

Abstract
Human disease specific neuronal cultures are essential for generating in vitro models for human neurological diseases. However, the lack of access to primary human adult neural cultures raises unique challenges. Recent developments in induced pluripotent stem cells (iPSC) provides an alternative approach to derive neural cultures from skin fibroblasts through patient specific iPSC, but this process is labor intensive, requires special expertise and large amounts of resources, and can take several months. This prevents the wide application of this technology to the study of neurological diseases. To overcome some of these issues, we have developed a method to derive neural stem cells directly from human adult peripheral blood, bypassing the iPSC derivation process. Hematopoietic progenitor cells enriched from human adult peripheral blood were cultured in vitro and transfected with Sendai virus vectors containing transcriptional factors Sox2, Oct3/4, Klf4, and c-Myc. The transfection results in morphological changes in the cells which are further selected by using human neural progenitor medium containing basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF). The resulting cells are characterized by the expression for neural stem cell markers, such as nestin and SOX2. These neural stem cells could be further differentiated to neurons, astroglia and oligodendrocytes in specified differentiation media. Using easily accessible human peripheral blood samples, this method could be used to derive neural stem cells for further differentiation to neural cells for in vitro modeling of neurological disorders and may advance studies related to the pathogenesis and treatment of those diseases.
Keywords: Developmental Biology, Issue 95, Hematopoietic progenitor cell, neural stem cell, blood, Sendai virus, neuron, differentiation
Introduction
In vitro neuronal cultures have been used as a fundamental tool for studies of neurological diseases. Primary animal (mostly rodent) neural cultures1,2 and human neural cell lines derived from gliomas or other tumors are the most commonly used in such studies. However, it has been recognized that there are significant differences between rodent and human cells. Many findings based on rodents cannot be translated to humans. Furthermore, with the rapid developments in analyzing mass genomic information and the relatively easy gene editing and whole genome sequencing, the trend is more and more geared to discovering disease prone genes and delineating their functions and roles in specific diseases, which makes the few human neuronal cell lines have only limited usage. Theoretically, human primary neural cultures derived from samples of patient nervous system are the best choice but they are impossible to obtain; hence alternative methods are necessary. In recent years, some approaches have been pursued, with two being the most distinguishable. Following the development of the technique of generating induced pluripotent stem cells (iPSC) using mouse and human somatic cells3,4, neural cells could be further differentiated from them5-7. However, generating and characterizing iPSC demands intensive labor, techniques, and time input, sometimes even prohibitively. Shortly after, another approach was developed to directly transform neuronal cells from somatic cells8,9. As the resulting neurons are non-proliferative, it limits its application in intensive studies and drug screening, which requires a large amount of cells. To take advantages of both techniques, direct derivation of neural stem/progenitor cells from somatic cells has been explored by several groups10-12, which bypasses the tedious process of iPSC generation and characterization but still provides a decent number of neural stem cells for later neural differentiation. We have previously shown that following the introduction of the Yamanaka transcription factors into hematopoietic progenitor cells, neural stem cells could be directly generated using a neural progenitor cell selecting medium13. Here, we report the method in detail.
Protocol
1. Enrichment of Hematopoietic Progenitor Cells from Adult Whole Blood
NOTE: Hematopoietic progenitor cells or CD34+ cells can be purified from peripheral blood mononuclear cells (PBMCs) derived from a variety of sources including cord blood, leukapheresis material and whole blood using density gradient centrifugation based methods. The method listed here uses whole blood as an example.
- Isolation of PBMCs from Whole Blood:
- Add 4.5 ml of lymphocyte separation medium to the SepMate tube by pipetting it through the central hole of the insert.
- Dilute the blood sample with an equal volume of sterile DPBS containing 2% human serum (v/v). For example, dilute 5 ml of sample with 5 ml of DPBS + 2% human serum.
- Add the diluted sample by pipetting it down the side of the tube. Take care not to pour the sample directly through the central hole.
- Centrifuge at 1,200 x g for 10 min at RT.
- Carefully remove the supernatant above the PMBC layer.
- Collect the PBMC layer (cloudy) into a new tube.
- Add 15 ml of Dulbecco’s Phosphate-buffered Saline (DPBS) containing 2% human serum into the tube and centrifuge at 150 x g for 10 min at RT with the brake off.
- Discard the supernatant and resuspend the pellet in 15 ml of DPBS containing 2% human serum. Count the number of cells.
- Centrifuge at 690 x g for 10 min at RT. Remove all the supernatant from the tube.
- Resuspend cells at 1 x 107 cells per 15 ml of Iscove’s modified Dulbecco’s medium (IMDM) containing 1% antibiotics (v/v) and 10% human serum (v/v).
- Incubate the cells O/N at 37 °C in 5% CO2 incubator before isolating CD34+ cells.
- Positive Selection of CD34+ cells from PBMCs using CD34 MultiSort Kit: NOTE: Work fast and use pre-cooled solutions to make cells cold and prevent capping of antibodies on the cell surface and non-specific cell labeling per the kit instruction.
- Collect all PMBCs in a 50 ml tube and count the number of total cells in the medium.
- Centrifuge the cells at 300 x g for 10 min. Remove all the supernatant completely.
- Resuspend up to 108 cells per 300 µl in bead buffer (DPBS containing 0.5% human serum (v/v) and 2 mM EDTA).
- Add 100 µl of FcR Blocking Reagent and add 100 µl of CD34 MultiSortMicroBeads.
- Mix well and incubate for 30 min in the refrigerator (2 - 8 °C).
- Wash cells by adding 2 ml of bead buffer per 108 cells and centrifuge at 300 x g for 10 min at RT. Remove supernatant completely.
- Resuspend up to 108 cells in 500 µl of buffer.
- Place MACS LS column in the magnetic field of a MACS Separator and prepare column by rinsing with 3 ml of bead buffer.
- Apply cell suspension onto the center of the column. Unlabeled cells will flow through which can be collected if preferred.
- Wash column three times with 3 ml of bead buffer.
- Remove the column from the separator and place it on a 15 ml tube.
- Add 5 ml of bead buffer onto the column and immediately flush out the magnetically labeled cells by pushing the plunger into the column.
- Add 20 µl of MultiSort Release Reagent per 1 ml of cell suspension. Mix well and incubate for 10 min in the refrigerator (2 - 8 °C).
- Add 1 - 2 ml of bead buffer per 107 cells and centrifuge the cells at 300 x g for 10 min at RT. Remove supernatant completely.
- Resuspend cells in 50 µl of bead buffer per 107 cells.
- Add 30 µl of MultiSort Stop Reagent per 107 cells and incubate for 15 min in the refrigerator.
- Add 5 ml of bead buffer and centrifuge the cells at 300 x g for 10 min at RT. Discard the supernatant.
2. Derivation of Induced Neural Stem Cells from CD34+ Cells
- CD34+ cells culture: NOTE: Freshly prepared medium should be used within 7 days when stored in the refrigerator. Significant CD34 cells loss could be observed after 24 hr but the significant proliferation of cells should be noticeable, especially after day 5. If the cell number decreases continuously or there is no proliferation, it implies either bad quality of medium or CD34 cells and the cells should not be used for next step.
- Resuspend and culture CD34+ cells in CD34 maintaining medium (StemSpan SFEM medium containing human thrombopoietin (TPO, 100 ng/ml), fms-like tyrosine kinase 3 (Flt-3) ligand (100 ng/ml) and stem cell factor (SCF, 100 ng/ml), interleukin-6 (IL-6, 20 ng/ml) and interleukin-7 (IL-7, 20 ng/ml)) in a 24-well plate (up to 3 x 105/well in 1 ml of medium per well) at 37 °C in 5% CO2 incubator.
- After 24 hr, collect the floating cells and centrifuge the cells at 110 x g at RT to get rid of any attached cells and cell debris. Discard the supernatant and resuspend the cells in 1 ml of fresh CD34 maintaining medium and seed in a new 24-well plate (up to 1 x 105/well) at 37 °C in 5% CO2 incubator for 5 additional days.
- Replace half of the medium every the other day by carefully aspirating the top half of supernatant while the CD34+ cells are floating on the bottom of the wells (Figure 1A).
- Sendai virus transfection:
- On day 5, collect CD34 cells and centrifuge the cells at 170 x g for 10 min at RT. Discard the supernatant.
- Resuspend the cells in fresh medium at 1 x 105/well in a 24-well plate in 1 ml of medium.
- Thaw a cytotune-iPS Sendai reprogramming kit by first immersing the bottoms of the tubes in a 37 °C water bath for 10 sec and then completely thaw at RT. Briefly centrifuge the tubes and put them on ice. Prepare the Sendai virus mixture by mixing the recommended volume of each virus listed on the Certificate of Analysis (COA) for the corresponding lot. MOI 15 is recommended.
- Add Sendai virus mix to each well of CD34 cells. Mix the wells by gently shaking. Incubate the cells at 37 °C in 5% CO2 incubator.
- Observe the transfection efficiency after 24 hr. Notice that cell aggregates and sphere formation are indicators for successful transfection (Figure 1B).
- Change half of the medium by carefully transferring the top half of the medium to another well. If there are cell spheres in the new well, add the same amount of fresh medium.
- Change medium every the other day by repeating step 2.2.6.
- Neural stem cell induction: NOTE: Depending on cell conditions, about 5 or 7 days after transfection, monolayer adherent cells will reach 30 - 50% confluence (Figure 1C).
- Remove the supernatants containing floating cells and spheres and then add 1 ml of neural progenitor medium (Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12), containing 1x N2 supplement, 0.1% (w/v) bovine serum albumin (BSA), 1% (v/v) antibiotics, 20 ng/ml of basic fibroblast growth factor (bFGF), and 20 ng/ml of epidermal growth factor (EGF)) into the adherent cells.
- Optionally, dissociate the cell spheres by gently pipetting and centrifuge the cells at 170 x g for 10 min. Remove the supernatant and resuspend the cells in 1 ml of neural progenitor medium in a new 24-well plate coated with matrigel per instruction as back up.
- Change the medium every other day till the cells reach 60 - 80% confluent, usually after 1 week.
- Discard the supernatant and add in 1 ml of neural stem cell medium (serum free medium, NSC SFM). Dissociate the cells by using a cell scraper followed by very gentle pipetting.
- Remove the cells and seed all of them into one well of a 6-well plate. Add another 1 ml of neural stem cell medium to make 2 ml media for each well. Incubate the cells at 37 °C in 5% CO2 incubator.
- Change medium every the other day.
- When cells reached 60% confluence, dissociate and replate the cells at 1:3 ratio following the steps in 2.3.4 and 2.3.6.
- Freezing of neural stem cells:
- Prepare neural stem cell freezing medium by adding 20% dimethyl sulfoxide (DMSO) into the neural stem cell medium. Keep the freezing medium on ice.
- Dissociate the cells in a 6-well plate, using cell scraper followed by gently pipetting. Count the cells. Centrifuge the cells at 170 x g for 10 min at RT. Discard the supernatant.
- Resuspend the cells in neural stem cell medium at 1 x 106/ml. Add the same volume of freezing medium to the cells.
- Add 1 ml (5 x 105 total cells) of cells in a cryovial. Freeze the cells using a Mr. Froster freezing container following the instruction.
3. Neural Cell Differentiation
- Neuronal cells differentiation:
- When neural stem cells reach 80% confluence, detach the neural stem cells using a rubber policeman and then dissociate by gently pipetting.
- Count the cells and adjust the concentration to 1 x 105/ml in neural stem cell medium.
- For neuronal differentiation, plate the cells on poly-D-lysine/laminin coated cover slips in 24-well plates at 1 x 105/well. Incubate the cells at 37 °C in 5% CO2 incubator.
- Replace the medium with neural differentiation medium (DMEM/F12 containing 1x N2 supplement, 1x B27 supplement, 300 ng/ml cAMP, 0.2 mM vitamin C, 10 ng/ml brain derived neurotrophic factor (BDNF) and 10 ng/ml glial cell derived neurotrophic factor (GDNF)) after 24 hr. Incubate the cells at 37 °C in 5% CO2 incubator.
- Change neural differentiation medium twice a week for 2 weeks.
- Astroglial differentiation:
- Dissociate the neural stem cells by pipetting.
- Count the cells and adjust the cell concentration to 5 x 104 cells/ml. Seed the cells on 24-well plates. Incubate the cells at 37 °C in 5% CO2 incubator.
- Replace the medium with DMEM/F12 with 10% fetal bovine serum (FBS) and incubate the cells at 37 °C in 5% CO2 Incubator.
- Change medium twice a week for 2 weeks. If cells reach confluence before 2 weeks, replate the cells following steps 3.2.1 and 3.2.2.
- Oligodendrocyte differentiation:
- Dissociate the neural stem cells by pipetting.
- Count the cells and seed 1 x 105 cells on poly-L-Ornithine coated 24-well plates in 1 ml of oligodendrocyte differentiation medium consisting of DMEM/F12 containing N2 supplement, 10 ng/ml of platelet derived growth factor-AA (PDGF-AA), 2 ng/ml of neurotrophin-3 (NT-3), 2 ng/ml of sonic hedgehog (Shh), and 3 nM of triiodothyronine (T3). Incubate the cells at 37 °C in 5% CO2 incubator.
- Change medium twice a week for 2 weeks.
- Replace the medium with DMEM/F12 with 1xN2 supplement and 3 nM of T3 and culture the cells for an additional week for myelin basic protein (MBP) production.
4. Immunofluorescence Staining
Replace media with 4% paraformaldehyde (PFA). Incubate for 10 min at RT.
Discard PFA and wash with PBS containing 0.1% Triton X-100 (PBS-T) 3 times, 5 min each time.
Permeabilize cells with PBS containing 0.5% Triton X-100 for 10 min at RT.
Block the cells with PBS-T containing 4% goat serum for 20 min at RT.
Incubate the cells with corresponding primary antibodies diluted in PBS-T containing 0.1% BSA for 2 hr at RT or O/N in cold room.
Wash the cells for 3 times with PBS-T, 5 min each time. Incubate the cells with corresponding secondary antibodies coupled with a fluorochrome using 1:400 dilution in PBS-T containing 0.1% BSA for 1 hr on a shaker in the dark.
Wash the cells with PBS-T containing 4',6-diamidino-2-phenylindole (DAPI) for 10 min at RT.
Wash the cells two more times with PBS-T for 5 min each at RT.
Observe the labeled cells using a microscope and take photos (Figure 3).
Representative Results
The quality of CD34 cells is critical for the success of neural stem cell transduction. High quality CD34 cells proliferate during the first couple of days of culture and appear as non adherent, homogenously round cells, floating just above the bottom of culture vessels (Figure 1A). The successful infection results in cell aggregates, which expands over time (Figure 1B), while non specific cell aggregates may occur during cell culture, that expand and the associations are loose. Adherent cells usually appear around three to five days after transfection; they are mostly bipolar and will proliferate to make monolayers (Figure 1C). After a selection with neural progenitor cell medium most of the adherent cells are nestin and SOX2 positive but OCT4 negative (Figure 2). The induced neural stem cells can be further differentiated to neurons and glia (Figure 3).
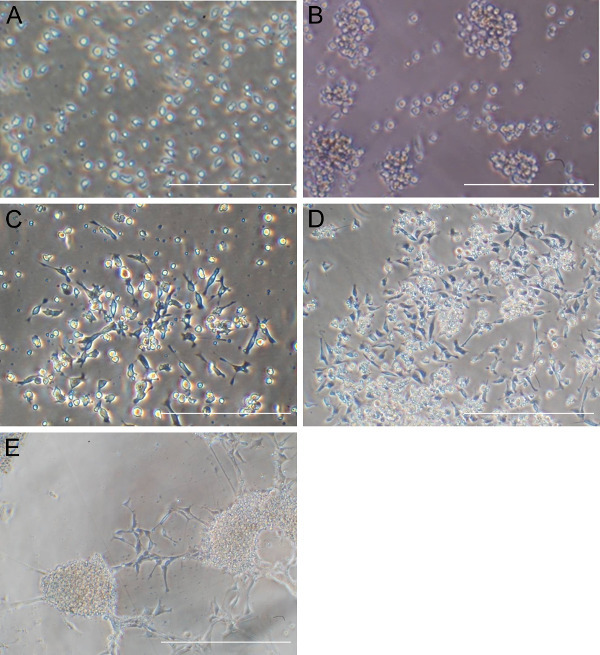 Figure 1. Morphological changes of CD34 cells after Sendai virus transfection. (A) Significant number of CD34 cells can be observed at the time of transfection. (B) Sphere like cell aggregates can be observed 24 hr after Sendai virus transfection which expands during time. (C) Mostly bipolar adherent cells appeared after 5 days of transfection. (D and E) Typical morphologies of neural stem cells are shown after induction with neural progenitor medium and expansion. Scale bar: 200 µm for (A) and (B), 400 µm for the others. Please click here to view a larger version of this figure.
Figure 1. Morphological changes of CD34 cells after Sendai virus transfection. (A) Significant number of CD34 cells can be observed at the time of transfection. (B) Sphere like cell aggregates can be observed 24 hr after Sendai virus transfection which expands during time. (C) Mostly bipolar adherent cells appeared after 5 days of transfection. (D and E) Typical morphologies of neural stem cells are shown after induction with neural progenitor medium and expansion. Scale bar: 200 µm for (A) and (B), 400 µm for the others. Please click here to view a larger version of this figure.
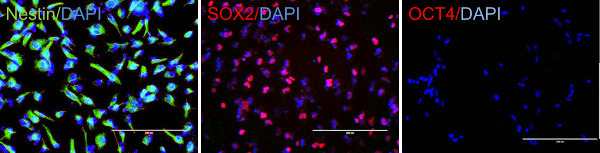 Figure 2. Induced neural stem cells express neural stem cell markers. Neural stem cells express nestin and SOX2 but not OCT4. Scale bar: 200 µm. Please click here to view a larger version of this figure.
Figure 2. Induced neural stem cells express neural stem cell markers. Neural stem cells express nestin and SOX2 but not OCT4. Scale bar: 200 µm. Please click here to view a larger version of this figure.
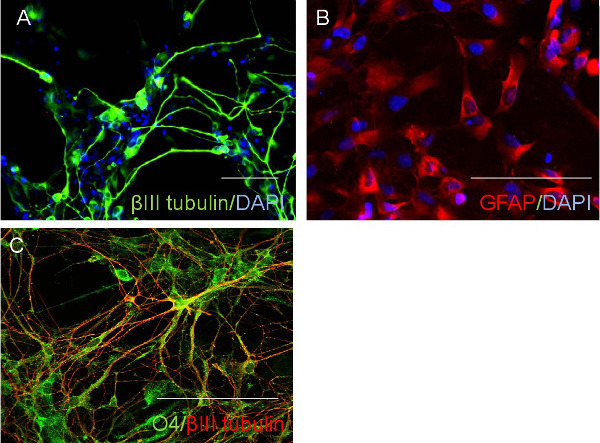 Figure 3. Neural stem cells differentiation. The neural stem cells may be differentiated to cells with neuronal marker (A), astroglial marker GFAP (B), and oligodendrocyte markers O4 (C). Scale bar: 100 µm. Please click here to view a larger version of this figure.
Figure 3. Neural stem cells differentiation. The neural stem cells may be differentiated to cells with neuronal marker (A), astroglial marker GFAP (B), and oligodendrocyte markers O4 (C). Scale bar: 100 µm. Please click here to view a larger version of this figure.
Discussion
A detailed protocol to directly generate neural stem cells from peripheral hematopoietic progenitor cells is provided.
Compared to fibroblast cells, human peripheral blood is more accessible. Using the presented protocol, more than 1 x 105 hematopoietic progenitor cells, or CD34 positive cells, can be enriched from 10 ml of whole blood. Although contamination with platelets is usually not preventable, it can be easily reduced by lower speed centrifugation and it does not interfere with neural stem cell generation. However, the quality and number of CD34 cells will determine the final production of neural stem cells. The quality of CD34 cells can be roughly judged by their response to CD34 culture medium. A positive response, meaning rapid proliferation of CD34 cells in the medium, is critical for the later successful transformation. If CD34 cells failed to proliferate, or even undergo significant cell loss, then the transformation will become difficult, resulting in less proliferative cells. Thus it is suggested to only start transformation with good quality CD34 cells. Although 1 x 105 CD34 cells are enough for an experienced researcher, 3 x 105 cells may be used for starters.
Using Sendai virus provides a non-integration, highly efficient and convenient way to introduce transcriptional factors into the targeting cells14,15. Originally used for generating iPSC, Yamanaka factors were also successfully used to generate neural stem cells directly from fibroblasts11. In a previous report using the presented protocol, Sendai virus containing Yamanaka factors could also be used to generate neural stem cells from cord blood or adult peripheral hematopoietic blood cells. Compared with using skin biopsy for culturing fibroblasts, blood is more accessible and convenient to get. Besides, it is reported that hematopoietic progenitor cells also express certain neural specific markers16, making it a better choice for generating neural stem cells. In fact, after Sendai virus infection, most of the attached monolayer cells expressed nestin, a neural stem cell marker, except the more aggregated compact colonies, some of which may give rise to iPSC at a later time point. Thus the attached monolayer of cells are collected and further incubated in neural progenitor cell medium, which has been widely used in primary neural progenitor cell culture, to facilitate the neural stem cell fate of the cells.
There is a delicate balance in maintaining the neural stem cells. On one hand, it is preferred that the cells to have better proliferating capability and to be passaged as many times as possible. On the other hand, the ease of differentiating them to neurons is also critical. Two different media are used to achieve this balance. The widely used neural progenitor medium is chosen for the neural stem cell identity induction/selection, which is the critical process in the first place. It can also be used at later passages to reinforce the neuronal differentiation capabilities. However, the neural stem cells are very fragile and sensitive so that prolonged exposure to neural progenitor cell medium may greatly inhibit cell proliferation and result in excessive differentiation. Thus neural stem cell medium is used to help cell recovery and to facilitate cell expansion. The neural progenitor medium is used for neural induction at passage 1, when the cells become confluent. If excessive inhibition of cell proliferation and toxicity is noticed, it is necessary to replace the medium early with neural stem cells medium. However, neural stem cell medium is less effective in neural selection; thus over time, especially with over-confluent neural stem cells or delayed passaging, contamination with non-neural cells or lack of neural differentiation could occur. Thus the cells need to be passaged regularly, when less than 60% confluent and at least once a week. Neural selection with neural progenitor cell medium is possible at later passages to reestablish the neural identity. Another factor that affects the stemness of the neural stem cells is the coating of the tissue culture plates. Coating with matrigel or poly-D-lysine can help cell attachment at first in case the cells are difficult to attach during replating. But long term exposure to the coating results in less passage numbers. This is because the coating enhances the cell differentiation process, and may result in more damage to the cells during dissociation.
As a summary, this protocol used Sendai virus containing Yamanaka factors to directly derive human neural stem cells within 3 - 4 weeks from adult hematopoietic progenitor cells obtained from accessible peripheral blood samples. The neural stem cells could be further differentiated to neurons and glia. This method bypasses the lengthy and complicated iPSC generation processes currently used and may be used for in vitro cell culture modeling for various neurological disorders, which may better represent the genetic differences in specific disorders, especially from individual patient samples.
Disclosures
The study is funded by NIH intramural funds.
Acknowledgments
The authors have no conflicts of interest to disclose.
References
- Azari H, et al. Purification of immature neuronal cells from neural stem cell progeny. PLoS ONE. 2011;6:e20941. doi: 10.1371/journal.pone.0020941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azari H, Sharififar S, Fortin JM, Reynolds BA. The neuroblast assay: an assay for the generation and enrichment of neuronal progenitor cells from differentiating neural stem cell progeny using flow cytometry. J Vis Exp. 2012. [DOI] [PMC free article] [PubMed]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Zhang N, An MC, Montoro D, Ellerby LM. Characterization of Human Huntington's Disease Cell Model from Induced Pluripotent Stem Cells. PLoSCurr. 2010;2:RRN1193. doi: 10.1371/currents.RRN1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, et al. Disease-Specific Induced Pluripotent Stem Cells. Cell. 2008;134(5):877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, et al. Neural differentiation of patient specific iPS cells as a novel approach to study the pathophysiology of multiple sclerosis. Stem Cell Research. 2012;8(2):259–273. doi: 10.1016/j.scr.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463(7284):1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfisterer U, et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proceedings of the National Academy of Sciences. 2011;108:10343–10348. doi: 10.1073/pnas.1105135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-T, et al. Direct Generation of Neurosphere-Like Cells from Human Dermal Fibroblasts. PLoS ONE. 2011;6:e21801. doi: 10.1371/journal.pone.0021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thier M, et al. Direct Conversion of Fibroblasts into Stably Expandable Neural Stem Cells. Cell Stem Cell. 2012;10(4):473–479. doi: 10.1016/j.stem.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Lujan E, Chanda S, Ahlenius H, Südhof TC, Wernig M. Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proceedings of the National Academy of Sciences. 2012;109:2527–2532. doi: 10.1073/pnas.1121003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, et al. Derivation of neural stem cells from human adult peripheral CD34+ cells for an autologous model of neuroinflammation. PLoS ONE. 2013;8:e81720. doi: 10.1371/journal.pone.0081720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin CH, et al. Recombinant Sendai virus provides a highly efficient gene transfer into human cord blood-derived hematopoietic stem cells. Gene Ther. 2003;10(3):272–277. doi: 10.1038/sj.gt.3301877. [DOI] [PubMed] [Google Scholar]
- Ban H, et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proceedings of the National Academy of Sciences. 2011;108:14234–14239. doi: 10.1073/pnas.1103509108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goolsby J, et al. Hematopoietic progenitors express neural genes. Proceedings of the National Academy of Sciences. 2003;100:14926–14931. doi: 10.1073/pnas.2434383100. [DOI] [PMC free article] [PubMed] [Google Scholar]