slide.
- Pipette another 10 µl of mount reagent onto the filter and place a coverslip over the filter.
3. Generating Microbial Metagenome
- Preparation of buffers and solutions
- Prepare 50 ml of 1x DNase buffer: 50 mM NaAc, 10 mM MgCl2, 2 mM CaCl2; adjust pH to 6.5. Filter sterilize (0.22 µm) and store at room temperature.
- Prepare DNase I enzyme to 1000 U/µl (in molecular grade water) from lyophilized bovine pancreas DNase I according to the activity defined by Dornase unit/mg dry weight.
- Prepare 100 ml of SE buffer: 75 mM NaCl, 25 mM EDTA; adjust pH to 7.5. Filter sterilize (0.22 µm) and store at room temperature.
- Sample Pre-treatment Prior to DNA Extraction
- Dilute the homogenate by adding 5 volumes of 0.22 µm-filtered 1x PBS. For example, add 10 ml of 1x PBS into 2 ml of sample.
- Add β-mercaptoethanol to 2% (v/v) final concentration. Rock the mixture (in the chemical hood) at room temperature for 2 hr.
- Spin the sample at 10 °C and 3,056 x g for 15 min, and discard supernatant.
- Resuspend the pellet in 10 ml of molecular grade water (or 0.22 µm filtered water), and incubate at room temperature for 15 min.
- Repeat steps 3.2.3 and 3.2.4 once.
- Spin at 10 °C and 3,056 x g for 15 min, and discard supernatant.
- Resuspend the pellet in 5 ml 1x DNase buffer and add 15 µl DNase I (1,000 U/µl) per ml of sample.
- Incubate at 37 °C with repeated mixing for 2 hr.
- Inactivate the DNase activity at 65 °C for 15 min.
- Spin at 10 °C and 3,056 x g for 15 min, and discard supernatant. Resuspend the pellet in 10 ml SE buffer.
- Repeat step 3.2.10.
- Spin at 10 °C and 3,056 x g for 15 min, and discard supernatant.
- Resuspend the pellet in 2 ml SE buffer, and transfer to two microfuge tubes.
- Pellet the cells in the microfuge tubes. Spin the tubes at 16,100 x g at room temperature for 15 min.
- Remove the supernatant and extract DNA from the pelleted cells using a genomic DNA extraction kit, Gram-positive variation protocol.
4. Generating Metatranscriptome
- Sample Pre-treatment
- Perform mechanical lysis of cells by bead beating in GITC-lysis buffer immediately after sample collection and homogenization. See step 1.4.
- Spin the mixture at 4 °C and 600 x g for 5 min to pellet the silica beads.
- Transfer the supernatant into a new tube.
- Add 200 μl of chloroform for every 750 μl of GITC-lysis buffer used, shake vigorously by hand for 15 sec, incubate at room temperature for 10 min, and spin at 4 °C and 3,056 x g for 15 min. During this 15 min spin, prepare for step 4.2.
- After the 15 min spin (a clear separation of aqueous phase-interphase-organic phase forms), extract the aqueous phase (without disrupting the interphase) into new RNase-free tube(s). NOTE: The aqueous phase contains the RNA. Keep the tubes on ice until the next step.
- Perform total RNA extraction and purification using commercially available column-based RNA purification kits or conventional isopropanol-based RNA precipitation.
- Silica Column-based RNA Purification
- Measure the total volume of the aqueous fraction obtained.
- Add appropriate volume of RNA-binding buffer to sample and mix well.
- Adjust mixture to appropriate binding condition according to manufacturer’s protocol. Mix well and do a short spin.
- Load the mixture into the RNA-column. For a large volume sample, use multiple loading and load each column up to 4x. Otherwise, consider using multiple columns for each sample.
- Wash the column appropriately according to the manufacturer’s protocol.
- Elute the RNA with at least 30 µl of RNase-free water. Double-elution will slightly increase the yield of RNA. However, this will dilute the RNA concentration.
- Measure the RNA concentration and proceed directly to DNase I treatment. Use the Bioanalyzer to check the quality of the RNA (recommended).
- RNA Precipitation
- Add an equal volume of isopropanol (e.g., 500 µl of isopropanol into 500 µl of aqueous fraction) and 2 µl of 10 µg/µl RNase-free glycogen to the sample.
- Incubate the mixture at room temperature for 10 min.
- Spin at 12,000 x g and 4 °C for 15 min.
- Carefully remove the supernatant, add 1 ml of RNase-free 75% ethanol. Spin the mixture at 7,500 x g and 4 °C for 5 min to make sure the pellet is intact.
- Carefully remove the ethanol.
- Repeat steps 4.2.2.4 and 4.2.2.5 once.
- Air dry the pellet for 10 min.
- Rehydrate the pellet in 50 µl of RNase-free water, incubate at 55 °C for 5 min and proceed directly to DNase treatment. Use the Bioanalyzer to check the quality of the RNA (Recommended).
- Store RNA in aliquots at -20 °C, or -80 °C for long-term storage.
Representative Results
Viral Metagenomes
CF sputum is exceptionally viscous and contains a high amount of mucin and free DNA (Figure 2A); the density gradient ultracentrifugation facilitates the elimination of host-derived DNA (Figure 2B). The results from a previous study9 showing eight viromes generated from the presented workflow are summarized here (Table 1). Seven samples (CF1-D, CF1-E, CF1-F, CF4-B, CF4-C, CF5-A, and CF5-B; Table 1) were processed as described in Section 2. The generated viromes contained little (0.02%-3.7%) human-derived sequences with only one exception (70%). CF4-A was omitted from the density gradient ultracentrifugation step (CF4-A) and the virome generated from this specific sample contained >97% human-derived sequences (Table 1). Figure 2 shows an example of the epifluorescence microscopy image of a typical CF sputum sample before (Figure 2A) and after (Figure 2C) density gradient ultracentrifugation. Clear viral-like particles (VLPs) were observed in the micrographs without large particles following the density gradient separation. After VLPs DNA extraction, bacterial contamination is often tested using 16S rDNA amplification prior to the sequencing of VLPs DNA.
Microbial Metagenomes
Seven sputum samples presented here were collected from a single CF patient across seven consecutive days. The patient started on oral antibiotic (Ciprofloxacin and Doxycycline) on Day 3 after the sputum was collected. The volume of each sputum sample collected from this patient was 15 ml throughout the 7 days; therefore, PBS was not added to the sample. The goal of this sampling event was to evaluate the protocols presented in this workflow by (i) evaluating the daily fluctuation of microbial community structure, and (ii) compare the microbial community structure and resolution between metagenomics and 16S rDNA sequencing. Therefore, total DNA and HL-DNA were extracted from each sample.
The HL-DNA concentration of each sputum sample following DNA extraction is presented in Table 2. The total yield of HL-DNA ranged from 210 ng to >5 μg. Illumina sequencing libraries were generated with a total starting material of 1 ng for each sample (Figure 3). The characteristics of the metagenomics data are presented in Table 2. All but one library yielded more than 1 million sequences and more than 85% high quality sequences were retained upon data preprocessing using the PRINSEQ29 software. All datasets were first preprocessed to remove duplicates and sequences of low quality (minimum quality score of 25), followed by further screening and removal of human-derived sequences using DeconSeq30. The amount of human-derived sequence contamination is highly dependent on the sample properties. Here, the total amount of human-derived sequences ranged from 14-46% (Table 2). The preprocessed sequences were then annotated using the Metaphlan31 pipeline as well as MG-RAST32 server.
In addition to metagenomes, 16S rDNA amplicon libraries were generated from both the total DNA and HL-DNA via primers targeting approximately 300 bp of the V1-V2 variable region in the 16S rRNA gene33,34. PCR products from individual samples were normalized and pooled for sequencing using the Illumina 500-cycle paired-end sequencing performed on the MiSeq platform. Paired-end 16S rDNA amplicon sequences were sorted by sample via barcodes using a python script and the paired reads were assembled using phrap35,36. Assembled sequence ends were trimmed until the average quality score was ≥20 using a 5 nt window. Potential chimeras were then removed using Uchime37 against a chimera-free subset of the SILVA38 reference sequences. Taxanomy was assigned to the high quality reads with SINA39 (version 1.2.11) using the 418,497 bacterial sequences from the SILVA38 database. Sequences with identical taxonomic assignments were clustered to produce Operational Taxonomic Units (OTUs). This process generated 1,655,278 sequences for 16 samples (average size: 103,455 sequences/sample; min: 72,603; max: 127,113). The median Goods coverage score, a measure of completeness of sequencing, was ≥ 99.9%. The software package Explicet40 (v2.9.4, www.explicet.org) was used for analysis and figure generation. Alpha-diversity (intra-sample) and beta-diversity (inter-sample) were calculated in Explicet at the rarefaction point of 72,603 sequences with 100 bootstrap re-samplings.
The first question targeted by this study was whether hypotonic lysis preferentially selects for (i.e., preferentially retains or lyses) particular groups of microbes. After the first hypotonic lysis, re-suspended pelleted cells were subsampled from the first two samples (CF1-1A* and CF1-2A*) to compare with the same samples after the second hypotonic lysis (CF1-1and CF1-2). All samples were treated equally, i.e., treated with DNase I prior to DNA extraction, followed by DNA extraction and the sequencing pipeline. As shown in Figure 4, the microbial profiles of the subsamples are highly similar to the samples after two hypotonic lysis treatments. In addition, the second hypotonic lysis increases the fraction of non-human sequences by 6-17% within the metagenomes (Table 2).
To test for differences in microbial composition between metagenomic- and 16S rDNA-based profiling, and for changes before and after hypotonic lysis that might explain the differences previously seen between our studies and others, bacterial 16S rDNA sequencing libraries were generated from both the total DNA and HL-derived DNA (Figure 4B). At genus level, the taxonomy profiles of the common CF-associated bacteria such as Pseudomonas, Stenotrophomonas, Prevotella, Veillonella,and Streptococcus were highly similar between the 16S rDNA libraries and metagenomes generated from the HL-derived DNA. However, Rothia detection in the 16S rDNA libraries was not as abundant as with the metagenomic libraries. When comparing the 16S rDNA taxonomical profiles generated from total DNA and HL-derived DNA, Pseudomonas was differentially represented in the total DNA compared to the HL-derived DNA starting from Day 3.
Metatranscriptomes
Typically, the total RNA extracted from CF sputum is partially degraded and the size ranges from 25-4,000 bps (Figures 5A and 5C). Here, the representative results presented was previously published in Lim et al. 20129. The fraction of rRNA within the non-depleted metatranscriptomes ranges from 27-83%, and the relative abundance of rRNA varied across samples (Table 3; data extracted from Lim et al.9). However, depletion with Ribo-Zero kit decreased the rRNAs relative abundance of rRNA to 1-5% with the exception of sample CF1-F. The variation in the effectiveness of rRNA removal could reflect the quality of extracted RNA, or differences in the microbial community present and hence the accessibility of rRNAs for probes hybridization9. The electropherograms of a successful (Figure 5B) and unsuccessful (Figure 5D) rRNA removal procedure using the Ribo-Zero rRNA removal kit differ, at which rRNA peaks are visible in the unsuccessful removal.
The size range of cDNA libraries generated often reflects the size range of the starting RNA sample. The cDNA libraries presented here were generated with a whole transcriptome amplification kit (WTA2) upon rRNA depletion followed by Roche-454 sequencing library preparation9. The cDNA generated contain fragments ranging from 50-4,000 bps (Figures 5E and 5F) and is highly consistent across samples (Lim et al. 2012)9. The availability of other platform-specific RNA-Seq library preparation kits currently provide more alternative options for one to combine cDNA synthesis and sequencing library preparation in optimum conditions. One recommended option to date is the ScriptSeq Complete Gold Kit combining rRNA removal reagents recommended above and RNA-Seq library preparation kit.
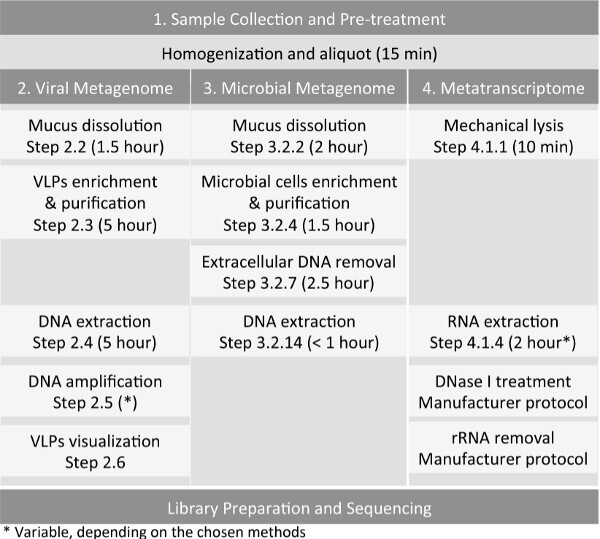 Figure 1: Workflow for the preparation of host-associated samples, such as sputum sample, for virome, microbiome, and metatranscriptome sequencing.
Figure 1: Workflow for the preparation of host-associated samples, such as sputum sample, for virome, microbiome, and metatranscriptome sequencing.
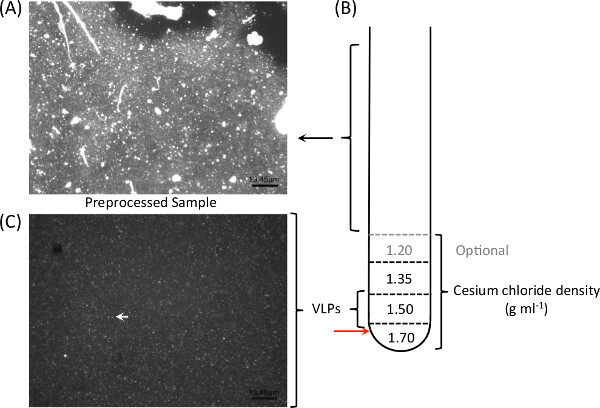 Figure 2: Cesium chloride density gradients ultracentrifugation facilitate the elimination of extracellular DNA and large particles (A), and allow for optimal isolation of viral-like particles from CF sputum. One milliliter of each gradient is layered on top of each other prior to loading the pre-treated sample (B). Following particles isolation and purification, epifluorescence microscopy with nucleic acid dyes such as SYBR Gold are used to verify the presence and purity of viral particles in samples. Clear viral-like particles (C; white arrow) were observed following the density gradient separation of CF sputum sample.
Figure 2: Cesium chloride density gradients ultracentrifugation facilitate the elimination of extracellular DNA and large particles (A), and allow for optimal isolation of viral-like particles from CF sputum. One milliliter of each gradient is layered on top of each other prior to loading the pre-treated sample (B). Following particles isolation and purification, epifluorescence microscopy with nucleic acid dyes such as SYBR Gold are used to verify the presence and purity of viral particles in samples. Clear viral-like particles (C; white arrow) were observed following the density gradient separation of CF sputum sample.
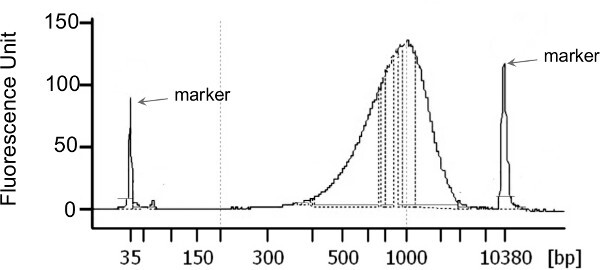 Figure 3: Example of the size distribution of Nextera XT libraries generated from 1 ng of HL-DNA that resulted in CF sputum microbiomes. Library normalization, pooling, and loading amount was done as described in the manufacturer protocol without any deviation.
Figure 3: Example of the size distribution of Nextera XT libraries generated from 1 ng of HL-DNA that resulted in CF sputum microbiomes. Library normalization, pooling, and loading amount was done as described in the manufacturer protocol without any deviation.
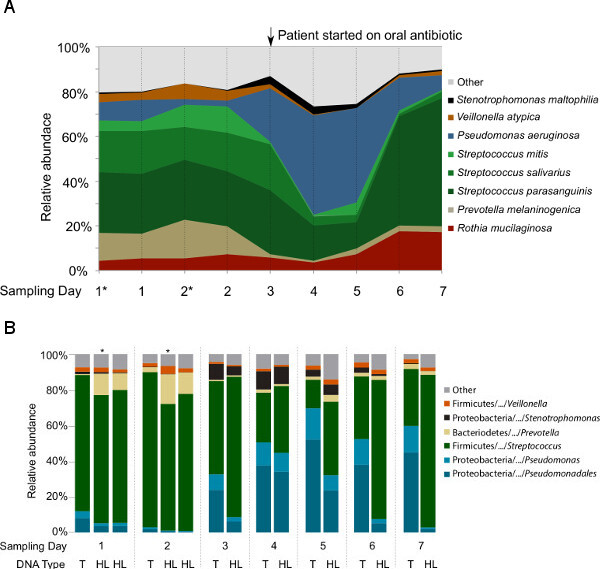 Figure 4: Taxonomic analysis of the microbial communities in nine samples collected longitudinally from one CF patient.(A) Microbial profiles based on the metagenomic libraries generated from hypotonic lysis method-based DNA. The species assignment was based on the Metaphlan pipeline following data preprocessing that remove duplicates and sequences with low quality and human sequence homology. In order to show that two-steps hypotonic lysis did not preferentially selects for particular groups of microbes, subsamples (*) after the first hypotonic lysis were included. (B) Microbial profiles based on the V1V2 region of 16S rRNA gene sequencing from total DNA (T) and hypotonic lysis method-based DNA (HL). These data have not been previously published.
Figure 4: Taxonomic analysis of the microbial communities in nine samples collected longitudinally from one CF patient.(A) Microbial profiles based on the metagenomic libraries generated from hypotonic lysis method-based DNA. The species assignment was based on the Metaphlan pipeline following data preprocessing that remove duplicates and sequences with low quality and human sequence homology. In order to show that two-steps hypotonic lysis did not preferentially selects for particular groups of microbes, subsamples (*) after the first hypotonic lysis were included. (B) Microbial profiles based on the V1V2 region of 16S rRNA gene sequencing from total DNA (T) and hypotonic lysis method-based DNA (HL). These data have not been previously published.
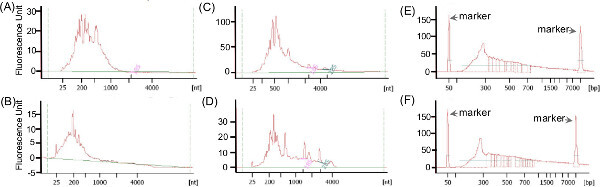 Figure 5:Examples of Agilent 2100 Bioanalyzer electropherograms of RNA (A-D) and cDNA (E-F) generated for the metatranscriptomic libraries, using RNA pico and high-sensitivity dsDNA chips respectively. (A) and (C) show the examples of electropherograms before rRNA removal procedures. The electropherograms of a successful (B) and unsuccessful (D) rRNA removal procedure using total rRNA Removal kit differ slightly, at which rRNA peaks are visible in the unsuccessful removal. The size range of cDNA (E-F) generated using the whole-transcriptome amplification kit (Sigma-Aldrich) is similar to the size range of the starting rRNA-depleted RNA, and highly consistent across the two different samples. Please click here to view a larger version of this figure.
Figure 5:Examples of Agilent 2100 Bioanalyzer electropherograms of RNA (A-D) and cDNA (E-F) generated for the metatranscriptomic libraries, using RNA pico and high-sensitivity dsDNA chips respectively. (A) and (C) show the examples of electropherograms before rRNA removal procedures. The electropherograms of a successful (B) and unsuccessful (D) rRNA removal procedure using total rRNA Removal kit differ slightly, at which rRNA peaks are visible in the unsuccessful removal. The size range of cDNA (E-F) generated using the whole-transcriptome amplification kit (Sigma-Aldrich) is similar to the size range of the starting rRNA-depleted RNA, and highly consistent across the two different samples. Please click here to view a larger version of this figure.
| CF1-D | CF1-E | CF1-F | CF4-A | CF4-B | CF4-C | CF5-A | CF5-B | |
| Total number of reads | 224,859 | 87,891 | 106,189 | 93,301 | 140,020 | 1,558 | 272,552 | 217,438 |
| Preprocessed readsa | 109,389 | 73,624 | 67,070 | 82,011 | 68,617 | 1,137 | 215,808 | 158,432 |
| 49% | 84% | 63% | 88% | 49% | 73% | 79% | 73% | |
| Number of bases | 47,239,573 | 33,351,525 | 28,922,479 | 27,667,695 | 29,386,841 | 243,986 | 95,205,805 | 69,581,811 |
| Mean read length | 432 | 453 | 431 | 337 | 428 | 215 | 441 | 439 |
| Host sequencesb | 240 | 526 | 28 | 79,774 | 13 | 797 | 585 | 5,859 |
| 0.21% | 0.71% | 0.04% | 97.27% | 0.02% | 70.10% | 0.27% | 3.70% | |
| Viral hitsc | 7,214 | 23,550 | 4,070 | 737 | 4,642 | 22 | 6,466 | 5,981 |
| 6.59% | 31.99% | 6.07% | 0.90% | 6.77% | 1.93% | 3.00% | 3.78% | |
| Unassigned Readsd | 103,888 | 60,490 | 32,780 | 1,935 | 68,440 | 311 | 105,612 | 119,551 |
| 94.97% | 82.16% | 48.87% | 2.36% | 99.74% | 27.35% | 48.94% | 75.46% | |
| a Reads after data pre-processing by PRINSEQ29. | ||||||||
| b Human reads identified by DeconSeq30 plus reads with a best BLASTn hit (NCBI nucleotide database) to the phylum Chordata. | ||||||||
| c tBLASTx hits against in-house viral genome database. The percentage was calculated using the total number of preprocessed reads. | ||||||||
| d Reads with no BLASTn hit against the NCBI nucleotide database. The percentage was calculated using the total number of preprocessed reads. Some reads with no BLASTn hit against the NCBI nucleotide database were identified as viral at protein level in the tBLASTx analysis. |
Table 1: Library characteristics of eight viromes generated from sputum samples using presented workflow. This table is extracted from Lim et al. (2012)9. Seven samples (CF1-D, CF1-E, CF1-F, CF4-B, CF4-C, CF5-A, and CF5-B) were processed as described in Section 2 and generated viromes that contained little (0.02% - 3.7%) human-derived sequences with one exception (70%). CF4-A was omitted from the density gradient ultracentrifugation step (CF4-A) and generated virome that contained > 97% human-derived sequences.
| Sample | Concentration | Total Yield | Total No. Reads | Total No. Reads (Processedb) | Non-human Sequences |
| (ng/μl) | (ng) | (Rawa) | (%) | ||
| CF1-1A* | 2.3 | 230 | 1,098,454 | 937,688 | 691,541 |
| 74% | |||||
| CF1-1 | 13 | 1,300 | 2,212,756 | 1,958,910 | 1,574,520 |
| 80% | |||||
| CF1-2A* | 2.1 | 210 | 672,878 | 588,106 | 407,530 |
| 69% | |||||
| CF1-2 | 5.2 | 520 | 1,944,012 | 1,697,010 | 1,455,174 |
| 86% | |||||
| CF1-3 | 28.8 | 2,880 | 1,048,304 | 896,756 | 560,852 |
| 63% | |||||
| CF1-4 | 24.1 | 2,410 | 1,154,922 | 984,702 | 621,098 |
| 63% | |||||
| CF1-5 | 33.6 | 3,360 | 1,029,622 | 888,630 | 481,548 |
| 54% | |||||
| CF1-6 | 43.2 | 4,320 | 1,434,016 | 1,256,504 | 725,858 |
| 58% | |||||
| CF1-7 | 57.8 | 5,780 | 1,000,174 | 872,036 | 565,376 |
| 65% | |||||
| * 1 ml of sample was subsampled from CF1-1 and CF1-2 following the first hypotonic lysis step (Step 3.1.5) before the second hypotonic lysis procedure. The cells were spun down as described in 3.1.7 and proceed through the remaining protocol without any modification. | |||||
| a Unprocessed Illumina reads from a 2 x 300 bp MiSeq sequencing run. | |||||
| b Reads were assessed, trimmed, and removed based on quality and length as described in the discussion. |
Table 2:Characteristics of microbiomes generated from sputum samples using presented workflow. The DNA concentration of each sample in 100 μl elution buffer (5 mM Tris/HCl, pH 8.5) and the characteristics of sequence data are presented. A total of 1 ng was used to generate individual library using the Nextera XT library preparation kit.
| Sample | CF1-D | CF1-F | CF4-B | CF4-C | ||||
| Treatment | None | Ribo-Zero | None | Ribo-Zero | None | Ribo-Zero | None | Ribo-Zero |
| Preprocessed reads | 2,088 | 1,991 | 40,876 | 25,238 | 19,728 | 32,737 | 31,791 | 36,172 |
| Mean read length | 275 | 245 | 262 | 270 | 233 | 259 | 240 | 267 |
| Total rRNA reads | 1,737 | 91 | 29,499 | 17,267 | 5,285 | 291 | 16,371 | 1,761 |
| 83.20% | 4.60% | 72.20% | 68.40% | 26.80% | 0.90% | 51.50% | 4.90% | |
| Microbial rRNA | 1,414 | 32 | 19,978 | 12,035 | 23 | 227 | 6,916 | 1,076 |
| 67.70% | 1.60% | 48.90% | 47.70% | 0.10% | 0.70% | 21.80% | 3.00% | |
| Eukaryota rRNA | 323 | 59 | 9,520 | 5,232 | 5,262 | 64 | 9,455 | 683 |
| 15.50% | 3.00% | 23.30% | 20.70% | 26.70% | 0.20% | 29.70% | 1.90% | |
| % rRNA removed* | 0% | 95% | 0% | 5% | 0% | 97% | 0% | 91% |
| Non-rRNA reads | 351 (16.8%) | 1,900 (95.4%) | 11,377 (27.8%) | 7,971 (31.6%) | 14,443 (73.2%) | 32,446 (99.1%) | 15,420 (48.5%) | 34,411 (95.1%) |
| Total NR hits | 102 (4.9%) | 691 (34.7%) | 3,327 (8.1%) | 2,857 (11.3%) | 4,938 (25.0%) | 10,751 (32.8%) | 5,905 (18.6%) | 15,766 (43.6%) |
| Eukaryotic | 74 | 407 | 2,790 | 2,524 | 4,614 | 10,227 | 4,553 | 8,274 |
| Bacterial | 26 | 283 | 520 | 312 | 287 | 471 | 1,326 | 7,442 |
| Unassigned reads | 249 (11.9%) | 1,209 (60.7%) | 8,050 (19.7%) | 5,114 (20.3%) | 9,505 (48.2%) | 21,695 (66.3%) | 9,515 (29.9%) | 18,645 (51.5%) |
| *The amount of rRNA removed expressed as a percentage of the amount present in the non-depleted aliquot. |
Table 3: Library characteristics of the metatranscriptomes with and without rRNA depletion. The data is extracted from Lim et al. (2012)9, which has additional comparison of other rRNA removal kits and the effect of cDNA nebulization prior to sequencing library preparation.
Discussion
Viral Metagenomics
Viral particles are concentrated using polyethylene glycol (PEG) precipitation or small volume concentrators. In some cases, concentration may not be needed, but pre-filtration or low speed centrifugation steps are used to remove eukaryotic and microbial cells. Viral lysates will be further enriched and purified using density gradient ultracentrifugation9,41 or small size filters (e.g., 0.45 μm) to remove eukaryotic and large microbial cells25. Density gradient ultracentrifugation is typically performed with dense but inert solutions such as sucrose or cesium chloride to isolate and concentrate viral particles41. Physical separation is based on the size and buoyant density of viral particles. Therefore, proper choice of filter pore size and the rigorous preparation of gradients are essential to isolate specific viral communities, as the success of the physical recovery of VLPs determines the community isolated41 (i.e., viral particles that do not pass through the filter or fall within the extraction density will not be detected in the metagenome). After viral isolation and concentration, there may be contaminating non-viral genomic material present in the sample both in the form of free nucleic acids and microbial and eukaryotic cells. Therefore, it is critical to verify the purity of viral particles in samples (Figures 1A and 1B). A chloroform treatment is commonly used to lyse remaining cells, followed by nuclease treatment to degrade free nucleic acids prior to nucleic acid extraction.
A caveat to the presented workflow was the use of density gradient separation to isolate viral particles as it may exclude enveloped viral particles that may be too buoyant to enter the CsCl gradient. An alternative “catch-all” method is to omit the density gradient separation and isolate the community DNA from the 0.45 μm – filtrates treated with chloroform and DNase I. This approach is also appropriate to accommodate small sample volumes such as those from swabs or blood plasma. However, this may result in chloroform-resistant bacterial contamination and higher amount of DNase I-resistant extracellular DNA.
Current sequencing protocols require 1 ng to 1 μg of nucleic acids for sequencing library preparation whereby higher DNA yields provide a wider choice of sequencing options. The DNA concentration of generated viromes often ranges from below the detection limit to more than 200 ng/μl. The amount of viral nucleic acids recovered may be insufficient for direct sequencing library preparation. In such cases, nucleic acid amplification is essential. Linker amplification shotgun libraries (LASLs)2,42,43 and whole genome amplification based on multiple displacement amplification (MDA) are the two methods most commonly used to generate sufficient DNA for sequencing. MDA methods such as those based on Phi29 DNA polymerase are known to suffer from amplification biases, and may preferentially amplify ssDNA and circular DNA, resulting in non-quantitative taxonomical and functional characterization44,45. An optimized version of the LASLs approach has been shown to introduce only minimal biases, promotes higher sensitivity (for small amounts of starting material), and is easily adapted for different sequencing platforms43. However, the approach has many steps, requires specialized equipment to minimize DNA loss, and is limited to dsDNA templates. In our laboratory, this approach has been successfully adapted to amplify detectable and undetectable amount of DNA extracted from bronchoalveolar lavage-, coral- and sea water-derived VLPs (unpublished and Hurwitz et al.46).
Developing data analysis pipelines has classically been one of the most challenging aspects of viral metagenomics analysis due to the highly diverse and largely unknown nature of the viral communities. While there are an estimated 108 viral genotypes in the biosphere, to date current viral databases contain ~ 4,000 viral genomes, which is about 1/100,000th of this approximate total viral diversity. Therefore, similarity-based searches (such as BLAST47) for taxonomic and functional assignment in viral metagenomes possess inherent challenges. Many sequences fail to have significant similarities to genomes in the database, and therefore, are classified as unknown. Even though homology-based searches are the most important applications for assigning taxonomy and function to sequence data, alternative approaches based on database-independent analysis have been developed48-50. Fancello et al.51 provide a complete review of computational tools and algorithms used in viral metagenomics.
Microbial Metagenomics
Typically, the total amount of DNA extracted from hypotonic lysis-treated microbial communities (HL-DNA) range from 20 ng to 5 μg. The yield is highly dependent on the patient’s health status and the amount of sputum sample collected, which explains the variations seen in the total yield of HL-DNA extracted in this study (Table 2). The critical steps to generate good quality sequence data rely on the quality of sequencing libraries generated. Figure 2 shows a typical size range for the sequencing libraries generated from CF sputum-derived microbial DNA using an enzymatic-based DNA fragmentation procedure. The optimal library size is dependent on the choice of sequencing platform and application, and therefore, the fragmentation procedure can be optimized, if necessary, through alternative approaches such as sonication and nebulization. In addition to the presented representative results, the success of the presented method on CF sputum collected from multiple patients across multiple time points is also illustrated in Lim et al. (2012)9 and Lim et al. (2014)10.
Previous studies9,10 suggest that every patient harbors a unique set of microbial community that shifts over time, thereby reflecting the persistence of the major players within the community while fluctuations are likely due to perturbations such as antibiotic treatments. Whether these fluctuations occur daily even without external perturbations or due to sampling procedure and sample processing, is still in question. Based on the HL-DNA metagenomic and 16S rDNA amplicon analysis, the 7-day longitudinal sampling shows that the daily fluctuation of microbial profiles without antibiotic perturbation (Day 1, 2, and 3) was minimal (Figures 3A and 3B). Upon introduction of oral antibiotics immediately after the Day 3 sampling, changes in the community profile became apparent on Day 4. While the antibiotic ciprofloxacin targets a broad spectrum of known bacterial pathogens such as P. aeruginosa, Staphylococcus aureus, and Streptococcus pneumonia, the treatment increased the relative abundance of P. aeruginosa while decreasing the Streptococcus spp. and P. melaninogenica. By Day 6, the community slowly recovered to the initial starting community structure. The results suggest that fluctuations of microbial profiles within a single patient are more likely due to community perturbations in the airways.
Given the consistency between the microbial profiles of 16S rDNA libraries and metagenomes from HL-derived DNA, we ruled out the biases originating from the 16S rRNA primers used in this study. One possible explanation for the differences seen across 16S rDNA taxonomical profiles generated from total DNA and HL-derived DNA (Figure 3B) may be the presence of high amounts of Pseudomonas spp. extracellular DNA after the antibiotic treatment. This is supported by the findings that these differences were most apparent at Day 7, three days after the antibiotics treatment, which targets Pseudomonas spp. in addition to others. Ciprofloxacin is commonly used as the first-line treatment in patients with CF and chronic P. aeruginosa infection even though its spectrum of activity includes most CF-associated pathogens. We hypothesized that the antibiotic treatment eradicates susceptible communities including Streptococcus spp. and hence creating a niche filled by resistant P. aeruginosa. Pseudomonas aeruginosa may gain resistance through increasing its biofilm communities and extracellular DNA has been shown to be the main structural support of its biofilm architecture52. Even as the community structure recovered, extracellular DNA may have remained in the CF sputum. Therefore, these data suggest that hypotonic lysis and the washing steps presented in this workflow potentially benefit in not only removing the human-derived DNA, but also microbial-derived extracellular DNA that may misrepresent the actual microbial profiles.
Metatranscriptomics
A high quality metatranscriptome should contain relatively few ribosomal RNA (rRNA) sequences and represent an unbiased sampling of the community transcripts (mRNA). Due to the short half-life and limited amount of mRNA, it is critical that the protocol, as presented here, minimizes sample handling to maximize the number of transcripts recovered.
In recent years, several approaches for rRNA depletion have been developed and adapted in commercially available kits. These include MICROBEnrich, Ribo-Zero, and sample-specific subtractive hybridizations53 that are based on oligonucleotide hybridization, and the mRNA-ONLY kit that is based on exonuclease enzymatic activity targeting RNA containing a 5’ monophosphate. In addition, several approaches for mRNA enrichments such as the MessageAmp II-Bacteria Kit that preferentially polyadenylates and amplifies linear RNA are also available. Some of these methods (e.g., mRNA-ONLY, MICROBExpress and the MessageAmp) are used concurrently for optimal efficiency. However, the efficacy of all of these approaches are limited, especially when working with partially degraded rRNA, as often observed in total RNA extracted from CF samples. Polyadenylation-dependent RNA amplification cannot be used to generate metatranscriptomes consisting of both eukaryotic and prokaryotic mRNA. In addition, the poly(A) tail added to the sequences may reduces the amount of useful sequence data. Regions with homopolymer stretches will tend to have lower quality scores, causing a significant number of reads to be filtered out by sequencing and post-sequencing software, and the average useful read length after trimming off poly (A) tails will be reduced significantly54.
Dealing with complex CF microbial communities and partially degraded RNA (Figures 4A and 4C), our previous study showed that the hybridization-capture method by the Ribo-Zero Gold kit was more effective in removing both human and microbial rRNA compared to the combine treatments using other kits9 (Table 3). The resultant data allows concurrent analysis of both human host and microbial transcripts. Depending on the yield and quality of RNA, as well as ultimate choice of sequencing platform, many of these processes including the cDNA synthesis step can be streamlined with sequencing library generation. For example, Ribo-Zero treated RNA can be used to make metatranscriptome sequencing libraries using ScriptSeq RNA-Seq Library Preparation kit.
Metagenomic analysis of animal-associated communities provides a comprehensive representation of the overall functional entity that includes the host and its associated communities. The workflow presented here is adaptable to a variety of complex animal-associated samples, especially those that contain thick mucus, high amounts of cell debris, extracellular DNA, protein and glycoprotein complexes, as well as host cells in addition to the desired viral and microbial particles. Even though viral and microbial particles may be lost at every step, particles isolation and purification are essential to minimize the amount of host DNA. While the metagenomics data provides metabolic potentials of the communities examined, metatranscriptomics complement this by revealing the differential expression of encoded functions9. A comprehensive assessment of the genomics and transcripts data has yielded new insights to the dynamics of community interactions and facilitates the development of improving therapies9,10,55.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the National Institute of Health (1 R01 GM095384-01) awarded to Forest Rohwer. We thank Epicentre, an Illumina company for providing early access to Ribo-Zero Epidemiology kits. We thank Mark Hatay for the design and production of the ultracentrifugation tube holder. We thank Andreas Haas and Benjamin Knowles for critical readings and discussions of the manuscript, and Lauren Paul for assisting the filming process.
References
- Suau A, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Applied and Environmental Microbiology. 1999;65(11):4799–4807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, et al. Genomic analysis of uncultured marine viral communities. Proceedings of the National Academy of Sciences. 2002;99(22):14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor LM. The human microbiome project in 2011 and beyond. Cell Hos., & Microbe. 2011;10(4):287–291. doi: 10.1016/j.chom.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Charlson ES, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. American Journal of Respiratory and Critical Care Medicine. 2011;184(8):957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS ONE. 2012;7(10):e47305. doi: 10.1371/journal.pone.0047305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem B, et al. Identification of the Cystic Fibrosis gene: Genetic analysis. Science. 1989;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Kleven D, McCudden C, Willis M. Cystic Fibrosis: Newborn screening in America. Medical Laboratory Observer. 2008;40(7):16–27. [PubMed] [Google Scholar]
- Fodor AA, et al. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS ONE. 2012;7(9):e45001. doi: 10.1371/journal.pone.0045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, et al. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. Journal of Cystic Fibrosis: Official Journal of the European Cystic Fibrosis Society. 2012;12(2):154–164. doi: 10.1016/j.jcf.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, et al. Clinical insights from metagenomic analysis of sputum samples from patients with cystic fibrosis. Journal of Clinical Microbiology. 2014;52(2):425–437. doi: 10.1128/JCM.02204-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Research. 2010;38(22):e200. doi: 10.1093/nar/gkq873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenstein S, Tümmler B, Römling U. Pulsed field gel electrophoresis of bacterial DNA isolated directly from patients’ sputa. Nucleic Acids Research. 1995;23(4):722–723. doi: 10.1093/nar/23.4.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. Recombinant human DNase I reduces the viscosity of Cystic Fibrosis sputum. Proceedings of the National Academy of Sciences. 1990;87(23):9188–9192. doi: 10.1073/pnas.87.23.9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lethem M, James S, Marriott C, Burke J. The origin of DNA associated with mucus glycoproteins in Cystic Fibrosis sputum. European Respiratory Journal. 1990;3(1):19–23. [PubMed] [Google Scholar]
- Childs WC, Gibbons RJ. Use of percoll density gradients for studying the attachment of bacteria to oral epithelial cells. Journal of Dental Research. 1988;67(5):826–830. doi: 10.1177/00220345880670050601. [DOI] [PubMed] [Google Scholar]
- Lee J-L, Levin RE. Use of ethidium bromide monoazide for quantification of viable and dead mixed bacterial flora from fish fillets by polymerase chain reaction. Journal of Microbiological Methods. 2006;67(3):456–462. doi: 10.1016/j.mimet.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Breitenstein S, Tümmler B, Römling U. Pulsed field gel electrophoresis of bacterial DNA isolated directly from patients’ sputa. Nucleic Acids Research. 1995;23(4):722–723. doi: 10.1093/nar/23.4.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokili JL, Rohwer F, Dutilh BE. Metagenomics and future perspectives in virus discovery. Current Opinion in Virology. 2012;2(1):63–77. doi: 10.1016/j.coviro.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibby K. Improved bacteriophage genome data is necessary for integrating viral and bacterial ecology. Microbial Ecology. 2014;67(2):242–244. doi: 10.1007/s00248-013-0325-x. [DOI] [PubMed] [Google Scholar]
- Willner D, et al. Metagenomic analysis of respiratory tract DNA viral communities in Cystic Fibrosis and non-Cystic Fibrosis individuals. PloS One. 2009;4(10):e7370. doi: 10.1371/journal.pone.0007370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner D, Furlan M. Deciphering the role of phage in the cystic fibrosis airway. Virulence. 2010;1(4):309–313. doi: 10.4161/viru.1.4.12071. [DOI] [PubMed] [Google Scholar]
- Willner D, et al. Case studies of the spatial heterogeneity of DNA viruses in the cystic fibrosis lung. American Journal of Respiratory Cell and Molecular Biology. 2012;46(2):127–131. doi: 10.1165/rcmb.2011-0253OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomar L, Maltz M, Colston S, Graf J. Directed culturing of microorganisms using metatranscriptomics. mBio. 2011;2(2):e00012–e00011. doi: 10.1128/mBio.00012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, et al. Metatranscriptomic array analysis of “Candidatus Accumulibacter phosphatis”-enriched enhanced biological phosphorus removal sludge. Environmental Microbiology. 2010;12(5):1205–1217. doi: 10.1111/j.1462-2920.2010.02163.x. [DOI] [PubMed] [Google Scholar]
- Mokili JL, et al. Identification of a novel Human Papillomavirus by metagenomic analysis of samples from patients with febrile respiratory illness. PLOS ONE. 2013;8(3):e58404. doi: 10.1371/journal.pone.0058404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henig NR, Tonelli MR, Pier MV, Burns JL, Aitken ML. Sputum induction as a research tool for sampling the airways of subjects with Cystic Fibrosis. Thorax. 2001;56(4):306–311. doi: 10.1136/thorax.56.4.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GB, et al. Use of 16S rRNA gene profiling by terminal restriction fragment length polymorphism analysis to compare bacterial communities in sputum and mouthwash samples from patients with Cystic Fibrosis. J. Clin. Microbiol. 2006;44(7):2601–2604. doi: 10.1128/JCM.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A, et al. Unraveling the unseen players in the ocean - a field guide to water chemistry and marine microbiology. Journal of Visualized Experiments. In press doi: 10.3791/52131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLOS ONE. 2011;6(3):e17288. doi: 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature Methods. 2012;9(8):811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9(1):386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara N, et al. Prevention of virus-induced type 1 diabetes with antibiotic therapy. Journal of Immunology (Baltimore, Md.: 1950) 2012;189(8):3805–3814. doi: 10.4049/jimmunol.1201257. [DOI] [PubMed] [Google Scholar]
- Markle JGM, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (New York, N.Y.) 2013;339(6123):1084–1088. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Research. 1998;8(3):186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Research. 1998;8(3):175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (Oxford, England) 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research. 2013;41(Database issue):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Peplies J, Glöckner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics (Oxford, England) 2012;28(14):1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CE, et al. Explicet: graphical user interface software for metadata-driven management, analysis and visualization of microbiome data. Bioinformatics (Oxford, England) 2013;29(23):3100–3101. doi: 10.1093/bioinformatics/btt526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber RV, Haynes M, Breitbart M, Wegley L, Rohwer F. Laboratory procedures to generate viral metagenomes. Nat. Protocols. 2009;4(4):470–483. doi: 10.1038/nprot.2009.10. [DOI] [PubMed] [Google Scholar]
- Henn MR, et al. Analysis of high-throughput sequencing and annotation strategies for phage genomes. PLoS ONE. 2010;5(2):e9083. doi: 10.1371/journal.pone.0009083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaime MB, Deng L, Poulos BT, Sullivan MB. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: a rigorous assessment and optimization of the linker amplification method. Environmental Microbiology. 2012;14(9):2526–2537. doi: 10.1111/j.1462-2920.2012.02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz S, Allgaier M, Hugenholtz P. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat Meth. 2010;7(12):943–944. doi: 10.1038/nmeth1210-943. [DOI] [PubMed] [Google Scholar]
- Kim K-H, Bae J-W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Applied and Environmental Microbiology. 2011;77(21):7663–7668. doi: 10.1128/AEM.00289-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz BL, Deng L, Poulos BT, Sullivan MB. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environmental Microbiology. 2013;15(5):1428–1440. doi: 10.1111/j.1462-2920.2012.02836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Angly F, et al. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics. 2005;6(1):41. doi: 10.1186/1471-2105-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angly FE, et al. The GAAS Metagenomic Tool and Its Estimations of Viral and Microbial Average Genome Size in Four Major Biomes. PLoS Comput Biol. 2009;5(12) doi: 10.1371/journal.pcbi.1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutilh BE, et al. Reference-independent comparative metagenomics using cross-assembly: crAss. Bioinformatics (Oxford, England) 2012;28(24):3225–3231. doi: 10.1093/bioinformatics/bts613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancello L, Raoult D, Desnues C. Computational tools for viral metagenomics and their application in clinical research. Virology. 2012;434(2):162–174. doi: 10.1016/j.virol.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allesen-Holm M, et al. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Molecular Microbiology. 2006;59(4):1114–1128. doi: 10.1111/j.1365-2958.2005.05008.x. [DOI] [PubMed] [Google Scholar]
- Stewart FJ, Ottesen EA, DeLong EF. Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME J. 2010;4(7):896–907. doi: 10.1038/ismej.2010.18. [DOI] [PubMed] [Google Scholar]
- Frias-Lopez J, et al. Microbial community gene expression in ocean surface waters. Proceedings of the National Academy of Sciences. 2008;105(10):3805–3810. doi: 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, et al. Mechanistic model of Rothia mucilaginosa adaptation toward persistence in the CF lung, based on a genome reconstructed from metagenomic data. PLOS ONE. 2013;8(5):e64285. doi: 10.1371/journal.pone.0064285. [DOI] [PMC free article] [PubMed] [Google Scholar]