

Abstract
Giant Unilamellar Vesicles (GUVs) are a popular biomimetic system for studying membrane associated phenomena. However, commonly used protocols to grow GUVs must be modified in order to form GUVs containing functional transmembrane proteins. This article describes two dehydration-rehydration methods — electroformation and gel-assisted swelling — to form GUVs containing the voltage-gated potassium channel, KvAP. In both methods, a solution of protein-containing small unilamellar vesicles is partially dehydrated to form a stack of membranes, which is then allowed to swell in a rehydration buffer. For the electroformation method, the film is deposited on platinum electrodes so that an AC field can be applied during film rehydration. In contrast, the gel-assisted swelling method uses an agarose gel substrate to enhance film rehydration. Both methods can produce GUVs in low (e.g., 5 mM) and physiological (e.g., 100 mM) salt concentrations. The resulting GUVs are characterized via fluorescence microscopy, and the function of reconstituted channels measured using the inside-out patch-clamp configuration. While swelling in the presence of an alternating electric field (electroformation) gives a high yield of defect-free GUVs, the gel-assisted swelling method produces a more homogeneous protein distribution and requires no special equipment.
Keywords: Biochemistry, Issue 95, Biomimetic model system, Giant Unilamellar Vesicle, reconstitution, ion channel, transmembrane protein, KvAP, electroformation, gel assisted swelling, agarose, inside-out patch clamp, electrophysiology, fluorescence microscopy
Introduction
When studying the physical principles that govern living systems, bottom-up approaches allow an experimentalist to control system composition and other parameters that are not easily manipulated in cell-based systems1. For membrane-based processes, Giant Unilamellar Vesicles (GUVs, diameter ~ 1–100 µm) have proven to be a very useful biomimetic system2–7 as they are well suited for microscopy studies and micromanipulation8–10. While there are many different protocols to produce GUVs, most fall into two categories — emulsion based approaches11,12 and techniques based on rehydrating a lipid film13–16. In emulsion-based methods, the inner and outer leaflets of the GUV membranes are assembled sequentially from lipid monolayers at water/oil interfaces. This approach is ideal for encapsulating soluble proteins within the GUVs, and for forming GUVs with asymmetric leaflet lipid composition. However, GUVs formed from emulsions can retain traces of solvent that change the membrane’s mechanical properties17, and the approach is not especially well-suited to trans-membrane protein reconstitution.
Film rehydration methods rely on the fact that drying (dehydration) causes many lipid mixtures to form a multi-lamellar stack of membranes. If this stack is then placed in contact with an aqueous buffer, membranes in the stack will move apart as solvent flows between them and at the surface of the stack, individual membranes can detach to form GUVs13,18 (as well a veritable zoo of other lipidic objects). However, even for optimal buffer and lipid compositions, this classical “spontaneous swelling” method has a relatively low yield of defect-free GUVs. One widely used method to boost the yield of defect-free GUVs is “electroformation”, in which an alternating current (AC) field is applied during film rehydration. While the mechanism remains poorly understood, “electroformation” can give spectacular GUV yields (> 90% in favorable circumstances) for low salt concentration buffers (< 5 mM)14,19, and can even work in physiological buffers (~100 mM) using a higher frequency (500 Hz versus 10 Hz) AC field and platinum electrodes15. An alternative approach to boost the yield of defect-free GUVs is “gel-assisted swelling”, in which the lipid solution is deposited onto a polymeric gel substrate rather than the passive (e.g., glass, PTFE) substrates used in classical “spontaneous swelling”. When the resultant lipid/gel film is rehydrated, GUVs can rapidly form even for physiological buffers16,20.
All these methods can produce lipid-only GUVs which can be used to study membrane associated phenomena such as the interaction between soluble proteins and membranes. However, to incorporate a trans-membrane protein into GUVs, significant modifications are needed to ensure that the protein remains in a functional state throughout the reconstitution procedure. While solutions of lipids in organic solvents (e.g., chloroform, cyclohexane) are ideal for producing lipid films, trans-membrane proteins are typically only stable when their hydrophobic trans-membrane domain is embedded in a lipid bilayer, or surrounded by a detergent micelle (e.g., during protein purification). Thus, the starting material for a reconstitution is typically native membranes, purified protein in a detergent solution, or small unilamellar protein-containing vesicles (proteo-SUVs) and/or multi-lamellar vesicles (proteo-MLVs) formed by detergent removal in the presence of lipids. Most methods to incorporate these membrane proteins into GUVs fall into three categories.
Direct Insertion: Trans-membrane protein suspended in detergent is mixed with pre-formed, lipid-only, mildly detergent solubilized GUVs, and the detergent then removed using biobeads21. While conceptually simple, this method requires precise control of the detergent concentration, as too high a detergent concentration can dissolve the GUVs while too low a concentration can cause the protein to unfold or aggregate.
GUV/Proteo-SUV Fusion: Protein in proteo-SUVs is combined with pre-formed, lipid-only GUVs and fusion is facilitated with special fusogenic peptides22 or detergent21. Typically the extent of fusion is limited leading to GUVs with low protein density.
Dehydration/Rehydration: A protein-containing lipid film is formed by partial dehydration of a proteo-SUV (or proteo-MLV) solution and GUVs are then grown as for a pure lipid film. The obvious challenge is to protect the protein during the partial dehydration step23, but the method has been successfully used to reconstitute trans-membrane proteins such as Bacteriorhodopsin, Calcium-ATPase, Integrin and VDAC into GUVs7,23–25.
This article describes dehydration/rehydration protocols to make GUVs containing the voltage-gated potassium channel, KvAP, from the hyper-thermophilic Archaea, Aeropyrum pernix. KvAP has a high degree of homology to eukaryotic voltage dependent potassium channels26 and a known crystal structure27, making it a good model for studying the mechanism of voltage gating. Production of the proteo-SUVs has been described in detail previously and is not part of this tutorial26,28,29. Importantly, KvAP proteo-SUVs do not have to be produced for each GUV preparation, as they can be stored in small (e.g., 10 µl) aliquots at -80 °C for extended periods of time (> 1 year). Electroformation or gel-assisted swelling can then be used to grow GUVs from the KvAP proteo-SUVs (or proteo-MLVs).
The key steps for the electroformation protocol are illustrated in Figure 1. Droplets of a solution of SUVs containing the protein are deposited on platinum wires (shown in Figure 2). Partial dehydration of the SUV suspension leads to the formation of a lipid protein film through the fusion of SUVs. During rehydration, an AC field is applied to the electrodes to assist the lipid layers to delaminate and form GUVs. A 10 Hz field works well when using “low-salt” (< 5 mM) rehydration buffer28 and GUVs take several hours to grow. In contrast, physiological buffers (containing ~100 mM salt) work well with a lower voltage, 500 Hz AC field but require a prolonged (~12 hr) swelling period15. This method is based upon an earlier protocol using ITO slides24, but uses a custom chamber containing two platinum wires as shown in Figure 2 (see the discussion for design details and suggestions for simpler, improvised chambers).
Figure 3 illustrates the gel-assisted swelling method. The protocol works well with buffers with physiological salt concentrations, is rapid, and produces GUVs with a more homogeneous protein distribution. However, the yield of isolated, apparently defect-free GUVs (i.e., the GUV membrane is uniform at optical length-scales and does not enclose any objects) is lower, although it provides a sufficient number for patch-clamp and micro-manipulation experiments. This method was based on a protocol using agarose gel to produce lipid-only GUVs16 and requires less specialized equipment than the electroformation method.
The characterization of GUVs with fluorescence microscopy is described, as well as procedures using a standard patch-clamp set-up to measure KvAP activity in “inside-out” excised membrane patches.
Growing protein-containing GUVs can be more difficult than lipid-only GUVs. In particular, the final GUV yield can depend sensitively on exactly how the SUV solution is deposited and dehydrated to form the membrane stack. For someone without any previous experience with GUVs, it may be helpful to first grow lipid-only GUVs following a conventional protocol15,16 in which the membrane film is formed by depositing lipids from an organic solvent. Once the conventional protocol works well, SUV deposition and partial dehydration can then be mastered using lipid-only SUVs, which are also very helpful when adjusting the protocol for a new lipid composition. When GUVs grow reliably from lipid-only SUVs, it is then only a small step to produce protein-containing GUVs from proteo-SUVs.
Protocol
1. Solution Preparation
Prepare 5 ml of ‘SUV buffer’ containing 5 mM KCl, 1 mM HEPES (pH 7.4) or TRIS (pH 7.5), and 2 mM trehalose. Filter the buffer with a 0.2 µm syringe filter and divide into 1 ml aliquots which can be stored at -20 °C. NOTE: Additional details for reagents and instruments are given in the materials list.
Prepare 40 ml of GUV ‘Growth Buffer’ that will fill the GUV interior during film rehydration. For a ‘low salt’ growth, combine 5 mM KCl, 1 mM HEPES (pH 7.4) or 1 mM TRIS (pH 7.5), and ~400 mM sucrose. For a ‘physiological salt’ growth, combine 100 mM KCl, 5 mM HEPES (pH 7.4) or 5 mM TRIS (pH 7.5), and ~200 mM sucrose.
Prepare 40 ml of ‘Observation Buffer’ for the external solution in the experimental chamber by combining 100 mM KCl, 5 mM HEPES (pH 7.4) or 5 mM TRIS (pH 7.5), and ~200 mM glucose. NOTE: These buffers are only examples. See the discussion to adapt the buffers for other experiments.
Measure the osmolarities of the growth and observation buffers with an osmometer. Add granules of sucrose or glucose to match them to within 1% so that GUVs will not lyse or collapse when transferred from the growth chamber to the observation chamber. NOTE: In this concentration range, adding 1 mM of sucrose (13.7 mg per 40 ml) or glucose (7.2 mg per 40 ml) increases osmolarity by ~ 1 mOsm.
Filter the growth and observation buffers with a 0.2 µm filter and store them at 4 °C to inhibit bacterial growth.
Dissolve 50 mg of beta-casein in 10 ml of 20 mM TRIS (pH 7.5) buffer to form a 5 mg/ml beta-casein solution needed to passivate the surfaces of experimental chambers so that GUVs do not stick, spread and burst. Once the beta-casein is completely dissolved (up to several hours at 4 °C), filter (0.2 µm) it into 0.5 ml aliquots which can be flash frozen and stored at -20 °C for later use (thawed aliquots stored at 4 °C can typically be used for up to a 1 week).
2. SUV Preparation
Prepare and freeze aliquots of proteo-SUVs following the previously published detailed protocol28. Use KvAP fluorescently labeled with Alexa-488 maleimide, reconstituted into DPhPC SUVs (10 mg/ml) at a protein to lipid ratio of 1:10 (by mass). NOTE: Wild-type KvAP contains one cysteine per monomer located near the intra-cellular C-terminus (amino acid 247).
- Fluorescent, Lipid-only SUVs: NOTE: Handle chloroform under a fume hood wearing nitrile gloves and safety glasses. Avoid the use of any plastic as chloroform can dissolve them. Chloroform solutions can be stored in amber glass vials with Teflon caps and transferred using glass syringes. Take care to rinse all glassware at least 5 to 10 times with chloroform before and after pipetting lipids.
- Prepare 100 µl of 10 mg/ml DPhPC SUVs containing 0.5 mol% of the red fluorescent lipid, Texas Red-DHPE, by mixing 100 µl of DPhPC solution (10 mg/ml in chloroform) with 8.2 µl of Texas Red-DHPE solution (1 mg/ml in methanol) in a 1.5 ml amber glass vial.
- Dry the lipids down under a stream of nitrogen in a chemical hood while rotating the vial. When the film appears to be dry, place the lipids under a vacuum for 3 hr to remove any residual solvent.
- Add 100 µl of SUV buffer to the lipids, and vortex vigorously until no lipid remains stuck to the walls of the vial and the solution is uniformly milky.
- Sonicate the lipid solution to form SUVs. Adjust the vial position until the ultrasound causes the most movement and flow inside the vial, and take care not to heat the solution unnecessarily. Continue sonication until the solution becomes translucent, or when possible, transparent (2–5 min for tip sonication, ~20 min for bath sonication).
- Aliquot SUVs (e.g., 10 µl or 20 µl) and freeze (-20 °C) for later use. NOTE: Lipids, especially unsaturated lipids, can easily breakdown. Store lipid solutions at 20 °C (or 80 °C) under argon and use within 6 months. Lipid breakdown products can be detected with Thin Layer Chromatography.
3. GUV Growth by Electroformation
- Prepare the electroformation chamber.
- If the chamber has not been cleaned, remove the windows, wipe off all sealant and grease, extract the wires, and rinse and scrub the chamber with a tissue using water and ethanol (≥70%) alternately.
- Rub the wires well, submerge the wires and chamber in acetone, and sonicate for 5 min. Wipe everything with a tissue again using acetone. Put the chamber in ethanol and sonicate for 5 min.
- Assemble the chamber by inserting the wires through the holes, and rotate and wipe the wires to make sure they are clean. Put the chamber in distilled water, sonicate for 5 min and dry the chamber with a stream of nitrogen or air.
- Prepare 30 µl of 3 mg/ml SUV suspension in SUV buffer. To form protein-containing GUVs, combine 8 µl of proteo-SUVs (DPhPC 10 mg/ml KvAP 1:10), 2 µl of fluorescent SUVs (10 mg/ml DPhPC, 0.5 mol% TexasRed-DHPE) and 20 µl of SUV buffer in a 1.5 ml microcentrifuge tube for a final protein to lipid (mass) ratio of 1:12.5 and 0.1 mol% TexasRed-DHPE. Mix the solution vigorously.
- Alternatively, to practice the protocol with lipid-only SUVs, simply combine 10 µl of fluorescent SUVs (10 mg/ml DPhPC and 0.5 mol% TexasRed-DHPE) with 20 µl SUV buffer.
- Deposit the SUV Solution.
- Use a 2 µl pipette or 5 µl glass syringe to deposit small (<0.2 µl) droplets of the SUV solution on the wires. Approximately 1 µl of solution is needed to form a series of drops along 1 cm of wire. Make sure the drops are small enough and spaced far enough apart that they do not touch or fuse.
- Let the deposited SUVs dry for ~30 min in open air. When all the drops have settled, rotate the wire so the lipid deposits are easier to observe with the microscope. NOTE: If the SUVs do not dry sufficiently, they can just wash off the wires when the growth buffer is added, while drying too much can damage the protein. Because air humidity influences the rate of drying, the drying time and/or air humidity can be adjusted for optimal results30. The lipid film on the wires should be visible under a microscope.
- Assemble the chamber.
- Seal the Chamber Bottom: Use a syringe to apply vacuum grease to the bottom of the chamber around the three wells and press a 40 mm x 22 mm coverslip gently against it to seal the chamber bottom so that it adheres without a gap. Seal the sides of the chamber (where the wires exit) with sealing paste. Apply vacuum grease on top of the chamber outlining the three wells.
- Slowly add growth buffer until each well is filled to the top. Avoid any rapid movement of the solution in the wells as this can strip the lipid film off the electrodes.
- Close the chamber by pressing the top cover slide gently onto the grease, taking care not to dislodge the bottom coverslip. Use a tissue to remove any drops of buffer at the edges of the top cover slip. NOTE: This is a good time to examine the chamber under the microscope to confirm that the lipid film has remained on the wires.
Connect the signal generator to the wires using two alligator clips. Set the frequency (10 Hz/500 Hz sine wave for low/high salt buffer) and use a multimeter to measure and adjust the voltage across the wires to 0.7/0.35 V root mean square (Vrms) for the low/high salt buffer. Cover the chamber with aluminum foil to protect the fluorophores from light. Leave the GUVs to grow for 2 to 3 hr for the low salt buffer, and 12 hr or O/N for the high salt buffer.
Disconnect the chamber from the generator and carefully place it on an inverted microscope to evaluate GUV growth. Use slow, steady movements or fluid flow in the wells may prematurely detach GUVs from the wires. NOTE: GUVs on the wire edges are usually visible in phase contrast (40X long working distance objective), while GUVs anywhere on the lower half of the wires can be seen with epifluorescence. If no GUVs are visible, try rotating the wires to look at the upper surface. GUVs can be stored at 4 °C in a growth chamber for several days.
4. GUV Growth by Gel-assisted Swelling
Prepare 10 ml of a 1% agarose solution by mixing 100 mg of agarose with 10 ml of pure water. Heat it until it boils by placing it in a microwave at 480 W for ~20 sec. Stir to make sure the agarose is completely dissolved. NOTE: The solution can be stored at 4 °C and reheated when needed.
Plasma-clean (air plasma) a cover-slide for 1 min so that the agarose solution will spread nicely on it. Use the cover-slides within the next 15 min as the effect of plasma cleaning wears off quickly.
Apply 200 µl of warm agarose solution to each 22 x 22 mm2 slide so the solution wets the entire surface. Tilt the slide vertically and touch the lower edge to a tissue to remove excess liquid and leave just a thin smooth layer of agarose on the slide.
Place the slide on a hot plate or oven at 60 °C and leave it to dry for at least 30 min. The agarose film is hardly visible by eye. After the slides cool to RT, use them immediately, or store them for up to one week in a closed container at 4 °C.
Place the agarose-coated coverslip in a standard 3.5 cm Petri dish.
Prepare the SUV solution as in Section 3.2 and apply ~15 µl of the SUV solution (3 mg/ml lipid) in ~30 very small drops gently onto the agarose surface. Take care not to distort the agarose layer too much.
Place the slide under a gentle stream of nitrogen for about 10–15 min and follow the evaporation of the buffer by eye as the droplets dry.
As soon as the SUVs have dried, add growth buffer to cover the slide surface. For a small 3.5 cm Petri dish use ~1 ml of buffer.
Allow the swelling to proceed for ~30 min, and then examine the growth of GUVs in the chamber using an inverted microscope with phase-contrast or Differential Interference Contrast (DIC). NOTE: Epifluorescence observation is difficult due to the strong background of fluorophores in the gel and the auto-fluorescence of the agarose.
5. Harvesting and Observing GUVs
Passivate the observation chamber (e.g., small Petri dish or glass coverslip) so that GUVs do not stick, spread and burst on the chamber bottom. Cover the chamber bottom with beta-casein solution, incubate for 5 min, rinse out the casein solution with pure water, dry with a stream of air or nitrogen, and finally add observation buffer (e.g., ~5 mm depth for a small Petri dish).
- Harvest the GUVs. Cut the end of a 100 µl pipette tips so the opening is larger (~2 mm diameter), and aspire slowly as the shear stress of pipetting can easily destroy GUVs.
- For electro-formed GUVs, open the growth chamber by gently removing the top coverslip. Place the pipette tip directly above each wire and aspirate ~50 µl while moving the pipette tip along the wire to detach the GUVs. NOTE: It may help to rotate the wire to collect GUVs on the “other side” of the wire.
- For “gel-assisted swelling” GUVs, first tap the side of the petri dish a few times to help the GUVs detach from the coverslip surface. Position the pipette tip just above the coverslip and aspirate 50 µl while pulling the tip back over the surface. Directly transfer harvested GUVs to an observation chamber, or store in a 1.5 ml microcentrifuge tube at 4 °C for up to 1 week.
Place the observation chamber on an inverted microscope, add the GUVs to the observation chamber, and wait a few minutes for the GUVs to settle at the chamber bottom.
Survey the chamber with phase contrast or DIC to quickly locate smooth, spherical (‘defect free’) “GUV candidates”. Examine each “GUV candidate” in epifluorescence to exclude any containing smaller liposomes nested inside. Finally, check that the lipid fluorescence intensity is uniform and compatible with a single membrane (i.e., unilamellar). NOTE: In some bilamellar (or multi-lamellar) vesicles, the membranes are too close together to be resolved so they appear unilamellar in phase contrast or DIC images. However, these objects can be distinguished from actual unilamellar GUVs by their lipid fluorescence, which is two times (or more) brighter.
6. Patch-clamping GUVs
Make patch pipettes with a 1–2 µm tip diameter from standard borosilicate capillary glass using the program recommended for the pipette puller. NOTE: Special treatments such as fire polishing are not necessary, and pipettes can be used for several days after they have been pulled if they are kept in a closed box.
Passivate the chamber by incubating with a beta-casein solution (5 mg/ml) to ensure that GUVs do not adhere, spread and rupture on chamber surfaces. Rinse the casein off after 5 min.
Insert the ground electrode, fill the chamber with observation buffer, transfer 10 µl of the GUV suspension as described in step 5.2 and 5.3, and wait a few minutes for the GUVs to settle at the bottom.
Fill a fresh patch pipette with solution (observation buffer or another iso-osmotic solution) and mount it on the patch-clamp amplifier headstage.
Search through the chamber to locate a “defect-free” GUV as described in section 5.4, and check that it contains fluorescent protein.
Apply a constant positive pressure (> 100 Pa, or roughly 1 cm H2O in a manometer) to keep the patch pipette interior clean, and insert the patch pipette into the chamber. Bring the patch pipette into the field of view, apply test pulses to measure/compensate the pipette voltage offset and resistance, and examine the pipette under fluorescent illumination to confirm that the tip is clean.
Bring the patch pipette towards the GUV, and if necessary, simultaneously reduce the positive pressure so the outward flow from the patch pipette does not make the GUV “run away”. When the patch pipette is close to the GUV, apply a negative pressure (up to 5 cm H2O) to pull the GUV against the patch pipette. Monitor the resistance as the “tongue” of GUV membrane enters the patch pipette and the gigaseal forms.
If a gigaseal did not form, remove the patch pipette from the chamber and return to step 6.4. If the membrane patch formed a gigaseal, but the GUV remains attached to the pipette, excise the patch by pulling away from the GUV, bursting the GUV against the chamber bottom, or briefly moving the pipette out of solution.
When the inside-out membrane patch has been excised from the GUV and the gigaseal is stable, switch off the test pulses and apply a voltage protocol such as the one shown in Figure 13. NOTE: Figure 13 follows the standard electrophysiological convention for an inside-out patch in which current flowing into the patch-electrode is “positive”, and V = Vbath– Vpipette. Holding the patch at a negative potential (e.g., V = -100 mV) for ~30 sec places KvAP in the resting state, while steps (100 msec to 5 sec) to more positive potentials (e.g., V = 100 mV) can then drive it into conducting (i.e., open) active states.
After measurements on a membrane patch are finished, break the patch with a zap or pressure pulse and check that the voltage offset of the patch electrode has not drifted. Remove the patch pipette from the chamber, and return to step 6.4.
Representative Results
The growth of GUVs can be quickly evaluated by examining the growth chamber under the microscope. For electroformation, the GUVs tend to grow in bunches along the platinum wires, as shown in Figure 4. During gel-assisted swelling, GUVs appear as spherical structures that rapidly grow and fuse together (Figure 5).
Defect-free GUVs are more easily identified and evaluated after transferring to an observation chamber. Calibration measurements are needed to rigorously evaluate GUV quality, and a systematic quantification has been published previously28. However, as an empirical guide, “good” GUVs should be isolated (i.e., not in a cluster), have a single, smooth, spherical outer membrane, contain no objects (i.e., tubes, nested vesicles, etc.) inside, and have the “standard” lipid fluorescence level (brighter objects are typically bi- or multi-lamellar). Figure 6 shows DIC and epifluorescence images of a ‘defect-free’ GUV after transfer to an iso-osmotic glucose solution. The contrast in DIC is due to the difference in optical density between the sucrose filled GUV and the glucose containing bath solution. The refractive index contrast of KvAP-containing GUVs often decreases over time, even though KvAP itself should not be permeable to sucrose or glucose. The uniform protein fluorescence in the GUV membrane confirms that KvAP is incorporated in the GUV (i.e., it did not remain in the lipid film) and has not formed micron-scale (or larger) aggregates.
Figures 7, 8 and 9 show confocal images of lipid and protein fluorescence from GUVs produced by the lower-salt electroformation protocol, physiological salt electroformation protocol, and gel-assisted swelling protocol. The fluorescent lipid (magenta) and protein (green) signals have been scaled to the same average intensity, so that GUVs with a low/high number of proteins per unit area (protein density) have a magenta/green shade in the overlay images (right column), while GUVs with an average protein density are white. Isolated, defect-free GUVs were identified and the GUV size distribution is shown in Figure 10. Typically electroformation produces more defect-free GUVs than gel-assisted swelling, but the GUVs produced by electroformation are smaller. Figure 11 shows the protein density distribution inferred from the fluorescence of the GUVs. Electroformation with high-salt buffer produces GUVs in which the protein density varies greatly from GUV to GUV. The protein density of individual GUVs varies much less for electroformation with low salt buffer, while the protein density of GUVs produced by gel-assisted swelling is remarkably uniform.
The patch-clamp technique is a widely-used method for studying the function of voltage-gated ion channels, such as KvAP. In “inside-out” recordings, a clean glass “patch” pipette is used to excise a patch of membrane from a GUV. An electrode inside the patch pipette is then used to apply voltage, and measure the resultant current flowing through the membrane patch. The composition of the membrane patch can differ greatly from the rest of the cell/GUV31, but the “inside-out” configuration is still very useful for measuring the single channel conductance, ionic selectivity, and voltage-dependent gating. These three properties are an excellent way to establish that currents are not due to artifacts (e.g., gigaseal issues) or contaminants (e.g., bacterial porins from the purification), and there are functional KvAP channels in the GUVs.
Channel conductance is most easily measured in patches with only one or two active channels. In the example shown in Figure 12, no channel openings are observed when the membrane is held at -100 mV, whereas at +100 mV, individual channel openings can be clearly resolved. The current histogram shows two peaks corresponding to the closed and the open states, and fitting them with a double Gaussian function yields a single channel current of 10.9 ± 0.85 pA, corresponding to a conductance of 109.2 ± 8.5 pS (in 100 mM KCl). Note that the single channel conductance depends on the solution (especially potassium concentration) and membrane composition32,33.
Like many other K-channels, individual KvAP channels exhibit “chattering” bursts of rapid openings and closings. As demonstrated previously, potassium selectivity can be tested by using a different solution in the patch pipette (e.g., patch pipette solution 90 mM NaCl, 10 mM KCl)28.
Voltage-dependent gating is often studied in patches with multiple channels, so as to more easily obtain an ensemble average. While there is no obvious mechanism favoring the physiological (intracellular domain on GUV interior) and inverse (intra-cellular domain on the GUV exterior) insertion of KvAP in GUVs, in “inside-out” membrane patches the majority of functional channels have the “physiological” insertion28. Figure 13 shows the response of a membrane patch containing multiple (> 10) channels to a series of 5-second depolarizing steps. Between each step, the patch is held at -100 mV for 30 sec to allow channels with the “physiological” insertion to return to their resting state. When the potential is sufficiently negative (e.g., V < -60 mV) most of the current is due to the gigaseal leak, and the occasional openings of one or two channels which are likely to have the “inverse” insertion. For steps to more positive potentials, increasing numbers of channels are observed until there are so many that individual openings and closings can no longer be resolved. Thus, the open probability of the channel is clearly voltage-dependent. The kinetics of KvAP activation and inactivation differ considerably between Black Lipid Membranes (BLMs)26 and GUVs, but this is consistent with previous reports that Kv channel gating can be sensitive to the membrane composition and state34.
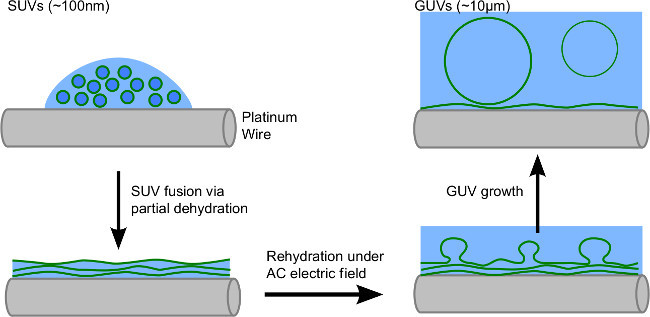 Figure 1.GUV Electroformation Schematic: Droplets containing SUVs are deposited onto an electrode. Partial dehydration of the solution causes the SUVs to fuse to form a stack of membranes. Buffer is then added and an AC electric field applied. As the film swells, individual membranes detach from the stack to form GUVs. (This figure has been modified from Aimon et al.28)
Figure 1.GUV Electroformation Schematic: Droplets containing SUVs are deposited onto an electrode. Partial dehydration of the solution causes the SUVs to fuse to form a stack of membranes. Buffer is then added and an AC electric field applied. As the film swells, individual membranes detach from the stack to form GUVs. (This figure has been modified from Aimon et al.28)
 Figure 2.GUV Electroformation Chamber. The chamber is milled out of a PTFE-block with three wells (10 mm diameter, 5 mm depth). Two 0.5 mm diameter platinum wires are separated by 3 mm (edge-to-edge distance) and are positioned close to the bottom of the chamber to facilitate imaging of the wires. Bottom and top cover slips are held in place with vacuum grease, and sealing paste prevents any leaks from the wire holes on the side. The AC generator is connected with alligator clips to the wires. The chamber is based on one developed by Ernesto Ambroggio and Luis Bagatolli. (This figure has been modified from Aimon et al.28)
Figure 2.GUV Electroformation Chamber. The chamber is milled out of a PTFE-block with three wells (10 mm diameter, 5 mm depth). Two 0.5 mm diameter platinum wires are separated by 3 mm (edge-to-edge distance) and are positioned close to the bottom of the chamber to facilitate imaging of the wires. Bottom and top cover slips are held in place with vacuum grease, and sealing paste prevents any leaks from the wire holes on the side. The AC generator is connected with alligator clips to the wires. The chamber is based on one developed by Ernesto Ambroggio and Luis Bagatolli. (This figure has been modified from Aimon et al.28)
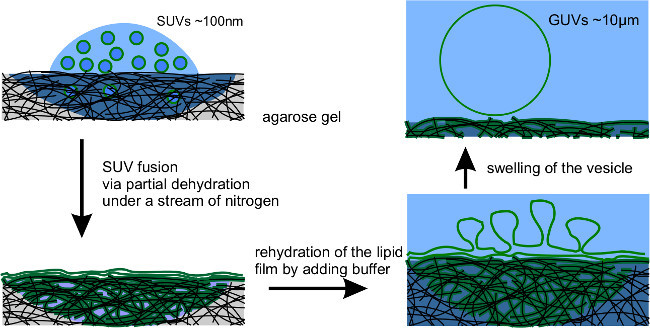 Figure 3.Gel-assisted Spontaneous Swelling Schematic: Droplets containing a SUV suspension are deposited onto an agarose gel. As the droplet dehydrates, the SUVs fuse to form a lipid film. When the growth buffer is added, the film rehydrates and GUVs form at the surface. GUVs grow to a size of ~10 µm by swelling and fusing with neighboring GUVs.
Figure 3.Gel-assisted Spontaneous Swelling Schematic: Droplets containing a SUV suspension are deposited onto an agarose gel. As the droplet dehydrates, the SUVs fuse to form a lipid film. When the growth buffer is added, the film rehydrates and GUVs form at the surface. GUVs grow to a size of ~10 µm by swelling and fusing with neighboring GUVs.
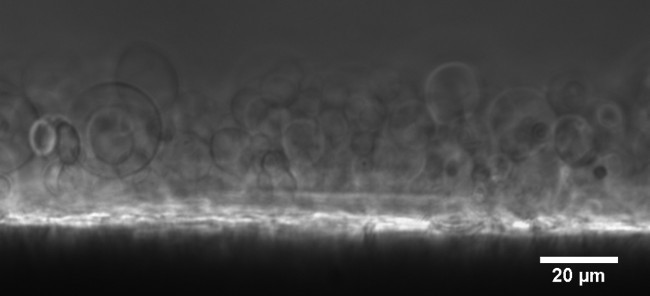 Figure 4.Representative image of DPhPC GUVs containing KvAP growing on the platinum wire in a high salt buffer. The GUVs resemble bunches of grapes along the wire. Phase Contrast image using a 40X LWD objective. Please click here to view a larger version of this figure.
Figure 4.Representative image of DPhPC GUVs containing KvAP growing on the platinum wire in a high salt buffer. The GUVs resemble bunches of grapes along the wire. Phase Contrast image using a 40X LWD objective. Please click here to view a larger version of this figure.
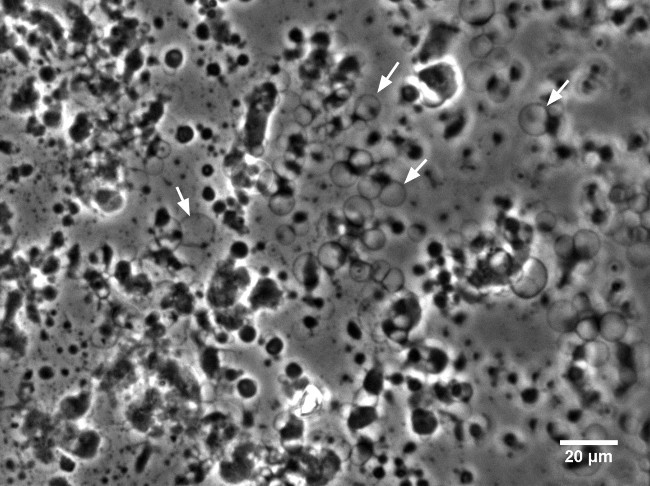 Figure 5.DPhPC GUVs containing KvAP swelling on agarose gel. The GUVs are visible as faint spheres with a diameter of ~10 µm. The dark/bright spots are lipid/agarose aggregates from which the vesicles swell. Phase Contrast image with 40X LWD objective. Please click here to view a larger version of this figure.
Figure 5.DPhPC GUVs containing KvAP swelling on agarose gel. The GUVs are visible as faint spheres with a diameter of ~10 µm. The dark/bright spots are lipid/agarose aggregates from which the vesicles swell. Phase Contrast image with 40X LWD objective. Please click here to view a larger version of this figure.
 Figure 6. Images of a defect-free GUV (Egg-PC:Egg-PA 9:1 by mass) containing KvAP labeled with Alexa-488. left: DIC, right: Alexa-488 epifluorescence. Excitation: 470/50 nm, emission: 545/75 nm. Note the uniform fluorescence from KvAP with no visible aggregates. Please click here to view a larger version of this figure.
Figure 6. Images of a defect-free GUV (Egg-PC:Egg-PA 9:1 by mass) containing KvAP labeled with Alexa-488. left: DIC, right: Alexa-488 epifluorescence. Excitation: 470/50 nm, emission: 545/75 nm. Note the uniform fluorescence from KvAP with no visible aggregates. Please click here to view a larger version of this figure.
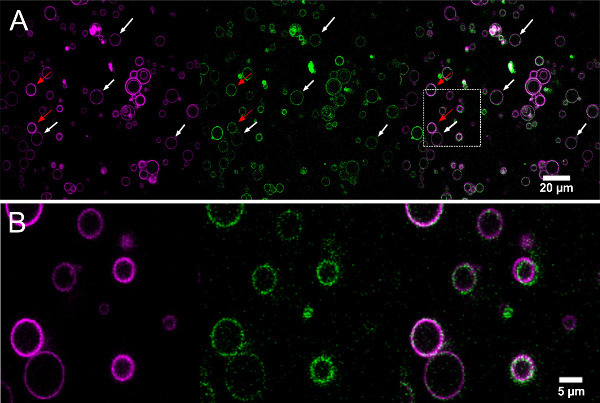 Figure 7.Confocal images of lipid (magenta) and protein (green) fluorescence from electroformed GUVs grown in a low salt buffer (Egg-PC:Egg-PA 9:1 by mass). (A) The white arrows mark (likely) GUVs, while the red arrow marks a potentially bi-lamellar vesicle with higher lipid fluorescence. Note that the fluorescence intensity is brighter in the center of this image because of the extremely large field of view. (B) Zoom showing a small group of GUVs. Left (magenta): TexasRed-DHPE excitation: 543 nm laser line, emission: 605/70 nm. Center (green): KvAP labeled with Alexa-488 excitation: 488 nm laser line, emission: 515/30 nm. Right: overlay. Please click here to view a larger version of this figure.
Figure 7.Confocal images of lipid (magenta) and protein (green) fluorescence from electroformed GUVs grown in a low salt buffer (Egg-PC:Egg-PA 9:1 by mass). (A) The white arrows mark (likely) GUVs, while the red arrow marks a potentially bi-lamellar vesicle with higher lipid fluorescence. Note that the fluorescence intensity is brighter in the center of this image because of the extremely large field of view. (B) Zoom showing a small group of GUVs. Left (magenta): TexasRed-DHPE excitation: 543 nm laser line, emission: 605/70 nm. Center (green): KvAP labeled with Alexa-488 excitation: 488 nm laser line, emission: 515/30 nm. Right: overlay. Please click here to view a larger version of this figure.
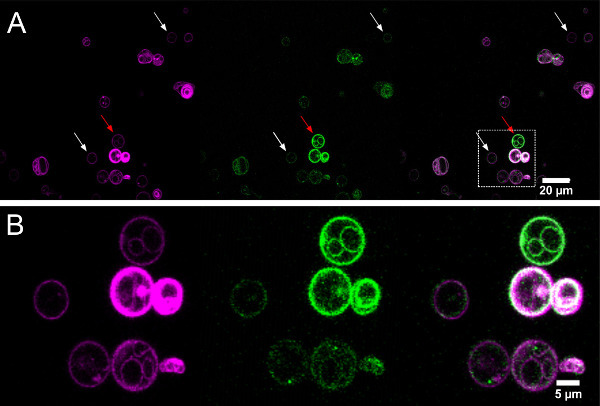 Figure 8.Confocal images of lipid (magenta) and protein (green) fluorescence from electroformed GUVs grown in physiological salt concentration (Egg-PC:Egg-PA 9:1 by mass). (A) The GUVs (white arrows show likely unilamellar examples) are more sparse compared to the low salt protocol and can have extremely different protein concentrations (red arrow). (B) Zoom showing a small group of GUVs. Left (magenta): TexasRed-DHPE excitation: 543 nm laser line, emission: 605/70 nm middle (green): KvAP labeled with Alexa-488 excitation: 488 nm laser line, emission: 515/30 nm. Right: Overlay. Please click here to view a larger version of this figure.
Figure 8.Confocal images of lipid (magenta) and protein (green) fluorescence from electroformed GUVs grown in physiological salt concentration (Egg-PC:Egg-PA 9:1 by mass). (A) The GUVs (white arrows show likely unilamellar examples) are more sparse compared to the low salt protocol and can have extremely different protein concentrations (red arrow). (B) Zoom showing a small group of GUVs. Left (magenta): TexasRed-DHPE excitation: 543 nm laser line, emission: 605/70 nm middle (green): KvAP labeled with Alexa-488 excitation: 488 nm laser line, emission: 515/30 nm. Right: Overlay. Please click here to view a larger version of this figure.
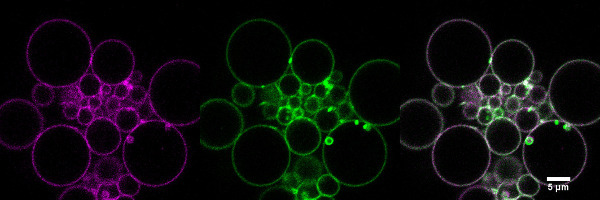 Figure 9. Confocal image of lipid (magenta) and protein (green) fluorescence of GUVs (DPhPC) formed by gel-assisted swelling with a physiological salt concentration buffer. GUVs show a more homogeneous protein density than electroformed GUVs prepared with physiological buffer. Left (magenta): BPTR-Cer 0.1% excitation: 543 nm laser line, emission: 605/70 nm. Middle (green): KvAP labeled with Alexa-488 excitation: 488 nm laser line, emission: 515/30 nm. Right: merge of the two channels. Please click here to view a larger version of this figure.
Figure 9. Confocal image of lipid (magenta) and protein (green) fluorescence of GUVs (DPhPC) formed by gel-assisted swelling with a physiological salt concentration buffer. GUVs show a more homogeneous protein density than electroformed GUVs prepared with physiological buffer. Left (magenta): BPTR-Cer 0.1% excitation: 543 nm laser line, emission: 605/70 nm. Middle (green): KvAP labeled with Alexa-488 excitation: 488 nm laser line, emission: 515/30 nm. Right: merge of the two channels. Please click here to view a larger version of this figure.
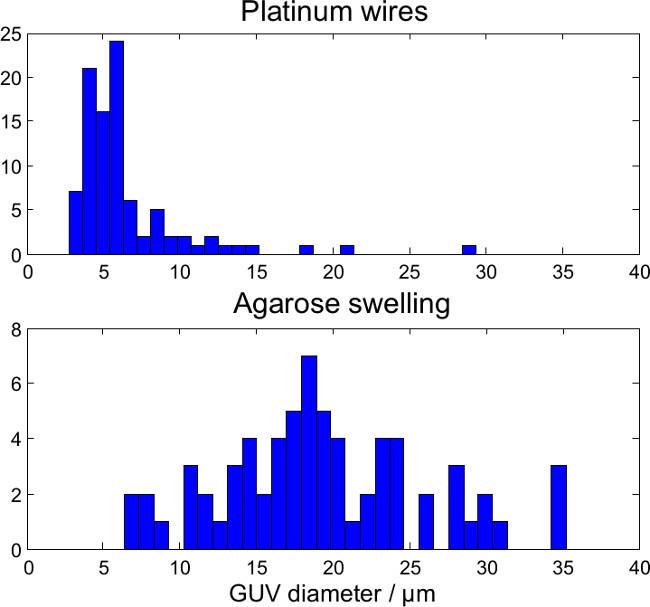 Figure 10.
Size distribution of defect-free proteo-GUVs (DPhPC) grown by electroformation in low salt buffer (top, N = 94) or gel-assisted agarose swelling (bottom, N = 68).
Figure 10.
Size distribution of defect-free proteo-GUVs (DPhPC) grown by electroformation in low salt buffer (top, N = 94) or gel-assisted agarose swelling (bottom, N = 68).
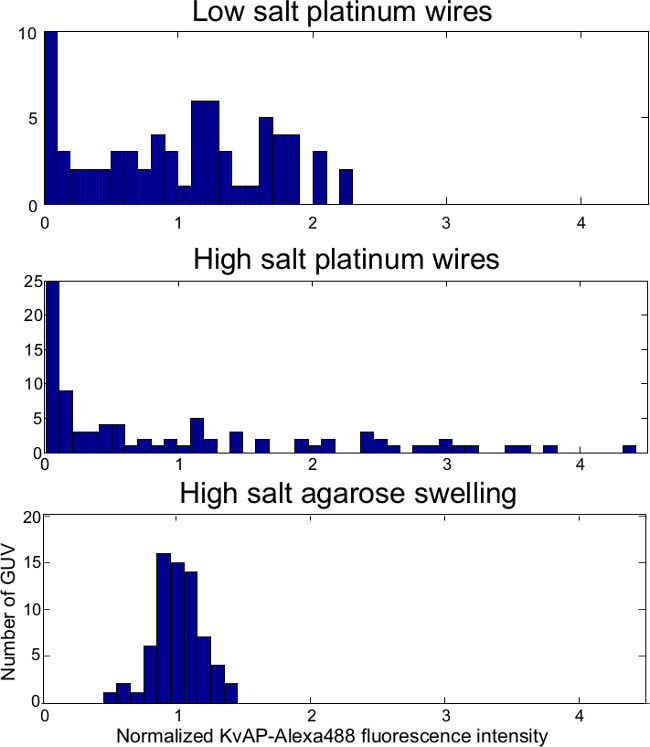 Figure 11.GUV protein density histograms: The protein density (number of proteins per unit area) of GUVs electroformed with low salt buffer (5 mM KCl, DPhPC) varies less than with physiological salt concentration (100 mM KCl, Egg-PC:Egg-PA 9:1 by mass). The protein density of GUVs (DPhPC) grown by gel-assisted swelling in physiological salt buffer shows the least variation. Protein density is proportional to KvAP-A488 fluorescence intensity for these concentrations28, and in each histogram the fluorescence intensities are normalized by the mean of the distribution. (The middle panel has been modified from Aimon et al.28)
Figure 11.GUV protein density histograms: The protein density (number of proteins per unit area) of GUVs electroformed with low salt buffer (5 mM KCl, DPhPC) varies less than with physiological salt concentration (100 mM KCl, Egg-PC:Egg-PA 9:1 by mass). The protein density of GUVs (DPhPC) grown by gel-assisted swelling in physiological salt buffer shows the least variation. Protein density is proportional to KvAP-A488 fluorescence intensity for these concentrations28, and in each histogram the fluorescence intensities are normalized by the mean of the distribution. (The middle panel has been modified from Aimon et al.28)
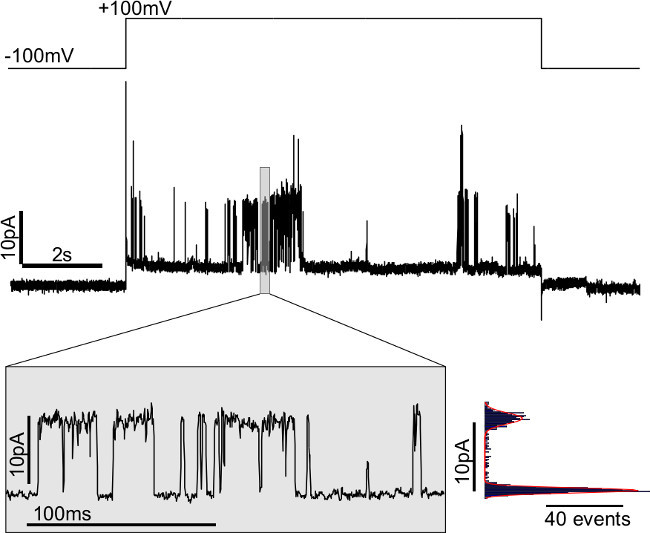 Figure 12.Single channel activity of GUV Membrane Patches (DPhPC 'inside-out' voltage convention). The GUV was grown in 100 mM KCl, 5 mM HEPES pH 7.4 on platinum wires. Single channels open after applying 100 mV potential on the patch. To the right of the inset is a histogram used to calculate the single channel conductance. The red line is a fit to a double Gaussian function with maxima at 4.70 ± 0.27 pA and 15.62 ± 0.58 pA, corresponding to a conductance of 109.2 ± 8.5 pS. The trace was filtered at 10 kHz with a 4-pole Bessel filter and recorded at 50 kHz.
Figure 12.Single channel activity of GUV Membrane Patches (DPhPC 'inside-out' voltage convention). The GUV was grown in 100 mM KCl, 5 mM HEPES pH 7.4 on platinum wires. Single channels open after applying 100 mV potential on the patch. To the right of the inset is a histogram used to calculate the single channel conductance. The red line is a fit to a double Gaussian function with maxima at 4.70 ± 0.27 pA and 15.62 ± 0.58 pA, corresponding to a conductance of 109.2 ± 8.5 pS. The trace was filtered at 10 kHz with a 4-pole Bessel filter and recorded at 50 kHz.
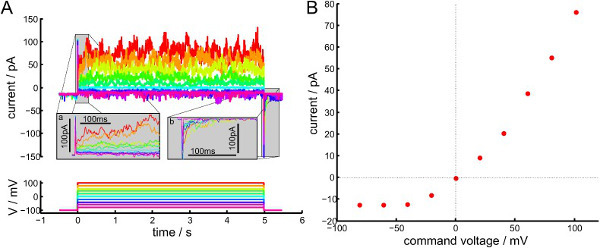 Figure 13.Voltage-dependent gating channels in the membrane patch from a GUV formed on agarose in a high salt buffer (DPhPC, 'inside-out' voltage convention). (A) Response of patch membrane current to a transient step in voltage. Pipette and bath solutions both contained 100 mM KCl, and the membrane was held for 30 sec at -100 mV between successive voltage steps. On a close inspection one can see that the trace contains 1 or 2 channels that seem to open with negative voltages. Inset a) shows a zoom of the delayed opening and inset b) the delayed closing of the channel. Currents were filtered with a 4-pole Bessel filter at 10 kHz and recorded at 51.3 kHz. Offline the trace was down-sampled to 513 Hz. The negative capacitance transient is cut off at -150 pA. (B) Average current (0.25 sec < t < 5 sec) versus step voltage. Currents at positive voltages are larger because the channel open probability is voltage-dependent. Please click here to view a larger version of this figure.
Figure 13.Voltage-dependent gating channels in the membrane patch from a GUV formed on agarose in a high salt buffer (DPhPC, 'inside-out' voltage convention). (A) Response of patch membrane current to a transient step in voltage. Pipette and bath solutions both contained 100 mM KCl, and the membrane was held for 30 sec at -100 mV between successive voltage steps. On a close inspection one can see that the trace contains 1 or 2 channels that seem to open with negative voltages. Inset a) shows a zoom of the delayed opening and inset b) the delayed closing of the channel. Currents were filtered with a 4-pole Bessel filter at 10 kHz and recorded at 51.3 kHz. Offline the trace was down-sampled to 513 Hz. The negative capacitance transient is cut off at -150 pA. (B) Average current (0.25 sec < t < 5 sec) versus step voltage. Currents at positive voltages are larger because the channel open probability is voltage-dependent. Please click here to view a larger version of this figure.
Discussion
Biomimetic model systems are an important tool for studying the properties and interactions of proteins and membranes. Compared to other reconstituted systems like BLMs or supported lipid membranes, GUV based systems present several opportunities including considerable control of membrane composition, tension and geometry, as well as being truly oil-free. However, incorporating transmembrane proteins, such as KvAP, into GUVs requires significant adaptations of conventional protocols for lipid-only GUVs. The electroformation protocol presented here was previously characterized and used for studying the biophysical principles of membrane protein distribution and dynamics in curved membranes2,4,35. This work demonstrates a new gel-assisted swelling protocol, adding to the set of methods for protein reconstitution in GUVs. Both protocols can produce defect-free GUVs containing high densities of KvAP, and measurements on inside-out patches confirm that these GUVs contain functional potassium-selective, voltage-dependent channels.
The two approaches have different strengths and weaknesses. When low salt conditions can be used, electroformation offers a good compromise between GUV yield and uniform protein density. Electroformation still gives reasonable yields with physiological salt concentrations, but the protein density can vary greatly between GUVs (see Figures 8 and 11). The density variations seem to be linked to the duration of electroformation, as low-salt buffer growth can also have substantial density variations if the growth continues much longer than 2 hr. In contrast, GUVs produced by gel-assisted swelling have a remarkably uniform protein density, even for physiological buffers. However, the fraction of multi-lamellar vesicles is greater, and agarose auto-fluorescence complicates quantification of low protein densities16. Using polyvinyl alcohol in place of agarose has been reported to improve the gel-assisted GUV yield when lipids were deposited from chloroform20, but we were unable to produce GUVs using polyvinyl alcohol with SUV solutions. If a lower yield of defect-free GUVs is acceptable, gel-assisted swelling has a clear advantage for experiments that require a uniform protein distribution.
Electroformation and gel-assisted swelling also have quite different equipment requirements. The gel-assisted swelling protocol uses little specialized equipment except for the plasma cleaner, which is not essential as there are many alternative methods to produce clean, hydrophilic glass. In contrast, electroformation requires a custom chamber with wire electrodes. Platinum wire is expensive, but for this protocol GUVs grew more readily with platinum than titanium wires, and GUVs did not grow on ITO slides when using physiological salt concentrations. The diameter of the electrodes (0.5 mm) is a compromise between electrode surface and price. The chamber shown in Figure 2 is based on a design by Luis Bagatolli15 and Ernesto Ambroggio, and was machined from Polytetrafluoroethylene (PTFE) to allow cleaning with most solvents. However, polyacetal or polyvinyl chloride (PVC) should also work well. The ability to remove the platinum wires for aggressive cleaning is important, and when first learning the protocol, it is very helpful to be able to observe GUV growth in situ through the bottom cover slide. The smaller, closed wells prevent solution from sloshing backwards and forwards and also allow tests of several lipid compositions in parallel. However, a special custom chamber is not essential when starting out. For example, simple, single-well chambers can be improvised by tacking two wires down to the bottom of a small petri dish, or using sealing wax to sandwich them between two glass slides, or simply poking them through the cap of a small vial.
Both the electroformation and gel-assisted swelling protocols should produce a sufficient number of defect-free GUVs for micro-manipulation and patch-clamp experiments. However, the GUV yield depends sensitively on the formation of the bilayer stack when the SUV solution is partially dehydrated. If difficulties are encountered in growing GUVs (i.e., no or few GUVs are visible in the growth chamber), it can be very helpful to grow lipid-only GUVs using a lipid/chloroform solution in place of SUVs15,16 (and a low salt buffer). If GUVs do not grow well from a lipid/chloroform film then something fundamental is wrong (e.g., incorrect lipid solutions or buffers, grease or dirt on the electrodes, incorrect voltage or frequency, etc.). However, if GUVs grow from lipid/chloroform films but not the SUV films, then the issue is likely to be with the partial dehydration.
The partial dehydration step is most easily optimized using lipid-only SUVs because there is no risk of “denaturing” lipids by excessive dehydration. For electroformation, it can be helpful to examine the wires after the SUV droplets have been allowed to dry to check there is bright lipid fluorescence at each spot where a droplet was deposited. If the lipid fluorescence disappears after the chamber is filled with growth solution, then either the growth solution has to be added more carefully, or the SUVs need to dehydrate for longer so the film attaches more firmly to the wire. During the growth, make sure neither the wires nor solution move within the chamber (e.g., when putting the chamber on the microscope) to avoid stripping GUVs off the wires. When GUVs grow well, they are usually easy to see in phase contrast images. However, smaller GUVs are often clearer using lipid fluorescence, which is also helpful for seeing how the membrane stack has changed during the growth. Examine all surfaces of the wires (inside, outside, top and bottom) in all wells as well as the chamber floor before discarding the growth, because yields can vary considerably from one spot to another.
The handling and storage of GUVs is simple compared to cells, but GUVs are quite sensitive to osmotic stress, shear forces and adhesion. Smooth, gentle motions are important when harvesting or transferring GUVs, and it is important to passivate surfaces to prevent GUVs from adhering and exploding. However, for patch clamp experiments the chamber must be thoroughly rinsed after passivation so that the passivation solution cannot coat the patch pipettes and prevent gigaseal formation. For passivation, the beta-casein treatment is simple and effective, and compared to bovine serum albumin, which has a lipid transport function, beta-casein has more limited interactions with lipid bilayers and should be preferred when working with GUVs36. By varying the incubation time, it is possible to get GUVs to adhere without exploding. However, the GUVs will not adhere to the cover slide as firmly as cells, and so care must still be taken during any procedure that can induce flow in the chamber (e.g., buffer perfusion, moving the chamber).
Patch-clamp recording is a classical method for studying voltage-gated ion channels like KvAP and this protocol is derived from standard techniques for obtaining “inside-out” patches from adherent cells. A standard patch-clamp set-up in which the patch pipette descends obliquely (e.g., 30–60 degrees from horizontal) into a small Petri dish should work well. However, clearer images of the patch pipette and gigaseal region can be obtained using a chamber in which cover-slips form the chamber top and bottom (~ 1 mm separation) so the patch pipette can enter horizontally from the side. Because large patch pipette pressures are not needed, the pressure can conveniently be controlled with a syringe and monitored with any simple pressure meter (e.g., improvised water manometer). It can be helpful to first practice patch clamp experiments with lipid-only GUVs grown using a lipid/chloroform solution and low-salt buffer. Because the yield of defect-free GUVs is very high, there will be plenty of perfect GUVs to work with even if many are destroyed during the harvesting and transfer to the experimental chamber.
With a little practice, excised membrane patches with stable gigaseals can be readily obtained from DPhPC GUVs and KvAP-DPhPC GUVs. To achieve a high success rate, it helps to carefully select round, but slightly-fluctuating, defect-free GUVs and check that the patch pipette is clean (looking for lipids in fluorescence) before attempting to form the gigaseal. When a membrane “tongue” enters the patch pipette, the gigaseal typically forms quickly (< 1 sec) without any need for strong suction or specific holding voltage. While a bad seal may improve as the membrane crawls further into the patch pipette, often the seal remains poor because the pipette interior was contaminated and it is necessary to start over with a fresh patch pipette and GUV. DPhPC forms very stable membrane patches (tens of minutes) with an excellent seal resistance even at high voltages (e.g., ±150 mV). SOPC:cholesterol (3:1 by mole) can also form very stable patches but can require higher suction to seal, while Egg-PC patches seem to break more easily.
For longer patch-clamp sessions it may be necessary to adjust the osmolarity of the chamber solution. If the osmolarity is too much lower than the GUV growth buffer, GUVs become swollen, tense and spherical and it may not be possible to aspirate the GUV membrane far enough into the patch pipette to form a gigaseal. This can often be fixed by simply waiting 10 or 20 min as evaporation from the chamber increases the external solution osmolarity until the GUVs deflate and begin to fluctuate. Conversely, if the chamber is left open for too long the osmolarity of the external solution can increase until GUVs deflate, tubulate and bud. This can be avoided by blocking evaporation from the chamber (e.g., with mineral oil) or periodically adding distilled water to replace the water that has evaporated.
Because proteins can be excluded from excised membrane patches31, the number of active channels in an excised patch is not simply related to the density of protein in the GUV membrane. Fluorescence measurements suggest the concentration of KvAP in the patch membrane is much lower than the GUV28 and it can be quite easy to obtain patches containing only a small number of channels. However, if patches contain too many channels for single channel recording, the obvious steps of using patch pipettes with smaller tips and/or lowering the protein-to-lipid density in the SUV mix are effective. In contrast, to perform ensemble measurements on patches containing many channels, it can be helpful to start with a relatively high protein density (e.g., 1:10, protein to lipid by weight), use protein fluorescence to select GUVs with a high protein density, use larger patch pipettes (e.g., 2 to 3 micron tip diameter) and aspirate quickly to try to form the seal rapidly. Clearly, the “whole-cell”-type configuration (i.e., 'whole-GUV') would be ideal to characterize all the channels in a GUV, but unfortunately the “whole-GUV” configuration poses several technical issues37.
The solutions, lipids and protein concentrations in this tutorial are merely provided as a starting point, and can be altered to suit the needs of a particular experiment following a few considerations.
All solutions should include a pH buffer such as HEPES or TRIS to ensure proteins are not exposed to extremes of pH. The SUV buffer should have as low a concentration of solutes as the protein will tolerate (e.g., 5 mM salt or lower), as solute concentrations increase during the partial dehydration step and high salt concentrations can denature the protein or cause the lipid film to rapidly delaminate during the rehydration step. Small amounts of sugars such as trehalose (e.g., 1 mM to 5 mM) are thought to protect the protein during dehydration23. While trehalose has been implicated in anhydrobiosis and is believed to protect membrane and proteins against dessication38, sucrose or glucose may work equally well.
For the growth buffer, the salt concentration is especially important as this will influence the parameters for optimal GUV growth (e.g., electroformation voltage, frequency and duration). In contrast, the primary constraint for the observation buffer is that it should have the same osmolarity as the growth buffer. The inclusion of sucrose and/or glucose in the “growth buffer” and “observation buffer” can be helpful to ensure GUVs sediment to the bottom of the observation chamber, while a difference in refractive index between GUV interior and exterior helps with phase contrast or DIC microscopy. Electrophysiologists often include calcium or magnesium ions to the bath and/or patch pipette solutions to enhance gigaseal formation with cell membranes, but these do not appear to be essential for GUVs. Indeed, divalent ions such as magnesium and calcium can induce lipid phase separation and facilitate adhesion, so if these problems arise it may be helpful to add 1 mM EDTA.
Clearly, a key attraction of reconstituted systems when compared to cells is the ability to control lipid composition. DPhPC GUVs grow well and form stable excised membrane patches, and these protocols have also worked effectively for lipid mixtures containing phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidic acid (PA), phosphatidylserine (PS) and cholesterol. However, GUV growth is sensitive to both lipid composition and buffers15, and so protocol parameters (e.g., amount of lipid deposited, electroformation voltage/frequency) may need to be adjusted for lipid mixtures containing high concentrations of PE, charged lipids (PG, PA, PS), or cholesterol. When starting out, Egg-PC, DOPC or DPhPC are a good first choice, and it is also very helpful to include a fluorescent lipid to observe GUV growth and to distinguish GUVs from multilamellar vesicles with two or more bilayers. Lipid mixtures can be prepared by combining SUV suspensions of stock solutions, as the lipids mix during the partial dehydration step (provided the temperature is higher than any individual phase transition temperature). Using higher SUV concentration (e.g., 10 mg/ml) stock solutions allows considerable flexibility, and these can then be diluted to 3 mg/ml before dehydrating the suspension.
If attempting to adapt these protocols to other trans-membrane proteins, it is very important to be able to both directly observe the incorporation of protein into the GUVs and test protein function. While not an issue with KvAP, there is always the possibility that during the rehydration step the trans-membrane protein will remain in the membrane stack leading to the formation of lipid-only GUVs. Fluorescent labeling of the protein is very convenient as it provides a rapid and unequivocal way to observe protein incorporation into GUVs and also check for aggregation within GUVs. It is also very important to test protein function in the GUVs to confirm that the protein was not damaged during the reconstitution process. For ion channels like KvAP, patch-clamp measurements can establish the presence of functional channels in the GUVs. However, a fluorescently-labeled, high-affinity ligand (e.g., toxin, substrate or anti-body) would also be very helpful for testing the state of proteins in GUVs.
In summary, this article demonstrates how to produce proteo-GUVs containing the voltage gated potassium channel KvAP and characterize them using fluorescence microscopy and electrophysiology. Hopefully these methods can be adapted to new classes of membrane proteins and provide a foundation for more complex in-vitro systems for studying and building up living matter from its fundamental components.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Susanne Fenz for discussing the possibility of reconstituting proteins by agarose swelling, Feng Ching Tsai for current measurements, and present and former members of the Bassereau group for support and assistance. The project was funded by the Agence Nationale de la Recherche (grant BLAN-0057-01), by the European Commission (NoE SoftComp), by the Université Pierre et Marie Curie (grant from the FED21, Dynamique des Systèmes Complexes). M.G. was supported by an Institut Curie International PhD Fellowship, S.A. by a fellowship from the Fondation pour la Recherche Médicale, G.E.S.T. by a Marie Curie Incoming International Fellowship from the European Commission and a grant from the Université Pierre et Marie Curie. The publication fees were covered by the Labex ‘CelTisPhyBio’ (ANR-11-LABX0038).
References
- Schwille P. Bottom-Up Synthetic Biology: Engineering in a Tinkerer’s World. Science. 2011;333(6047):1252–1254. doi: 10.1126/science.1211701. [DOI] [PubMed] [Google Scholar]
- Aimon S, et al. Membrane Shape Modulates Transmembrane Protein Distribution. Developmental Cell. 2014;28(2):212–218. doi: 10.1016/j.devcel.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux A, et al. Membrane curvature controls dynamin polymerization. Proceedings of the National Academy of Sciences. 2010;107(9):4141–4146. doi: 10.1073/pnas.0913734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanov YA, et al. Mobility in geometrically confined membranes. Proceedings of the National Academy of Sciences. 2011;108(31):12605. doi: 10.1073/pnas.1102646108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorre B, et al. Curvature-driven lipid sorting needs proximity to a demixing point and is aided by proteins. Proceedings of the National Academy of Sciences. 2009;106(14):5622–5626. doi: 10.1073/pnas.0811243106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faris MDEA, et al. Membrane Tension Lowering Induced by Protein Activity. Physical Review Letters. 2009;102(3):038102. doi: 10.1103/PhysRevLett.102.038102. [DOI] [PubMed] [Google Scholar]
- Streicher P, et al. Integrin reconstituted in GUVs: A biomimetic system to study initial steps of cell spreading. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2009;1788(10):2291–2300. doi: 10.1016/j.bbamem.2009.07.025. [DOI] [PubMed] [Google Scholar]
- Walde P, Cosentino K, Engel H, Stano P. Giant Vesicles: Preparations and Applications. ChemBioChem. 2010;11(7):848–865. doi: 10.1002/cbic.201000010. [DOI] [PubMed] [Google Scholar]
- Liu AP, Fletcher DA. Biology under construction: in vitro reconstitution of cellular function. Nature Reviews Molecular Cell Biology. 2009;10(9):644–650. doi: 10.1038/nrm2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sens P, Johannes L, Bassereau P. Biophysical approaches to protein-induced membrane deformations in trafficking. Current Opinion in Cell Biology. 2008;20(4):476–482. doi: 10.1016/j.ceb.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Pautot S, Frisken BJ, Weitz DA. Production of Unilamellar Vesicles Using an Inverted Emulsion. Langmuir. 2003;19(7):2870–2879. [Google Scholar]
- Stachowiak JC, et al. Unilamellar vesicle formation and encapsulation by microfluidic jetting. Proceedings of the National Academy of Sciences. 2008;105(12):4697–4702. doi: 10.1073/pnas.0710875105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves JP, Dowben RM. Formation and properties of thin-walled phospholipid vesicles. Journal of Cellular Physiology. 1969;73(1):49–60. doi: 10.1002/jcp.1040730108. [DOI] [PubMed] [Google Scholar]
- Angelova MI, Soléau S, Méléard P, Faucon F, Bothorel P. Preparation of giant vesicles by external AC electric fields. Kinetics and applications. Trends in Colloid and Interface Science VI. 1992;89:161–176. [Google Scholar]
- Bagatolli LA, Pott T. Giant Unilamellar Vesicle Electroformation. Methods in Enzymology. 2009;465:161–176. doi: 10.1016/S0076-6879(09)65009-6. [DOI] [PubMed] [Google Scholar]
- Horger KS, Estes DJ, Capone R, Mayer M. Films of Agarose Enable Rapid Formation of Giant Liposomes in. Solutions of Physiologic Ionic Strength. Journal of the American Chemical Society. 2009;131(5):1810–1819. doi: 10.1021/ja805625u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campillo C, et al. Unexpected Membrane Dynamics Unveiled by Membrane Nanotube Extrusion. Biophysical Journal. 2013;104(6):1248–1256. doi: 10.1016/j.bpj.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok R, Evans E. Thermoelasticity of large lecithin bilayer vesicles. Biophysical Journal. 1981;35(3):637–652. doi: 10.1016/S0006-3495(81)84817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathivet L, Cribier S, Devaux PF. Shape change and physical properties of giant phospholipid vesicles prepared in the presence of an AC electric field. Biophysical Journal. 1996;70(3):1112–1121. doi: 10.1016/S0006-3495(96)79693-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger A, et al. Gel-Assisted Formation of Giant Unilamellar Vesicles. Biophysical Journal. 2013;105(1):154–164. doi: 10.1016/j.bpj.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezi M, Di Cicco A, Bassereau P, Levy D. Detergent-mediated incorporation of transmembrane proteins in giant unilamellar vesicles with controlled physiological contents. Proceedings of the National Academy of Sciences. 2013;110(18):7276–7281. doi: 10.1073/pnas.1303857110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahya N, Pecheur EI, de Boeij WP, Wiersma DA, Hoekstra D. Reconstitution of membrane proteins into giant unilamellar vesicles via peptide-induced fusion. Biophysical Journal. 2001;81(3):1464–1474. doi: 10.1016/S0006-3495(01)75801-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeven MK, et al. Distribution, lateral mobility and function of membrane proteins incorporated into giant unilamellar vesicles. Biophysical Journal. 2005;88(2):1134–1142. doi: 10.1529/biophysj.104.053413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard P, Prost J, Bassereau P. Passive or Active Fluctuations in Membranes Containing Proteins. Physical Review Letters. 2005;94(8):088102. doi: 10.1103/PhysRevLett.94.088102. [DOI] [PubMed] [Google Scholar]
- Betaneli V, Petrov EP, Schwille P. The Role of Lipids in VDAC Oligomerization. Biophysical Journal. 2012;102(3):523–531. doi: 10.1016/j.bpj.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruta V, Jiang Y, Lee A, Chen J, MacKinnon R. Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature. 2003;422(6928):180–185. doi: 10.1038/nature01473. [DOI] [PubMed] [Google Scholar]
- Jiang Y, et al. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423(6935):33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- Aimon S, et al. Functional Reconstitution of a Voltage-Gated Potassium Channel in Giant Unilamellar Vesicles. PLoS ONE. 2011;6(10):e25529. doi: 10.1371/journal.pone.0025529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Zheng H, Shi L, Jiang Q-X. Reconstitution of a Kv Channel into Lipid Membranes for Structural and Functional Studies. Journal of Visualized Experiments. 2013. [DOI] [PMC free article] [PubMed]
- Baykal-Caglar E, Hassan-Zadeh E, Saremi B, Huang J. Preparation of giant unilamellar vesicles from damp lipid film for better lipid compositional uniformity. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2012;1818(11):2598–2604. doi: 10.1016/j.bbamem.2012.05.023. [DOI] [PubMed] [Google Scholar]
- Suchyna TM, Markin VS, Sachs F. Biophysics and Structure of the Patch and the Gigaseal. Biophysical Journal. 2009;97(3):738–747. doi: 10.1016/j.bpj.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sinauer Associates, Inc; 2001. [Google Scholar]
- Finol-Urdaneta RK, McArthur JR, Juranka PF, French RJ, Morris CE. Modulation of KvAP unitary conductance and gating by 1-alkanols and other surface active agents. Biophysical journal. 2010;98(5):762–772. doi: 10.1016/j.bpj.2009.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, MacKinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proceedings of the National Academy of Sciences. 2008;105(49):19276–19281. doi: 10.1073/pnas.0810187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quemeneur F, et al. Shape matters in protein mobility within membranes. Proceedings of the National Academy of Sciences of the United States of America. 2014. [DOI] [PMC free article] [PubMed]
- Parc AL, Leonil J, Chanat E. αS1-casein, which is essential for efficient ER-to-Golgi casein transport, is also present in a tightly membrane-associated form. BMC Cell Biology. 2010;11(1):1–15. doi: 10.1186/1471-2121-11-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten M, Toombes GES, Aimon S, Bassereau P. Studying Voltage Dependent Proteins with Giant Unilamellar Vesicles in a “Whole Cell” Configuration. Biophysical Journal. 2013;104(2):466a. [Google Scholar]
- Crowe LM, Reid DS, Crowe JH. Is trehalose special for preserving dry biomaterials. Biophysical Journal. 1996;71(4):2087–2093. doi: 10.1016/S0006-3495(96)79407-9. [DOI] [PMC free article] [PubMed] [Google Scholar]