

Abstract
Inherent processes of Drosophila pupal development can shift and distort the eye epithelium in ways that make individual cell behavior difficult to track during live cell imaging. These processes include: retinal rotation, cell growth and organismal movement. Additionally, irregularities in the topology of the epithelium, including subtle bumps and folds often introduced as the pupa is prepared for imaging, make it challenging to acquire in-focus images of more than a few ommatidia in a single focal plane. The workflow outlined here remedies these issues, allowing easy analysis of cellular processes during Drosophila pupal eye development. Appropriately-staged pupae are arranged in an imaging rig that can be easily assembled in most laboratories. Ubiquitin-DE-Cadherin:GFP and GMR-GAL4–driven UAS-α-catenin:GFP are used to visualize cell boundaries in the eye epithelium 1-3. After deconvolution is applied to fluorescent images captured at multiple focal planes, maximum projection images are generated for each time point and enhanced using image editing software. Alignment algorithms are used to quickly stabilize superfluous motion, making individual cell behavior easier to track.
Keywords: Developmental Biology, Issue 95, Drosophila melanogaster, pupal eye, pattern formation, ommatidial development, live cell imaging, motion stabilization, fluorescence microscopy
Introduction
The compound Drosophila eye is characterized by the stereotyped arrangement of its ~750 ommatidia separated by a honeycomb-lattice of accessory pigment cells 4,5. These pigment cells are patterned by a coordinated combination of events: local cell movements, cell growth, changes in cell shape, and apoptosis. Live visualization of this epithelium allows one to study the molecular mechanisms underpinning these events in a physiologically relevant and unperturbed three-dimensional context.
In contrast to previous protocols 6,7, the technique outlined here incorporates an efficient method for stabilizing extraneous tissue movement that cannot be uncoupled from the imaging process. This method enhances studies of cell behavior in the developing Drosophila pupal eye epithelium – a tissue that grows, rotates, and shifts over the course of imaging. In addition the motion-stabilization technique described here will be useful for studying cells in other contexts where extraneous movement occurs.
To visualize cell boundaries in the Drosophila retinal field, transgenic fly lines were generated that express ubi-DE-Cadherin:GFP as well as UAS-α-catenin:GFP under control of the eye-specific driver GMR-GAL4 1-3. The use of two GFP-tagged membrane markers allows for the visualization of cell boundaries at lower intensity light. This minimizes tissue damage and photobleaching on repeated exposure to high-energy wavelengths, enabling increased frame rate and movie duration. To enhance efficacy of RNAi transgenes, UAS-Dcr-2 was also incorporated into a second fly line 8.
In a third update from previous protocols, a simpler imaging rig that is easily assembled in most laboratories is described. This apparatus obviates the requirement to have a specialized imaging rig generated by a university’s ‘machine shop’ or similar service. This imaging rig is similar to that used to image other pupal tissues 9,10.
Presented here is a simple live-cell imaging protocol that can be used to directly assess the morphogenetic events that contribute to eye patterning from ~17 to 42 hr after puparium formation (hr APF). Specifically, this protocol enables one to determine the consequences of modifying gene expression during pupal development.
Protocol
Figure 3 shows a summary of the experimental procedure.
1. Tissue Preparation
To determine the consequences of modified expression of a gene of interest, set up the following cross in duplicate and maintain at 25 °C: UAS-transgene (males)X GMR-GAL4; UAS-α-cat:GFP, ubi-DE-Cad:GFP ; + / SM5-TM6b (virgin females) NOTE: To obtain control tissue cross UAS-lacZ males to GMR-GAL4; UAS-α-cat:GFP, ubi-DE-Cad:GFP females. β-Galactosidase generates no defects in cell behavior in the retina.
To enhance efficacy of RNAi transgenes, cross UAS-RNAi males to GMR-GAL4, UAS-Dcr-2; UAS-α-cat:GFP, ubi-DE-Cad:GFP; + / SM5-TM6b virgin females. NOTE: Occasional defects in cell behavior in retinae expressing ectopic Dcr-2 have been observed (data not shown). Vigilance is recommended if using this approach.
Select white pre-pupae of appropriate genotypes and place in 1.5 ml microcentrifuge tubes. If the pupae express Dcr-2, select either male or female pupae: expression of UAS-Dcr-2, an insert on the X chromosome, is stronger in males. Pupae should be sexed at 0 hr APF before pigmentation of the pupal case – the gonads of males are visible as large translucent globes on the lateral sides of the pupa (approximately one-third from the posterior).
Incubate microcentrifuge tubes containing pupae, in a clean plastic tip-box at 25 °C. To prevent desiccation of pupae place a 10 cm2 piece of tissue, soaked in distilled water, into the tip-box. NOTE: if the humidity in the tip-box becomes too high the pupal case may become soggy and difficult to dissect in subsequent steps.
Incubate for appropriate time. To capture cell behaviors associated with patterning the eye, prepare pupae for imaging at around 17 hr APF, 20 hr APF or 24 hr APF. See Representative Results for further discussion on when specific cell behaviors are best observed.
Place a piece of fresh double-sided tape onto a black sylgard dissection dish. Position a pupa, dorsal side up, with the head of the pupa adhered to the double-sided tape (Figure 1A).Try not to adhere the thoracic and abdominal regions of the pupa to the double-sided tape. With forceps carefully lift and remove the operculum to expose the pupal head. Tear along the side of the pupal case to expose the area around one eye.
Gently remove the pupa from the adhesive tape. If the pupa remains adherent, apply a small volume of distilled water to the junction between the pupal case and tape to weaken adhesion.
2. Mounting
Construct a small 15 mm2 frame from blotting paper and punch a hole in the middlethat is 5.5 mm in diameter (Figure 1B). Immerse in distilled water and place in the center of a microscope slide. Using a 30 cc syringe, squeeze out a uniform ring of petroleum jelly to surround the blotting paper frame. Ensure that the petroleum jelly ring is marginally higher than the width of the pupa and that the diameter of the ring is marginally less than the width of a cover slip.
Add a small bead of petroleum jelly directly onto the slide, in the hole punched into the blotting paper(Figure 1C).Carefully arrange the pupa, on its side, supported by the petroleum jelly bead (Figure 1D).
Place a coverslip on top of the petroleum jelly ring so that it contacts the epithelium above the eye neuroepithelium (Figure 1D). Gently compress the preparation to seal the coverslip against the petroleum jelly and generate a small flat contact surface between the pupal eye region and the coverslip.
3. Fluorescence Imaging
Image the pupal preparation using a fluorescent microscope. Every 7 min capture serial sections through the apical domain of the eye neuroepithelium, in the region of the adherens junctions. NOTE: a) Appropriate serial sections are usually 4-5 per z-stack comprised of 0.2 μm z-steps. b) Irregularities in the topology of the epithelium, including mild bumps and folds, may make it challenging to acquire in-focus images of large regions of tissue. While imaging, one may find part of an area of interest to be in-focus, and a neighboring region to be out-of-focus. Including a small droplet of distilled water between the pupal eye and coverslip can eliminate some acute tissue folds but not the mildly uneven topology characteristic of the early stages of pupal eye morphogenesis. In these instances oversample the tissue by extending the z-stack until in-focus slices are acquired for the entire field. c) Capturing serial sections every 7 min should enable live-imaging for 3 to 4 hr without a significant reduction in the intensity of GFP fluorescence that may compromise image quality. Shorter time-intervals may reduce the total time of imaging.
Continue imaging for 3 to 4 hr. Do not use an automatic focus and time function that is available on many fluorescent microscopes since pupal growth and pumping of the hemolymph moves the position of the retina and vigilant re-focusing is required every 14-21 min. NOTE that imaging beyond 4 hours may slow or stall morphogenesis of the eye.
Use appropriate deconvolution software to reduce background and enhance contrast of the serial section images. For each z-stack file, perform the following in LAS AF software (see Table of Materials): Navigate to the Tools panel, select 3D Deconvolution and click Apply.
Generate a maximum projection (MP) image for each deconvoluted stack file: Navigate to the Tools panel, select 3D Projection and click Apply. NOTE: The Maximum Projection algorithm may fail to generate a uniformly in-focus image for tissue that has been oversampled. A technique for sharpening out-of-focus regions is outlined in step 5.2.
4. Post-imaging Pupal Rescue and Phenotype Verification
To address whether artifacts were introduced or the pupa harmed during imaging, carefully remove the pupa from the imaging rig: using forceps remove the coverslip and transfer the pupa to a clean vial of food. Store at room temperature or 25 °C until the adult fly emerges.
Carefully examine the phenotype of the adult eye and compare to the eyes of adult flies emerging from the original fly cross.
5. Image Processing
- Automatic Image Alignment: NOTE: The live Drosophila tissue will shift and grow throughout the imaging process. Consequently, the centers of images gathered at successive time points may not correspond to the same point in the Drosophila tissue. In addition the entire retinal disc rotates about 30° between ~21 and 23 hr APF. To highlight individual cell behaviors and reduce distractions caused by organismal growth,align each MP image.While this can be performed manually,the image editing software used here (see Table of Materials) has built-in algorithms that can expedite the process.
- From the File tab of the main menu, open Scripts and select Load Files into Stack.
- Import the MP image files into the Load Layers panel and select OK. Make sure that the Attempt to Automatically Align Source Images option is not selected. NOTE: If this option is selected, the software may use an alignment algorithm that distorts the image data.
- Once all MP files have loaded into the Layers pane, make sure that all images are in chronological order such that the earliest time point is at the top of the layer stack. If the layers are not in the correct order, drag and drop them until they are ordered correctly.
- Select Auto-Align Layers from the Edit tab of the main menu. Choose Reposition and click OK.
- If the Auto-Align algorithm fails to orient the frames to a relevant focal point, make minor adjustments using the Move tool.
- Composite Sharpening: NOTE: Out-of-focus regions of an initial MP image can be sharpened by ‘cropping’ the corresponding deconvoluted stack in LAS AF software. Cropping a stack file allows one to manually restrict the interval of a z-stack that is subjected to the Maximum Projection algorithm.
- Locate the Crop function in the Tools panel (under the Process tab). Manually restrict the initial and final slices such that they span only the adhesion belt within the initially out-of-focus region. Click Apply to generate a new stack file that is optimized for this region.
- Generate a new Maximum Projection for the locally optimized stack and export as a TIFF file.
- In the image editing software (see Table of Materials), open Scripts (located in the File tab of the main menu) and select Load Files into Stack.
- Import the initial MP image file and locally optimized MP file for a particular time point into the Load Layers panel and select OK. Make sure that the Attempt to Automatically Align Source Images option is not selected.
- Select both layers in the Layers pane and then choose Auto-Blend Layers (located in the Edit tab of the main menu) to automatically mask the appropriate regions of the two images, yielding a uniformly in-focus retinal field. Save this composite image as a TIFF file.
- Repeat steps 5.2.1 through 5.2.5 for all suboptimal MP images.
- Further Adjustments: Rotate, crop, and Adjust the Levels of the Movie Layers:
- With all of the layers selected, use the Transform tool (Edit > Free Transform) to rotate the images so that the dorsal-ventral axis of the retina is aligned to the y-axis of the movie frame.
- If necessary, use the Crop tool to reduce the field of view to a desired size and shape.
- Use level adjustment (of individual frames) to help enhance contrast between the cell membrane and cell body.
- For stylistic purposes and to emphasize specific cell behaviors, introduce false color to highlight points of interest in each frame.
- Animation: Convert the Images into a Movie:
- Open the Animation pane from the Windows tab of the main menu. Select Make Frames from Layers from the menu located in the upper right hand corner of the Animation pane.
- Note that the layers are added to the animation pane in sequential order beginning from the bottom of the stack. To reverse the order of the frames, first select all of the frames and then select Reverse Frames from the Animation pane menu.
- Select a frame delay time for each frame using the dropdown menu below the frame thumbnail. NOTE: The frame delay for Movies 1 to 4 presented in this paper is 0.8 sec.
- From the File tab of the main menu, open Export and select Render Video. Choose QuickTime Export under File Options and select a desired video format. Under Render Options set Frame Rate to Custom, enter a frame rate of 15, and click Render.
Representative Results
Live-imaging of the pupal eye is a successful strategy to observe cell behaviors that contribute to patterning of the neuroepithelium. The role of specific proteins can be readily assessed by expressing transgenes that modify protein levels during eye development. To do this GMR-GAL4 is used to drive transgene expression behind the morphogenetic furrow. This offers the advantage of not perturbing earlier events that establish the eye field during the first two larval instars. In addition many RNAi transgenes require significant time to effectively reduce protein levels (data now shown). Hence driving RNAi transgenes with the GMR-GAL4 driver line often enables third larval instar eye development and eversion of the eye discs during metamorphosis to proceed unscathed and significantly reduces protein levels only later, during pupal eye morphogenesis. If larval development is however perturbed by a highly efficient RNAi transgene, fly crosses can be maintained at 18 °C to reduce transgene expression and pupae transferred to 25 °C at 0 hr APF. To address the efficacy of RNAi transgene expression, transcript or protein levels should be assessed in stage-matched pupae using standard techniques (e.g. quantitative-PCR, immunofluorescence, immunoblotting).
During larval development, the photoreceptor cores of each ommatidium are recruited as a wave that travels from the posterior to anterior side of the eye disc 11. This developmental gradient was retained in the pupal eye. Indeed, when imaged at 22.5 hr APF, patterning of GMR-GAL4, UAS-Dcr-2; UAS-α-cat:GFP, ubi-DE-Cad:GFP; + / SM5-TM6b retinae was markedly more mature in the posterior region (Figure 2). The phenotype of these retinae was comparable to that of wild-type fly strains (data not shown). Traditionally pupal eye morphogenesis is described according to developmental time (e.g. hr APF). However, because of the pronounced developmental gradient we propose describing eye patterning according to the following events:
1° wrapping (Figure 2 and Movie 1): Initially as many as eight cells directly contacted the photoreceptor bundles. Two cells - usually the anterior and posterior cells - were recruited as primaries (1°s). These 1s rapidly encircled the photoreceptor bundles, connecting to each other to form stable junctions. Undifferentiated interommatidial cells (ICs) lay in a disorganized manner between each ommatidial grouping. Concomitant with 1° cell recruitment, apoptosis pruned approximately 5% of the IC pool as previously described (not annotated in Movie 1) 12.
IC intercalation (Figure 2 and Movie 2): As the 1°s wrapped and sealed about each photoreceptor bundle, local cell movements reorganized the ICs grouped at the dorsal and ventral sides of each ommatidium. The ICs intercalated from multiple (usually two) rows into a single row. The target position of moving ICs could be confidently predicted in wild type tissue by observing the directional projection of large ‘cellular extensions’. Growth of the 1° cells accompanied and likely contributed to IC intercalation 13. Many ICs will go on to differentiate as secondary cells (2°s) (not shown).
3° competition (Figure 2 and Movie 3): Following intercalation, three ICs competed to occupy the tertiary (3°) niche. In this dynamic battle, cells frequently occupied the 3° niche for as little as 5 to 10 min before being displaced. Eventually a single cell established a stable 3° niche.
Final pruning (Figure 2 and Movie 4): A surge of apoptosis 3,15-18 reduced the number of ICs to the minimum required to efficiently generate a neat hexagonal IC lattice across the compound eye 5,14: three 3°s and six 2°s around each ommatidium. As previously noted, usually those excess ICs lying closest to the bristles were eliminated by apoptosis 7. We observed cell death concomitant with IC intercalation and 3° niche establishment and propose that pruning of excess cells contributed to the efficiency of these events. In addition, subtle re-positioning of bristle organules was often observed as ICs were pruned.
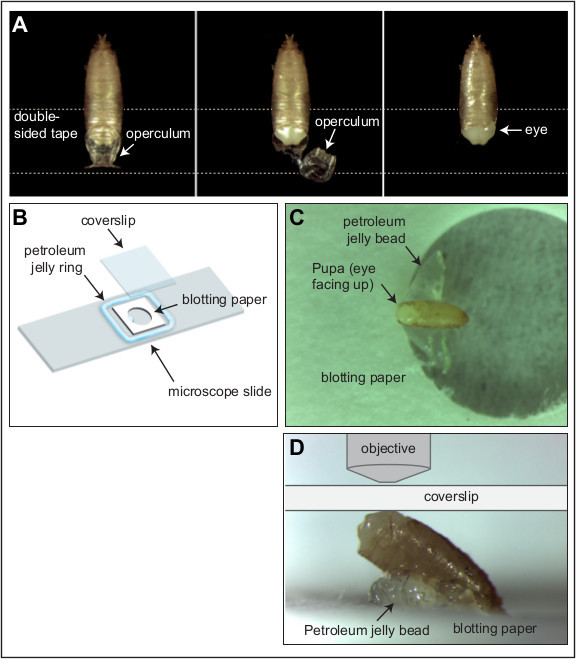 Figure 1:Pupal dissection and imaging.(A) The operculum is removed from a Drosophila pupa (shown here at 23 hr APF) to expose the head of the animal. Dotted line represents a piece of double-sided tape. (B) Illustration of the simple live-imaging rig. (C and D) With head exposed, the pupa is positioned at about 35° resting on the petroleum jelly bead. (D) Higher magnification image of a pupa positioned atop the petroleum jelly bead. The position of the coverslip and objective are illustrated (not drawn to scale). Please click here to view a larger version of this figure.
Figure 1:Pupal dissection and imaging.(A) The operculum is removed from a Drosophila pupa (shown here at 23 hr APF) to expose the head of the animal. Dotted line represents a piece of double-sided tape. (B) Illustration of the simple live-imaging rig. (C and D) With head exposed, the pupa is positioned at about 35° resting on the petroleum jelly bead. (D) Higher magnification image of a pupa positioned atop the petroleum jelly bead. The position of the coverslip and objective are illustrated (not drawn to scale). Please click here to view a larger version of this figure.
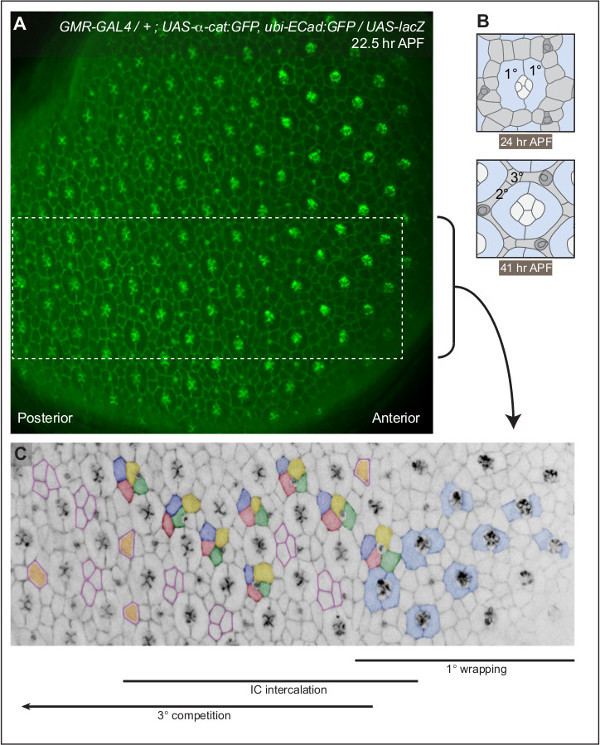 Figure 2:A gradient of development across the pupal eye. (A) A single image of a pupal eye imaged at 22.5 hr APF. Adherens junctions are labeled by DE-Cad:GFP and αCat:GFP. Boxed region is annotated in (C). (B) Illustrations of mid-stage and fully-patterned ommatidia, at 24 and 41 hr APF respectively. Two 1°s (blue) encircle a central grouping of four cone cells (white). By 41 hr APF the patterning of interommatidial cells (light grey) is complete: three 3°s occupy alternate vertices and a single 2° forms each side of the hexagon. Three bristle organules (dark grey) surround each ommatidium. (C) Annotation of a small region of the eye (from A). Two 1° cells (examples pseudo-colored light blue) expand to encircle each photoreceptor bundle. ICs (green, yellow, red and dark blue) intercalate into single rows. At each alternative vertex three cells compete (outlined in pink) to occupy the 3° niche (colored orange). See Movies 1-4 to observe these events unfolding. Please click here to view a larger version of this figure.
Figure 2:A gradient of development across the pupal eye. (A) A single image of a pupal eye imaged at 22.5 hr APF. Adherens junctions are labeled by DE-Cad:GFP and αCat:GFP. Boxed region is annotated in (C). (B) Illustrations of mid-stage and fully-patterned ommatidia, at 24 and 41 hr APF respectively. Two 1°s (blue) encircle a central grouping of four cone cells (white). By 41 hr APF the patterning of interommatidial cells (light grey) is complete: three 3°s occupy alternate vertices and a single 2° forms each side of the hexagon. Three bristle organules (dark grey) surround each ommatidium. (C) Annotation of a small region of the eye (from A). Two 1° cells (examples pseudo-colored light blue) expand to encircle each photoreceptor bundle. ICs (green, yellow, red and dark blue) intercalate into single rows. At each alternative vertex three cells compete (outlined in pink) to occupy the 3° niche (colored orange). See Movies 1-4 to observe these events unfolding. Please click here to view a larger version of this figure.
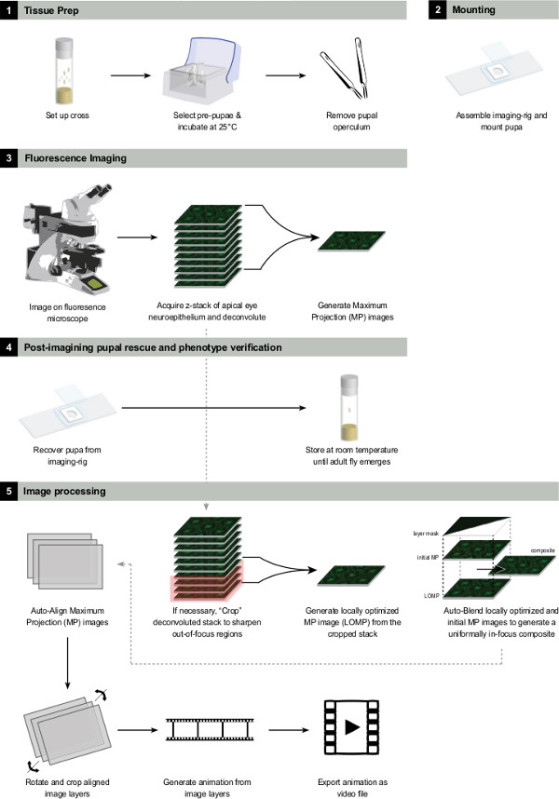 Figure 3:
Summary of the experimental procedure.
Please click here to view a larger version of this figure.
Figure 3:
Summary of the experimental procedure.
Please click here to view a larger version of this figure.
Movie 1: Primary wrapping. 1° cells are selected from the pool of cells surrounding each fledgling ommatidium. As two 1°s encircle each ommatidium and connect, adherens junctions rapidly form (pink lines). The straight 1°:1° cell junctions expand and 1°s begin to grow, insulating photoreceptor bundles from the surrounding ICs. Initially the photoreceptors glow brightly, dense GFP marking elaborate adherens junctions. Morphological changes gradually draw these photoreceptor junctions below the plane of imaging and X-shaped junctions between each set of four apical cone cells atop each ommatidium become clearer. Original images on left; pseudo-coloring and annotations introduced into panels on right. Eye imaged from 21 hr APF.
Movie 2: Interommatidial cell intercalaction. The interommatidial cells (ICs) highlighted in red, blue, yellow and green initially form a quadrant separating a dorsal (top) and ventral (bottom) ommatidium by a distance of two cells. The red and yellow ICs stretch ventrally and dorsally respectively (from t = 42 min), making contact with target 1°s by 56 min. The blue and green ICs similarly extend to target opposite ommatidium. ‘New’ 1°:IC junctions rapidly expand laterally. At 112 min the original quadrant of ICs has fully intercalated to generate a single row of cells. Original images on left; pseudo-coloring and annotations introduced into panels on right. Eye imaged from 23 hr APF.
Movie 3: Tertiary competition. At alternate vertices two or three cells (outlined in pink) compete to occupy the 3° niche. Appearing to ‘push’ their neighbors aside, cells generate large extensions that reach toward opposite ommatidia. Occupying the 3° niche (colored orange) is often fleeting and cells either retract or are pushed out of position by their neighbors. By the end of this 217 min movie, many 3° niches appear established. Original images on left; pseudo-coloring and annotations introduced into panels on right. Eye imaged from 23 hr APF.
Movie 4: Final Pruning. Cells in the vicinity of bristle organules are removed by apoptosis. Examples are highlighted in purple, red, yellow and blue. Over the course of imaging, removal of cells left just one 2° along each horizontal arm of the IC hexagon. Pruning of excess cells on the diagonal sides of the IC hexagon was not captured in this movie. In this genotype minor errors in the placement of bristle groups were often observed (compare with Figure 2B). Two of the three bristle groups maneuvered into corner niches during imaging: re-positioning appeared to be facilitated by removal and movement of neighboring ICs. Original images on left; pseudo-coloring and annotations introduced into panels on right. Eye imaged from 26 hr APF.
Discussion
The description of wild-type development (above) forms the basis for comparisons of patterning events in RNAi or overexpression genotypes. Comparisons of live tissue development are invaluable when determining precisely which cell behaviors are regulated by a protein of interest. Further, and not described here, live cell imaging enables one to make qualitative and/or quantitative descriptions of the role of a gene of interest in the events that pattern the eye. By 30-32 hr APF most pigment cells have acquired stable positions and apoptosis has pruned excess cells from the retinal field. Following this, only subtle changes are observed as the pigment cells adopt their characteristic shapes: the 3°s become hexagonal and, as 1°s subtly expand, the width of the neighboring rectangular 2° cells decreases. Subsequent generation of pigment and lens material likely hampers successful imaging of adherens junctions.
Though a valuable strategy, several pitfalls must be borne in mind when imaging the live pupal eye. First, removing the anterior pupal case renders the animal particularly vulnerable to dehydration, overheating, and injury during mounting and imaging. Insults such as these can confound data by inducing atypical tissue development that is not relevant to the genotype under examination. Second, the protocol described here requires that a glass coverslip make direct contact with the pupal head epithelium. We have observed that retinal development stalls if excessive pressure is applied when pushing the coverslip against the eye. Indeed exogenous mechanical forces have been shown in other systems to influence developmental processes including tissue architecture 19,20. For these reasons it is important to very gently place the coverslip against the eye. Additionally, imaged pupae should be rescued from the imaging rig and allowed to emerge as adults: artifacts introduced during imaging can often be detected by comparing the imaged-eye of these adults with eyes of age-matched flies from the original cross. Further, data from multiple imaging sessions must be compared to verify cell behaviors. Finally, standard immunofluoresence should be used to determine whether the arrangement, size and number of cells in stage-matched tissue are the same.
Ectopic Dcr-2 is frequently used to enhance the efficacy of RNAi. However in the presence of ectopic Dcr-2, IC intercalation, stabilization of the 3° niche and apoptosis were occasionally disrupted (data not shown). Thus Dcr-2 expression should be used with caution.
The protocol described here utilizes GFP to label adherens junctions and assess morphogenesis of pigment cells in the pupal eye. This strategy could easily be modified for other purposes (e.g. to assess photoreceptor morphogenesis). For a double-labeling approach, additional UAS transgenes that label other cellular components of interest (e.g. secretary vesicles, endoplasmic reticulum, nucleus) with non-GFP tags could be incorporated. However the intensity and longevity of these transgenes will require assessment: RFP, DsRed and mCherry, for example, have proven to be poor labels for imaging the pupal eye for the prolonged periods required to capture pigment cell patterning (data not shown). If investigating rapid cell behaviors (e.g. secretion) the protocol described here could easily be modified for confocal microscopy of suitable fluorescent proteins.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors thank David Larson for developing the original protocol for imaging the live Drosophila pupal eye. We thank our colleagues Richard Carthew, Shoichiro Tsukita and Matthew Freeman, who developed the transgenic flies that we combined to generate our live-imaging fly lines. Brittany Baldwin-Hunter gave helpful comments on the manuscript. This work was supported by start-up funds awarded to Ruth Johnson by Wesleyan University.
References
- Oda H, Tsukita S. Real-time imaging of cell-cell adherens junctions reveals that Drosophila mesoderm invagination begins with two phases of apical constriction of cells. J. Cell. Sci. 2001;114:493–501. doi: 10.1242/jcs.114.3.493. [DOI] [PubMed] [Google Scholar]
- Oda H, Tsukita S. Dynamic features of adherens junctions during Drosophila embryonic epithelial morphogenesis revealed by a Dalpha-catenin-GFP fusion protein. Dev. Genes Evol. 1999;209:218–225. doi: 10.1007/s004270050246. [DOI] [PubMed] [Google Scholar]
- Freeman M. Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell. 1996;87:651–660. doi: 10.1016/s0092-8674(00)81385-9. [DOI] [PubMed] [Google Scholar]
- Cagan R. Cell fate specification in the developing Drosophila. retina. Dev Suppl. 1993. p. 19.p. 28. [PubMed]
- Wolff T, Ready DF. Bate M, Arias AM. The Development of Drosophila melanogaster. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1993. Pattern formation in the Drosophila. retina; pp. 1277–1325. [Google Scholar]
- Larson DE, Liberman Z, Cagan RL. Cellular behavior in the developing. Drosophila pupal retina. Mech. Dev. 2008;125:223–234. doi: 10.1016/j.mod.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monserrate JP, Brachmann CB. Identification of the death zone: a spatially restricted region for programmed cell death that sculpts the fly eye. Cell Death Differ. 2007;14:209–217. doi: 10.1038/sj.cdd.4401947. [DOI] [PubMed] [Google Scholar]
- Lee YS, et al. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell. 2004;117:69–81. doi: 10.1016/s0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- Zitserman D, Roegiers F. Live-cell imaging of sensory organ precursor cells in intact Drosophila pupae. J. Vis. Exp. 2011. p. 2706. [DOI] [PMC free article] [PubMed]
- Classen AK, Aigouy B, Giangrande A, Eaton S. Imaging Drosophila pupal wing morphogenesis. Methods Mol. Biol. 2008;420:265–275. doi: 10.1007/978-1-59745-583-1_16. [DOI] [PubMed] [Google Scholar]
- Sato M, Suzuki T, Nakai Y. Waves of differentiation in the fly visual system. Dev. Biol. 2013;380:1–11. doi: 10.1016/j.ydbio.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Cordero J, Jassim O, Bao S, Cagan R. A role for wingless in an early pupal cell death event that contributes to patterning the Drosophila eye. Mech. Dev. 2004;121:1523–1530. doi: 10.1016/j.mod.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Larson DE, Johnson RI, Swat M, Cordero JB, Glazier JA, Cagan RL. Computer simulation of cellular patterning within the Drosophila pupal eye. PLoS Comput. Biol. 2010;6:e1000841. doi: 10.1371/journal.pcbi.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagan RL, Ready DF. The emergence of order in the Drosophila pupal retina. Dev. Biol. 1989;136:346–362. doi: 10.1016/0012-1606(89)90261-3. [DOI] [PubMed] [Google Scholar]
- Cagan RL, Ready DF. Notch is required for successive cell decisions in the developing Drosophila retina. Genes Dev. 1989;1099:1112. doi: 10.1101/gad.3.8.1099. [DOI] [PubMed] [Google Scholar]
- Miller DT, Cagan RL. Local induction of patterning and programmed cell death in the developing Drosophila retina. Development. 1998;125:2327–2335. doi: 10.1242/dev.125.12.2327. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Okano H, Kobayakawa Y, Hayashi S, Mikoshiba K, Tanimura T. The function of argos in regulating cell fate decisions during Drosophila eye and wing vein development. Dev. Biol. 1994;164:267–276. doi: 10.1006/dbio.1994.1197. [DOI] [PubMed] [Google Scholar]
- Yu SY, et al. A pathway of signals regulating effector and initiator caspases in the developing Drosophila eye. Development. 2002;129:3269–3278. doi: 10.1242/dev.129.13.3269. [DOI] [PubMed] [Google Scholar]
- Liu Z, et al. Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. 2010;107:9944–9949. doi: 10.1073/pnas.0914547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Inoue N, Nishimura K, Kinoshita N, Hosoya H, Yonemura S. Actomyosin tension is required for correct recruitment of adherens junction components and zonula occludens formation. Exp. Cell Res. 2006;312:1637–1650. doi: 10.1016/j.yexcr.2006.01.031. [DOI] [PubMed] [Google Scholar]