

Abstract
RNA or DNA folded in stable tridimensional folding are interesting targets in the development of antitumor or antiviral drugs. In the case of HIV-1, viral proteins involved in the regulation of the virus activity recognize several nucleic acids. The nucleocapsid protein NCp7 (NC) is a key protein regulating several processes during virus replication. NC is in fact a chaperone destabilizing the secondary structures of RNA and DNA and facilitating their annealing. The inactivation of NC is a new approach and an interesting target for anti-HIV therapy. The Nucleocapsid Annealing-Mediated Electrophoresis (NAME) assay was developed to identify molecules able to inhibit the melting and annealing of RNA and DNA folded in thermodynamically stable tridimensional conformations, such as hairpin structures of TAR and cTAR elements of HIV, by the nucleocapsid protein of HIV-1. The new assay employs either the recombinant or the synthetic protein, and oligonucleotides without the need of their previous labeling. The analysis of the results is achieved by standard polyacrylamide gel electrophoresis (PAGE) followed by conventional nucleic acid staining. The protocol reported in this work describes how to perform the NAME assay with the full-length protein or its truncated version lacking the basic N-terminal domain, both competent as nucleic acids chaperones, and how to assess the inhibition of NC chaperone activity by a threading intercalator. Moreover, NAME can be performed in two different modes, useful to obtain indications on the putative mechanism of action of the identified NC inhibitors.
Keywords: Immunology, Issue 95, HIV-1, Nucleocapsid protein, NCp7, TAR-RNA, DNA, oligonucleotides, annealing, Gel electrophoresis, NAME
Introduction
The nucleocapsid protein NCp7 (NC) of human immunodeficiency virus type 1 (HIV-1) is a small, basic protein that is tightly associated with genomic RNA in the mature infectious virus, playing a pivotal role in virus replication as a cofactor during reverse transcription, genome recognition, and packaging.1-3 NC acts as a nucleic acid chaperone catalyzing the destabilization of stable nucleic acid structures and the annealing of complementary sequences. Its nucleic acid aggregating activity resides primarily in the 11 amino acids N-terminal domain while the duplex destabilizing activity has been mapped to its zinc finger structures (12-55 peptide).4 The positively charged protein lowers the electrostatic barrier of the annealing reaction and increases the rate at which two separate complementary sequences come together.
Nucleic acids folded in stable conformation require the chaperone activities of NC to facilitate their annealing.5 This is particularly important in reverse transcription, where NC is critical in strand transfer events: in the minus strand transfer, the minus strand stop DNA, just retrotranscribed, must be transferred and annealed to a complementary sequence in the R region at the 3' end of the RNA genome template.6 Although thermodynamically favored, this reaction does not occur extensively in the absence of NC due to the presence of the stable structure of the trans activation responsive region (TAR) RNA in the R regions, that must be associated to its DNA copy (cTAR DNA).7 cTAR and TAR are, in fact, highly structured regions with a characteristic stem-loop conformation. NC protein denatures these hairpins, and promotes minus-strand transfer by increasing the rate of intermolecular annealing between the complementary nucleic acid strands. The mechanism of NC annealing of TAR and cTAR has been thoroughly investigated and described as TAR annealing assay in several research papers and the proposed scheme is depicted in excellent reviews.8-11 Summarizing, NC destabilizes the secondary structure of stable RNA such as TAR-RNA, destabilizes the secondary structure of its complementary sequence, cTAR-DNA, and promotes the annealing reaction of RNA/DNA leading to TAR/cTAR heteroduplex formation.10,11 As a result, the strand-transfer step during HIV replication is favored.12
NC is an attractive target for the development of new antiviral agents since the potential interference induced by small molecules towards NC would result in a reduction of the reverse transcription of the viral genome as a consequence of a compromised NC activity.2,13 This approach could ultimately lead to the development of successful anti-HIV agents. In the course of a screening for NC inhibitors14 we developed an assay relying on the well-known properties of nucleocapsid to efficiently destabilize and anneal complementary oligonucleotides.10,11 We called it Nucleocapsid Annealing-Mediated Electrophoresis (NAME) assay. NAME assay uses NC to mediate the annealing between stably structured copies of complementary nucleic acids, in our case of the oligonucleotide corresponding to the apical part of a TAR-RNA sequence with its complementary DNA sequence (cTAR) to yield the hybrid TAR/cTAR heteroduplex. The analysis of the TAR/cTAR annealing reaction in the presence of NC can be investigated by native polyacrylamide gel electrophoresis (PAGE).
This protocol shows how to perform the NAME assay to rapidly anneal at room temperature oligonucleotides folded in stable hairpin structures, how to analyze the outcome of the reaction and possible troubleshooting of the experiment. Radioactive labeling of the nucleic acid is not requested, and detection of oligonucleotide bands can be performed by conventional staining methods. The assay, that employs either the recombinant full-length protein or a truncated synthetic version of NC, allows the identification of threading intercalators inhibiting NC, an activity shown to correlate with strong binding to RNA and DNA .14
Protocol
1. Preparation of Material, Nucleic Acids and Proteins
- Nucleic Acids
- Autoclave the 1.5 ml tubes and store them in a sterile container. Perform every step using sterile filter tips to eliminate contaminating agents i.e. nucleases from laboratory consumables.
- Prepare Tris-HCl 10 mM buffer pH 7.5 in DEPC-treated water and filter the solution with a 0.22 µm pore size filter.
- NOTE: The oligonucleotide called “TAR” corresponds to the short (29-mer) RNA sequence 5’-GGCAGAUCUGAGCCUGGGAGCUCUCUGCC-3’ 15 while cTAR is its DNA complementary sequence 5’-GGCAGAGAGCTCCCAGGCTCAGATCTGCC-3’.
- Solubilize both TAR and cTAR in the Tris buffer above mentioned (1.1.2.) to make 100 µM stock solutions. Store cTAR stock solution at -20 °C (aliquots can be stored for months in these conditions). For long-term storage of RNA, make 20 µl aliquots of the TAR stock solution, dry each aliquot using a vacuum concentrator centrifuge and store them at -80 °C. Freshly before the use, resuspend each TAR aliquot in 20 µl DEPC-treated water. NOTE: Working TAR aliquots can be stored at -20 °C for two weeks.
- NC protein and (12-55)NC peptide
- Prepare the full-length recombinant NC protein as reported.16 Store the stock solution in aliquots at -20 °C. Determine the exact protein concentration with a UV-Vis Spectrophotometer using an extinction coefficient at 280 nm of 6,410 M-1 cm-1.
- Resuspend the synthetic (12-55)NC peptide in Tris-HCl 10 mM pH 7.5 and store the stock solution in aliquots at -20 °C. Determine the correct peptide concentration on a UV-Vis Spectrophotometer using an extinction coefficient at 280 nm of 5,700 M-1 cm-1. NOTE: The (12-55)NC peptide was obtained HPLC purified and lyophilized out of a solution containing two equivalents of Zinc chloride.
- Compound 1
- Weigh about 1 mg of the lyophilized compound 1 using an analytical balance and dissolve it in 100 µl of 100% DMSO, opportunely weighed, to obtain a high concentration (≈10 mM) stock solution. Determine the exact compound concentration on a UV-Vis Spectrophotometer using its extinction coefficient (at 354 nm: 11,387 M-1 cm-1). Store the stock solution in the dark at -20 °C prior to use.
2. Setting up of Gel Apparatus and Casting of the Gel
To set up the gel, rinse two plates (one long and one shorter) with 70% ethanol, let them dry, and then place two 1 mm spacers along the long edges of the longer plate; cover it with the short plate, and make sure to align the two plates at the bottom.
To cast the gel, follow the instructions provided by the supplier (different suppliers use slightly different apparatus; sandwich clamps and stacks are provided by each casting apparatus). In all cases, be sure that clamps, stacks and gaskets are clean, and remove traces of acrylamide left by previous users.
Place the assembled gel sandwich in the casting stand and follow specific instructions by the supplier. NOTE: Usually a clean silicone gasket at the bottom of the casting slot ensures a good seal and helps to avoid leaks when pouring the gel.
To check for leaks, pour distilled water using a pipet between the glass plates. Add water to fill up the sandwich and wait for some minutes to make sure that no leaks occur. If the sandwich is correctly assembled, remove the water and insert a filter paper between the two glasses to dry the glasses. Remove the paper and now the sandwich is ready for gel casting.
- Pour gel at room temperature (RT, 25 °C). Since polyacrylamide is highly neurotoxic, wear gloves. Use continuous 12% polyacrylamide gels in non-denaturing (native) conditions to perform NAME assay.
- Prepare the 12% gel solution: mix 12 ml of 40% acrylamide/bisacrylamide (19/1) solution, 4 ml TBE 10x buffer (10x: Tris-HCl 890 mM, Borate 890 mM, EDTA 20 mM) and 24 ml MilliQ water. Add 400 µl APS and 50 µl TEMED immediately before use. Mix the solution and rapidly use a pipet to pour the solution down between the glass plates. Introduce the comb between the glass plates, avoiding bubbles. Add gel solution to fill the sandwich completely.
Let the gel polymerize for 45 min to 1 hr. Immediately before sample loading, remove the comb slowly and rinse wells thoroughly with distilled water.
After the polymerization and wells rinsing steps, follow the specific instructions by the supplier to place the gel sandwich in the apparatus for vertical gel electrophoresis. If a cooling system is present, make sure to place the gel system in the correct position of the cooling core.
If the system is designed to handle more than one gel, but only one gel has to be run, attach a gel sandwich as buffer dam to the cooling core on the other side to form the complete upper buffer chamber. NOTE: if the dam is not placed correctly, the upper buffer will leak possibly stopping the gel run.
Prepare 2.5 L of TBE running buffer (1x: Tris-HCl 89 mM, Borate 89 mM, EDTA 2 mM) in deionized water. Set apart about 500 ml of TBE buffer for the upper buffer chamber and pour the remainder into the lower buffer chamber. Pour the remaining 500 ml TBE buffer into the upper buffer chamber.
Place the lid on the top of the lower buffer chamber, so that the anode and cathode are in the appropriate position.
Connect the cooling core, if present, to a water tap using hoses. Fill the core with tap water, which will act as a heat sink during electrophoresis, and let tap water circulate through the core during electrophoresis.
3. Nucleocapsid Annealing-mediated Electrophoresis (NAME) Assay
- Preparation of Buffers
- Prepare TNMg buffer (1x: Tris-HCl 10 mM, NaCl 20 mM, Mg(ClO4)2 1 mM, pH 7.5) in DEPC-treated water and filter the solution with a 0.22 µm pore size filter. NOTE: TNMg buffer can be stored at 4 °C up to 7 days. For best folding and annealing results, we recommend use of freshly made buffers.
- Prepare the Gel Loading Buffer containing SDS (GLBSDS: Tris-HCl 100 mM, EDTA 4 mM, 50% w/v glycerol, 2% w/v SDS, 0.05% w/v bromophenol blue) in DEPC-treated water. NOTE: GLB helps to track how far the oligonucleotides samples are running into the gel system and allows to sink the samples into the gel. Addition of SDS to the GLB is a critical step for the optimal success of NAME assay. If this step is not followed, it is not possible to distinguish nucleic acids band in the gel system (Figure 2).
- Controls preparation
- Dilute serially the oligonucleotides stock solutions and use freshly made 10 µM solutions to prepare 10 µl of a 1 µM solution of TAR. Prepare in the same way, separately, 10 µl of a 1 µM cTAR in TNMg 1x buffer. Heat each tube to 95 °C for 5 min and then leave to cool to RT in order for TAR and cTAR to assume their stem-bulge-loop structures.
- Prepare 1 µM solution of hybrid TAR/cTAR as control. Mix 1 µl of 10 µM TAR with 1 µl of 10 µM cTAR in TNMg 1x, in a total volume of 10 µl. Denature the oligonucleotides by heating the tube to 95 °C for 5 min and leave it to slowly cool to RT in order for TAR to anneal to its complementary sequence cTAR and to form the double-stranded hybrid TAR/cTAR.
- When the solutions are cooled to RT, perform a short-spin centrifugation. Add 3 µl of GLBSDS, pipette each solution inside the tube 3 times and put the tube on ice. Keep the control samples steady on ice until the loading step.
- Samples preparation to analyze NC protein and (12-55)NC peptide activity
- Use 2.5 µl of freshly made 4 µM oligonucleotide solution for each sample. Always prepare a little excess amount of each solution to prevent pipetting errors. Fold 32.5 µl of 4 µM solution of TAR and, separately, of cTAR in TNMg 1x buffer as reported for the controls preparation above mentioned (step 3.2.1.).
- Dilute the stock solution of both NC protein and (12-55)NC peptide to 80 µM solution with MilliQ water.
- When TAR and cTAR solutions are cooled to RT, perform a short-spin centrifugation of both tubes and start the preparation of the samples tubes.
- Prepare 12 empty 0.5 ml autoclaved tubes. Place 3 rows of 4 tubes in the rack for lab samples. Each row indicates respectively the samples to analyze the full-length NC protein, the samples without protein and the samples to analyze the (12-55)NC peptide activity.
- Always replace tips anytime a different reagent is used.
- Add in each tube 2.5 µl of folded TAR and 2.5 µl of folded cTAR. Add 4 µl DEPC-treated water to the samples for the analysis of NC and of (12-55)NC. Add 5 µl DEPC-treated water to the other samples.
- Add 1 µl of 80 µM NC or 1 µl of 80 µM (12-55)NC to the samples for, respectively, NC or (12-55)NC analysis.
- Add immediately 3 µl of GLBSDS to the time 0 min samples (reported as 0’ in Figure 1), pipetting the solution inside each tube 3 times, and finally put the tube on ice. Repeat this step for all samples and controls. Keep the samples steady on ice until the loading step.
- After 15 min, 30 min and 1 hr of incubation at RT, add 3 µl of GLBSDS, pipette the solution inside each tube 3 times and put the tube on ice. Repeat this step for all samples and controls, respectively, and keep on ice until the loading step.
- Remove the lid of the gel apparatus. Load 13 µl of each sample into the wells formed as previously described by casting the gel with a well-forming comb. Load the samples following the order reported in Figure 1.
- Replace the lid of the gel apparatus. Attach the leads of the gel apparatus to the power supply. Check that the electrodes are plugged into the correct slots in the power supply.
- Turn on the power and run the gel for 2 hr and 30 min at a constant voltage of 200 V, until the dye has migrated to 1.5 cm above the bottom of the gel.
- Sample preparation to analyze anthraquinone activity on NC and (12-55)NC
- Dilute the compound 1 stock solution. Prepare 10 µl of the 500 µM, 250 µM, 50 µM, 5 µM dilution of compound 1 in MilliQ water.
- Prepare control samples as reported above (paragraph 3.2.).
- Use 2.5 µl of freshly made 4 µM oligonucleotide solution for each sample. Always prepare a little excess amount of each solution to prevent pipetting errors. Fold 27.5 µl of 4 µM solution of TAR and, separately, of cTAR in TNMg 1x buffer as above mentioned (step 3.2.1.).
- Dilute the stock solution of both NC protein and (12-55)NC peptide to 80 µM solution in water.
- When TAR and cTAR solutions are cooled to RT, perform a short-spin centrifugation of both tubes and start the preparation of the samples.
- Prepare 20 empty 0.5 ml autoclaved tubes. Place 2 rows of 10 tubes each in the rack for lab samples.
- Always replace tips anytime a different reagent is used.
- Add every tube of the first row with 2.5 µl of folded TAR. Add every tube of the second row with 2.5 µl of folded cTAR.
- Add 2 µl of the appropriate compound1 dilution (increasing concentration from the 5 µM to the 500 µM dilution) to TAR and to cTAR. Replace compound with water in the samples for the analysis of NC and (12-55)NC in the absence of compound. Incubate the samples for 15 min at RT.
- After 15 min incubation, mix the cTAR samples (tubes of second row) with the correspondent TAR samples (tubes of first row).
- Add 1 µl of 80 µM NC or 1 µl of 80 µM (12-55)NC to the samples for NC or (12-55)NC analysis, respectively.
- After 15 min incubation, add 3 µl of GLBSDS, pipette each tube 3 times and put the tube on ice. Repeat this step, respectively, using the samples for NC and (12-55)NC analysis. Keep the samples steady on ice until the loading step.
- Remove the lid of the gel apparatus. Load 13 µl of each sample into the wells. Load the samples following the order reported in Figure 3B.
- Replace the lid of the gel apparatus. Attach the leads of the gel apparatus to the power supply. Check that the electrodes are plugged into the correct slots in the power supply.
- Turn on the power and run the gel for 2 hr and 30 min at a constant voltage of 200 V, until the dye has migrated to 1.5 cm above the bottom of the gel.
- Sample preparation: Oligo-preincubation mode vs NC-preincubation mode
- Dilute the compound1 stock solution. Prepare 10 µl of the 500 µM, 250 µM, 50 µM, 5 µM dilution of compound 1 in MilliQ water.
- Prepare control samples as reported above (paragraph 3.2.).
- Use 2.5 µl of 4 µM oligonucleotide solution for each sample. Always prepare a little excess amount of each solution to prevent pipetting errors. Fold 27.5 µl of 4 µM solution of TAR and, separately, of cTAR in TNMg 1x buffer as above mentioned (step 3.2.1.).
- Dilute the stock solution of both NC protein and (12-55)NC peptide to 80 µM solution in water.
- When TAR and cTAR solutions are cooled to RT, perform a short-spin centrifugation of both tubes and start the preparation of the samples.
- Always replace tips each time reagents are changed or if any other liquid or solution contained in the reaction tube is touched.
- From this step on make sure to clearly label and separate the samples for the two different pre-incubation procedures.
- Oligo-preincubation mode
- Prepare 10 empty 0.5 ml autoclaved tubes. Place 2 rows of 5 tubes in the rack for lab samples.
- Add in each tube of the first row 2.5 µl of folded TAR. Add in each tube of the second row 2.5 µl of folded cTAR.
- Add 2 µl of the appropriate compound1 dilution (increasing concentration from the 5 µM to the 500 µM dilution) to TAR and to cTAR. Replace compound with water in the samples for the analysis of NC in the absence of compound. Incubate the samples for 15 min at RT.
- After 15 min incubation, take the cTAR samples (second row) and put them with the correspondent TAR samples (first row).
- Add 1 µl of 80 µM NC to each sample and incubate the samples for 15 min at RT.
- After 15 min incubation, add 3 µl of GLBSDS, pipette each tube 3 times and put the tube on ice. Keep the samples steady on ice until the loading step.
- NC-preincubation mode
- Prepare 5 empty 0.5 ml autoclaved tubes and place them in the rack for lab samples.
- Add in each tube 4 µl of the appropriate compound 1 dilution (increasing concentration from the 5 µM to the 500 µM dilution). Replace compound with water in the samples for the analysis of NC in the absence of compound.
- Add in each tube 1 µl of 80 µM NC and incubate the samples for 15 min at RT.
- Mix 15 µl of folded TAR with 15 µl of folded cTAR in a tube and use this TAR + cTAR solution in the next step.
- After 15 min incubation, add to each sample 5 µl of the TAR + cTAR mix solution and incubate the samples for 15 min at RT.
- After 15 min incubation, add 3 µl of GLBSDS, pipette each tube 3 times and put the tube on ice. Keep the samples steady on ice until the loading step. NOTE: From step 3.5.8 on, analysis can be performed with all the samples (resume from 3.5.7).
- Remove the lid of the gel apparatus. Load 13 µl of each sample into the wells. Load the samples following the order reported in Figure 4.
- Replace the lid of the gel apparatus. Attach the leads of the gel apparatus to the power supply. Check that the electrodes are plugged into the correct slots in the power supply.
- Turn on the power and run the gel for 2 hr and 30 min at a constant voltage of 200 V, until the dye has migrated to 1.5 cm above the bottom of the gel.
4. Removing the Gel and Gel Staining
After electrophoresis, turn off the power supply, and disconnect the electrical leads. Turn off the water tap of the cooling system and disconnect hoses from the core.
Remove the lid, and, if the system was cooled, remove the cooling core and pour off the buffer from both chambers.
Gently disassemble the gel sandwich from the apparatus and remove all clamps from the glass plates. To remove the gel from the plates, push one of the spacers out to the side of the plates and twist it gently to pull up and away the upper glass plate. Grasp two corners of the gel and lift it off gently to remove it from the glass.
Place the gel in a suitable container with the desired nucleic acids staining solution for 30-45 min with stirring.
After staining, put the gel on a transilluminator imaging system to detect nucleic acids fluorescence signal in the gel system.
Representative Results
Figure 1 shows a representative result of the Nucleocapsid Annealing Mediated Electrophoresis (NAME) assay, performed i) with the full-length recombinant protein, ii) without the protein, i.e. mixing cTAR + TAR and incubating up to 1 hr at RT, and iii) the same assay performed with (12-55)NC, a synthetic peptide lacking the 11 amino acids of N-terminal domain. The pre-folded TAR and cTAR were mixed and the formation of the annealed heteroduplex TAR/cTAR was monitored in the presence of the full-length recombinant NC (left lanes of Figure 1), in the absence of protein (central lanes in Figure 1), and in the presence of the truncated (12-55)NC peptide (right lanes in Figure 1). Controls are: folded cTAR, folded TAR, and annealed hybrid (TAR/cTAR), obtained through thermal denaturation of the stably folded structures of TAR to cTAR followed by their slow annealing.14
The ability of NC to denature two stable nucleic acid sequences into the extended heteroduplex helix is clearly evident: the full-length NC protein leads immediately to the formation of the TAR/cTAR hybrid: 0’ indicates the minimal time passing between addition of NC to the samples and addition of gel loading buffer, which stops the reaction; complete formation is already achieved after 15’ incubation time. The increase in the intensity of the annealed heteroduplex parallels the decrease in the intensity of the folded cTAR and TAR oligonucleotides. The heteroduplex formation is achieved also in the presence of the truncated form, i.e. the (12-55)NC, confirming the biological activity of the synthetic peptide. In the absence of the protein or of the peptide the heteroduplex formation is not observed, and TAR and cTAR oligonucleotides will not anneal at room temperature into the heteroduplex (Figure 1). In all experiments, the fast formation of unstable intermediates (“intermediate complexes” in Figure 1) that decrease over time is observed, consistently with the mechanism proposed of strand exchange through the hairpins of cTAR and TAR till the correct nucleic acids folding.17
A possible troubleshooting of the experiment is the lack of SDS in the Gel Loading Buffer. In the absence of SDS in the GLB the outcome of the reaction is shown in Figure 2 and compared to the outcome with GLB + SDS. The pre-folded TAR and cTAR were mixed and incubated for 3 hr at RT of for 3 hr at 37 °C with the recombinant full-length NC protein. After incubation the samples were split in two: to one half gel loading buffer without SDS (GLB) was added, while to the other half GLB was with SDS (GLBSDS). Samples were loaded on the gel, and the position of the nucleic acids bands was visualized after the gel run by dye staining. When NC was present but the samples were loaded with GLB, all nucleic acids bands were shifted up: this is expected and indicates a strong binding between the protein and the nucleic acids. The results with GLBSDS are shown in the extreme right of the gel: the addition of SDS denatures the protein and releases the nucleic acids from the stable complex with the protein, allowing the comparison with the controls.
The NAME assay is used to assess the ability of threading intercalators to stabilize dynamic nucleic acid structures and inhibit NC chaperone activity.14 Threading intercalators are planar aromatic molecules substituted by bulky side chains located at the opposite sites of the ring system, such as the anthraquinone shown in Figure 3A.18 These intercalators are able to thread through the double helix of nucleic acids, finally locating their side chains in each groove of the double helix.19,20
Figure 3B shows the outcome of the NAME assay in the presence of compound 1, a known threading intercalator,18 in the presence of full-length NC or of the truncated peptide. In both cases, the formation of the TAR/cTAR hybrid by NC is reduced by increasing the concentration of the intercalator, while, at the same time, the amount of free TAR and cTAR increases. The truncated form of the protein lacking the N-terminal tail (12-55NC) is more sensitive to the inhibition of its activity: this difference was verified with all the compounds tested so far.
The NAME assay can be performed in two ways, either by pre-incubating the test compound with NC, followed by addition of the folded oligonucleotides (NC-preincubation mode), or by preincubating the test compound for 15 min with the folded nucleic acid substrates, followed by addition of NC and a further incubation for 15 min (oligo-preincubation mode). Although the overall incubation time is the same in the two different modes, the preincubation with the folded oligonucleotides before addition of the NC allows more time for the threading of the intercalators into the folded nucleic acid. This results in a stronger stabilization of their dynamic structures and into an easier inhibition of NC chaperone activities. The analysis of the NAME assay in the two modes therefore enhances differences in the mechanism of action of NC inhibitors, as shown in Figure 4: when compound 1 was analyzed by NAME in the oligo-preincubation mode (Figure 4, lanes at the left) it shows potent inhibition of NC-mediated annealing, whereas much lower inhibition is observed in the NC-preincubation mode (Figure 4, lanes at the right). Please note that for the determination of inhibitory concentrations while screening sets of related compounds with this assay and in these conditions, each experiment must be performed at least in triplicate using closely spaced concentrations (especially around the IC50 value).14
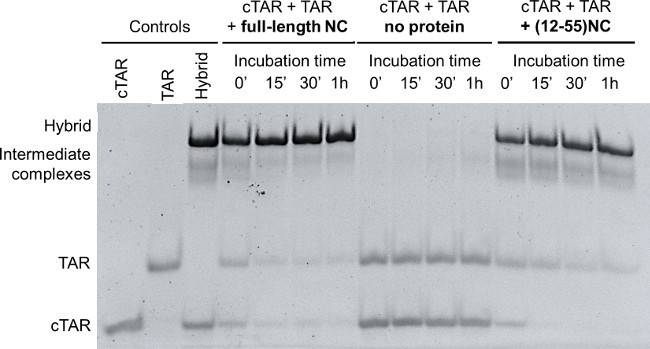 Figure 1: NAME assay with full-length NC and with the truncated (12-55)NC. Folded TAR and cTAR, each 1 µM, were incubated with recombinant NC or with (12-55)NC (oligos/NC=1/8) for 0 min, 15 min, 30 min and 1 hr. TAR and cTAR incubated in the absence of protein (0 min, 15 min, 30 min and 1 hr) were used as negative control. Folded TAR, folded cTAR and the hybrid TAR/cTAR obtained after thermal denaturation followed by in vitro annealing were used as controls. The reactions were then stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
Figure 1: NAME assay with full-length NC and with the truncated (12-55)NC. Folded TAR and cTAR, each 1 µM, were incubated with recombinant NC or with (12-55)NC (oligos/NC=1/8) for 0 min, 15 min, 30 min and 1 hr. TAR and cTAR incubated in the absence of protein (0 min, 15 min, 30 min and 1 hr) were used as negative control. Folded TAR, folded cTAR and the hybrid TAR/cTAR obtained after thermal denaturation followed by in vitro annealing were used as controls. The reactions were then stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
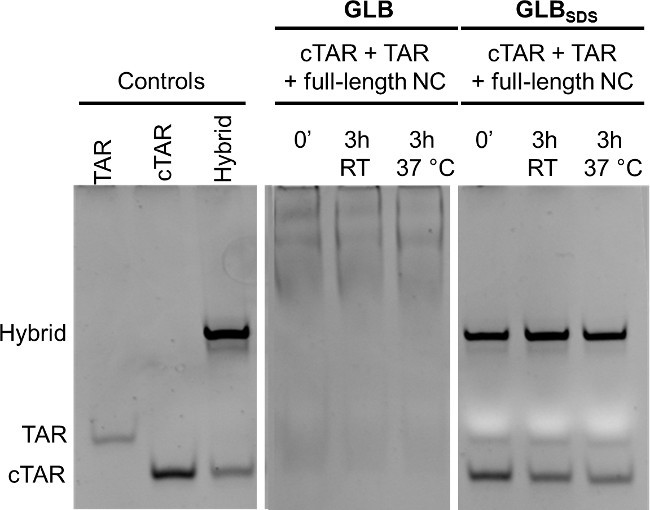 Figure 2: Effect of SDS in gel loading buffer (GLB) when analyzing TAR/cTAR annealing by NC. TAR and cTAR, prefolded in TNMg 1x, were incubated with full-length recombinant NC (oligos/NC = 1/8) for 3 hr at room temperature or at 37 °C. GLB or GLBSDS were then added to the samples incubated with NC. Controls: TAR, cTAR and the annealed hybrid TAR/cTAR (each 1 µM). Electrophoresis was performed using a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator. This figure has been modified from Figure S4 of: Sosic, A. et al. Design, synthesis and biological evaluation of TAR and cTAR binders as HIV-1 nucleocapsid inhibitors. MedChemComm 4, 1388-1393, doi:10.1039/c3md00212h (2013).
Figure 2: Effect of SDS in gel loading buffer (GLB) when analyzing TAR/cTAR annealing by NC. TAR and cTAR, prefolded in TNMg 1x, were incubated with full-length recombinant NC (oligos/NC = 1/8) for 3 hr at room temperature or at 37 °C. GLB or GLBSDS were then added to the samples incubated with NC. Controls: TAR, cTAR and the annealed hybrid TAR/cTAR (each 1 µM). Electrophoresis was performed using a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator. This figure has been modified from Figure S4 of: Sosic, A. et al. Design, synthesis and biological evaluation of TAR and cTAR binders as HIV-1 nucleocapsid inhibitors. MedChemComm 4, 1388-1393, doi:10.1039/c3md00212h (2013).
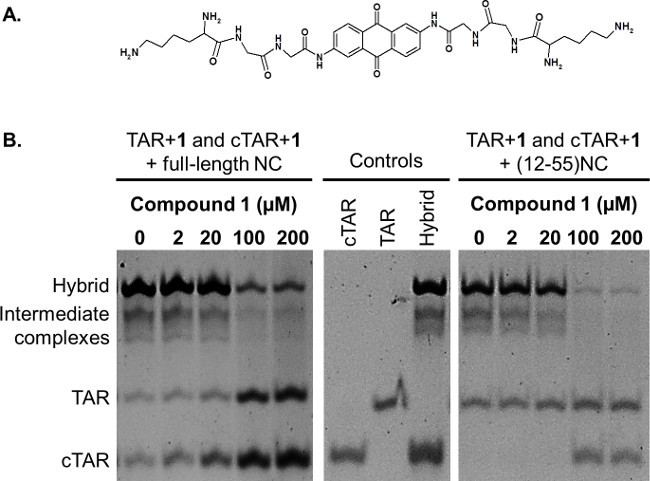 Figure 3: (A) Chemical structure of the threading intercalator 1. (B) Effect of threading intercalator 1 assessed by NAME assay. Folded TAR and cTAR, each 1 µM, were incubated in the presence of the indicated concentrations of the compound 1 with the full-length NC or with the (12-55)NC, each 8 µM. Folded TAR, folded cTAR and the extended heteroduplex TAR/cTAR were used as controls. The reactions were then stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
Figure 3: (A) Chemical structure of the threading intercalator 1. (B) Effect of threading intercalator 1 assessed by NAME assay. Folded TAR and cTAR, each 1 µM, were incubated in the presence of the indicated concentrations of the compound 1 with the full-length NC or with the (12-55)NC, each 8 µM. Folded TAR, folded cTAR and the extended heteroduplex TAR/cTAR were used as controls. The reactions were then stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
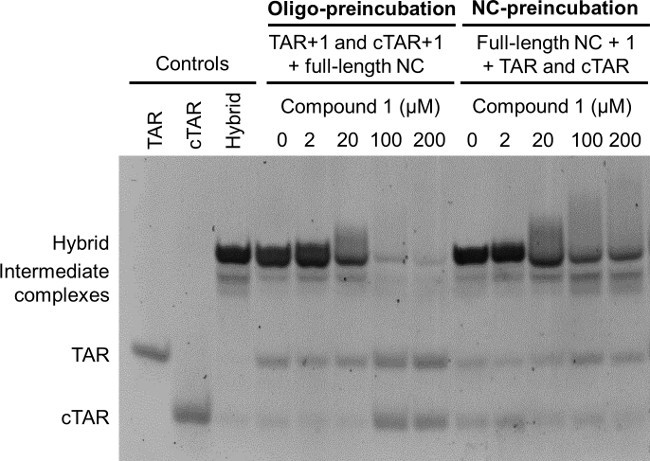 Figure 4: Oligo-preincubation versus NC-preincubation modes: effects on NAME assay. Oligo-preincubation mode: folded TAR and folded cTAR, each 1 µM, were preincubated with the indicated concentrations of the threading intercalator 1 for 15 min, and then for additional 15 min in the presence of the full-length NC (8 µM). NC-preincubation mode: the indicated concentrations of the threading intercalator 1 were preincubated with the full-length NC (8 µM) for 15 min, and then for additional 15 min in the presence of folded TAR and folded cTAR, each 1 µM. Folded TAR, folded cTAR and the extended heteroduplex TAR/cTAR were used as controls. All reactions were finally stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
Figure 4: Oligo-preincubation versus NC-preincubation modes: effects on NAME assay. Oligo-preincubation mode: folded TAR and folded cTAR, each 1 µM, were preincubated with the indicated concentrations of the threading intercalator 1 for 15 min, and then for additional 15 min in the presence of the full-length NC (8 µM). NC-preincubation mode: the indicated concentrations of the threading intercalator 1 were preincubated with the full-length NC (8 µM) for 15 min, and then for additional 15 min in the presence of folded TAR and folded cTAR, each 1 µM. Folded TAR, folded cTAR and the extended heteroduplex TAR/cTAR were used as controls. All reactions were finally stopped using GLBSDS. Samples were resolved on a 12% polyacrylamide gel in TBE 1x; after electrophoresis nucleic acids on the gel were stained and detected on a transilluminator.
Discussion
NAME is an assay that allows to rapidly assess the inhibition of the chaperone activity of the nucleocapsid protein of HIV-1, an interesting target in the search of novel anti-HIV drugs. NC anneals fast and at room temperature nucleic acids whose stable folding would otherwise require thermal denaturation at high temperature followed by careful annealing. The assay can be performed with either full-length NC or with the truncated (12-55)NC: in both cases the two nucleic acids (RNA and its complementary DNA copy) are annealed rapidly. This is consistent with literature observation with the truncated NC peptide: annealing is initiated by the efficient melting of the stem of cTAR followed by interaction of its complementary apical loop, which is a weak NC binding site, with the complementary sequence of TAR apical loop, ending up through several unstable intermediates to the extended annealed hybrid.21
NC is a very stable protein and few problems while performing NAME could occur. It is very important however to add freshly made SDS in the Gel Loading Buffer used to stop the reaction: if this is not achieved, the gel will not show the nucleic acid bands at the correct positions. In fact, NC is a nucleic acid binding protein, so that to correctly visualize TAR/cTAR heteroduplex by gel electrophoresis SDS must be present in the Gel Loading Buffer to denature the protein and release it from its tight complex with the nucleic acids. This is also a possible troubleshooting of the experiment, i.e. the formation of shifted bands during the gel run. This effect is particularly evident when the full-length NC was employed, as in Figure 2, and is linked to the presence of the highly basic N-terminal tail, having a strong aggregating effect on nucleic acids.
The presence of the terminal basic tail makes the annealing reaction more difficult to be inhibited, as shown in Figure 3 using compound 1, a representative threading intercalator, i.e. an intercalator particularly efficient at stabilizing the stem-loop structures of TAR and of cTAR. In fact, this type of interaction with nucleic acids is favored when bulges and loops interrupt the double helix continuity, allowing for an easier threading of the bulky substituents into the base pairs. Stabilization of TAR and cTAR by these nucleic acid binders was previously analyzed by the determination of the increase in the melting temperature of the two oligonucleotides.14 Moreover, the increase in melting temperature correlates with the inhibition of the annealing catalyzed by HIV-1 NC, leading to the reduced formation of the heteroduplex TAR/cTAR and indicating that threading intercalators are an interesting class of binders for dynamic structures of RNA such as the TAR element.14
The mechanism of action invoked for these compounds can be further validated by performing the NAME assay in different alternative formats, as shown in Figure 4: in the case of intercalators the inhibition of annealed heteroduplex is enhanced when the drug is incubated with the two folded oligos. Compounds acting with different mechanism of action, such as direct binders of the NC protein, would be less affected by the different preincubation mode. NAME assay performed in the two methods therefore could be used to screen libraries of compounds for the identification of NC inhibitors, and also to assess the putative mechanism of action of the positive hits while searching for anti-HIV drugs targeted at NC. Clearly, the use of these techniques alone when claiming for a specific mechanism of action of identified NC inhibitors is not sufficient, and other suitable biochemical assays are needed to sustain the working hypothesis.14
Finally, it must be stressed that NAME uses highly purified enzymes, either recombinant or synthetic, giving precious information on the “in vitro” inhibition of NC by the tested compounds. This is the “strength and weakness” of the assay: NAME can well complement virtual screening and molecular modeling approaches in the preliminary steps of drug discovery programs, enabling to rapidly build and analyze structure-activity relationships and eventually redirecting the synthesis and drug design. However, as other biochemical assays used in drug development projects in HIV, NAME will not give information on the effects of putative inhibitors in virus-infected cells.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by Ministero degli Affari Esteri (MAE, DRPG, Unità per la cooperazione scientifica e tecnologica, Grant: PGR Italy-USA) and by MIUR, Progetti di Ricerca di Interesse Nazionale (Grant: 2010W2KM5L_006).
References
- Druillennec S, et al. A mimic of HIV-1 nucleocapsid protein impairs reverse transcription and displays antiviral activity. Proc Natl Acad Sci USA. 1999;96:4886–4891. doi: 10.1073/pnas.96.9.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlix JL, Garrido JL, Morellet N, Mely Y, de Rocquigny H. Properties, functions, and drug targeting of the multifunctional nucleocapsid protein of the human immunodeficiency virus. Adv Pharmacol. 2007;55:299–346. doi: 10.1016/S1054-3589(07)55009-X. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Gorelick RJ. Nucleocapsid protein function in early infection processes. Virus Res. 2008;134:39–63. doi: 10.1016/j.virusres.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirambeau G, Lyonnais S, Gorelick RJ. Features, processing states, and heterologous protein interactions in the modulation of the retroviral nucleocapsid protein function. RNA Biol. 2010;7:724–734. doi: 10.4161/rna.7.6.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, et al. Probing nucleation, reverse annealing, and chaperone function along the reaction path of HIV-1 single-strand transfer. Proc Natl Acad Sci USA. 2007;104:12651–12656. doi: 10.1073/pnas.0700350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu VP, et al. Strand transfer events during HIV-1 reverse transcription. Virus Res. 2008;134:19–38. doi: 10.1016/j.virusres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- Guo J, Henderson LE, Bess J, Kane B, Levin JG. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J Virol. 1997;71:5178–5188. doi: 10.1128/jvi.71.7.5178-5188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godet J, Mely Y. Biophysical studies of the nucleic acid chaperone properties of the HIV-1 nucleocapsid protein. RNA Biol. 2010;7:687–699. doi: 10.4161/rna.7.6.13616. [DOI] [PubMed] [Google Scholar]
- Darlix JL, et al. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J Mol Biol. 2011;410:565–581. doi: 10.1016/j.jmb.2011.03.037. [DOI] [PubMed] [Google Scholar]
- Lapadat-Tapolsky M, Pernelle C, Borie C, Darlix JL. Analysis of the nucleic acid annealing activities of nucleocapsid protein from HIV-1. Nucleic Acids Res. 1995;23:2434–2441. doi: 10.1093/nar/23.13.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo M-N, Barany G, Rouzina I, Musier-Forsyth K. Mechanistic studies of mini-TAR RNA/DNA annealing in the absence and presence of HIV-1 nucleocapsid protein. J Mol Biol. 2006;363:244–261. doi: 10.1016/j.jmb.2006.08.039. [DOI] [PubMed] [Google Scholar]
- Beltz H, et al. Structural determinants of HIV-1 nucleocapsid protein for cTAR DNA binding and destabilization, and correlation with inhibition of self-primed DNA synthesis. J Mol Biol. 2005;348:1113–1126. doi: 10.1016/j.jmb.2005.02.042. [DOI] [PubMed] [Google Scholar]
- Mori M, et al. Nucleocapsid Protein: a Desirable Target for Future Therapies against HIV-1. Curr Top Microbiol Immunol. Forthcoming. [DOI] [PMC free article] [PubMed]
- Sosic A, et al. Design, synthesis and biological evaluation of TAR and cTAR binders as HIV-1 nucleocapsid inhibitors. MedChemComm. 2013;4:1388–1393. [Google Scholar]
- Tabarrini O, et al. Studies of Anti-HIV Transcription Inhibitor Quinolones: Identification of Potent N1-Vinyl Derivatives. Chemmedchem. 2010;5:1880–1892. doi: 10.1002/cmdc.201000267. [DOI] [PubMed] [Google Scholar]
- Hagan N, Fabris D. Direct mass spectrometric determination of the stoichiometry and binding affinity of the complexes between nucleocapsid protein and RNA stem-loop hairpins of the HIV-1 Psi-recognition element. Biochemistry. 2003;42:10736–10745. doi: 10.1021/bi0348922. [DOI] [PubMed] [Google Scholar]
- Ivanyi-Nagy R, Davidovic L, Khandjian EW, Darlix JL. Disordered RNA chaperone proteins: from functions to disease. Cell Mol Life Sciences: CMLS. 2005;62:1409–1417. doi: 10.1007/s00018-005-5100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotto G, et al. Tuning G-quadruplex vs double-stranded DNA recognition in regioisomeric lysyl-peptidyl-anthraquinone conjugates. Bioconjug Chem. 2011;22:2126–2135. doi: 10.1021/bc200389w. [DOI] [PubMed] [Google Scholar]
- Carlson CB, Vuyisich M, Gooch BD, Beal PA. Preferred RNA binding sites for a threading intercalator revealed by in vitro evolution. Chem Biol. 2003;10:663–672. doi: 10.1016/s1074-5521(03)00147-9. [DOI] [PubMed] [Google Scholar]
- Gooch BD, Beal PA. Recognition of duplex RNA by helix-threading peptides. J Am Chem Soc. 2004;126:10603–10610. doi: 10.1021/ja047818v. [DOI] [PubMed] [Google Scholar]
- Kanevsky I, et al. Structural determinants of TAR RNA-DNA annealing in the absence and presence of HIV-1 nucleocapsid protein. Nucleic Acids Res. 2011;39:8148–8162. doi: 10.1093/nar/gkr526. [DOI] [PMC free article] [PubMed] [Google Scholar]