

Abstract
Laser capture microdissection (LCM) is used to isolate a concentrated population of individual cells or precise anatomical regions of tissue from tissue sections on a microscope slide. When combined with immunohistochemistry, LCM can be used to isolate individual cells types based on a specific protein marker. Here, the LCM technique is described for collecting a specific population of dopamine neurons directly labeled with tyrosine hydroxylase immunohistochemistry and for isolation of the dopamine neuron containing region of the ventral tegmental area using indirect tyrosine hydroxylase immunohistochemistry on a section adjacent to those used for LCM. An infrared (IR) capture laser is used to both dissect individual neurons as well as the ventral tegmental area off glass slides and onto an LCM cap for analysis. Complete dehydration of the tissue with 100% ethanol and xylene is critical. The combination of the IR capture laser and the ultraviolet (UV) cutting laser is used to isolate individual dopamine neurons or the ventral tegmental area when using PEN membrane slides. A PEN membrane slide has significant advantages over a glass slide as it offers better consistency in capturing and collecting cells, is faster collecting large pieces of tissue, is less reliant on dehydration and results in complete removal of the tissue from the slide. Although removal of large areas of tissue from a glass slide is feasible, it is considerably more time consuming and frequently leaves some residual tissue behind. Data shown here demonstrate that RNA of sufficient quantity and quality can be obtained using these procedures for quantitative PCR measurements. Although RNA and DNA are the most commonly isolated molecules from tissue and cells collected with LCM, isolation and measurement of microRNA, protein and epigenetic changes in DNA can also benefit from the enhanced anatomical and cellular resolution obtained using LCM.
Keywords: Neuroscience, Issue 96, Laser capture microdissection, dopamine neuron, Immunohistochemistry, Tyrosine hydroxylase, Ventral tegmental area, PEN membrane glass slide.
Introduction
All tissue consists of a heterozygous population of cells. This is particularly relevant for brain tissue, consisting of various morphologically and/or neurochemically distinct neurons surrounded by various types of glial cells (oligodendrocytes, microglia and astrocytes). Additionally, distinct regions in the brain, such as areas of the cortex or brain stem nuclei, have specific functions. Therefore, the ability to isolate specific populations of cells or very small anatomically distinct areas, when combined with analytical techniques (i.e., Q-PCR, microarrays, RNA-sequencing, and proteomics), can markedly enhance the understanding of various biological processes. LCM is a technology that provides the capability to isolate very discrete anatomical areas or specific cells from tissue on a microscope slide providing a much more homogeneous source for further analysis of various molecules, such as RNA, microRNA, DNA and proteins.
Since LCM was first introduced, several different approaches have been used to microdissect cells from tissue 1-3 . The earliest approaches included direct attachment of tissue on a slide using an infrared (IR) laser and a thermoplastic film and a non-contact method using an ultraviolet (UV) cutting laser to detach a cell or tissue and collection into a tube. Subsequently, LCM has been successfully used on a variety of tissues and cell types and shown to be compatible with isolation of various molecules for downstream analysis including RNA, microRNA, DNA, and proteins, including enzymatic activity 1,4-7. Here LCM is demonstrated using the a LCM system which uses a cutting UV laser and a capture IR laser for cutting around cells or tissue and attaching it to an LCM cap, respectively. This LCM system uses both the IR capture laser to melt a plastic film on a cap over the cell or tissue of interest that attaches the cells to the plastic film and removes it from the tissue with the removal of the cap (Figure 1). Additionally, in combination with the IR laser, a UV laser is available to cut-out tissue or cells of interest from tissue mounted on slides with a membrane then isolated by attaching the tissue to the LCM cap using the IR laser (Figure 2). A brief overview of these techniques is found in Figures 1 and 2.
Several variations on this technique are described to either isolate specific cells using direct fluorescent immunohistochemistry and an indirect immunohistochemistry-guided technique that is used for the isolation of a region of tissue based on the expression of a specific protein. Specifically, the isolation of dopamine neurons from the substantia nigra and isolation of the ventral tegmental area and the subsequent isolation of RNA from fresh, frozen brain tissue are shown. As RNA is the most labile of molecules measured (as compared to DNA or protein), steps to help preserve RNA integrity are included and data shown on the quantity and quality of RNA obtained.
Protocol
NOTE: Brain tissue from mice was used in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and study protocols were approved by the Institutional Animal Care and Use Committee at Mississippi State University.
1. Preparation of Slides and Tissue
Use either Silane-prep slides or polyethylene napthalate (PEN) membrane glass slides.
For RNA isolation, dip slides in RNase decontamination solution, wash 3 times with RNase-free water then dehydrate with a graded series of RNase-free ethanol and vacuum dry for 30 min.
Cut tissue sections at 10 µm thickness using a cryostat and mount on the prepared RNase free slides. Keep the sections frozen during sectioning to preserve RNA quality. NOTE: Be sure that the tissue is approximately 4 mm away from the edges of the slide as the LCM cannot remove tissue that is too close to the edges of the slide. In order to remove tissue from a PEN membrane slide, ensure that the tissue is 4 mm from the left, top and bottom and 7 mm from the right side of the slide which are the dimensions of the membrane that is not attached to the slide.
For isolation of individual cell populations using direct immunohistochemistry, section tissue onto either the Silane-prep slides or PEN membrane slides. For isolation of regions of tissue using indirect immunohistochemistry guided techniques, mount one set of sections on a Silane-prep slide for immunohistochemistry and alternate sections on either a PEN membrane slide and/or a Silane-prep slide to be used for LCM.
2. Tissue Preparation for Laser Capture – Rapid Tyrosine Hydroxylase Immunohistochemistry for Direct Laser Capture of Immunoreactive Cells
Outline tissue with a hydrophobic pen and allow to dry.
Fix tissue in acetone-methanol (1:1) solution at -20 °C for 10 min. NOTE: Our experience has shown that the acetone-methanol fixation resulted in much more consistent immunohistochemistry than acetone or methanol alone.
Rinse slide in phosphate buffered saline (PBS) with 1% Triton (RNase free).
Cover sections with 100-200 µl PBS with 1% Triton with tyrosine hydroxylase antibody diluted 1:100 with 400 U/ml RNasin. Incubate for 5-10 min.
Rinse briefly in PBS twice and PBS-1% Triton.
Cover tissue with 100-200 µl of goat anti-Rabbit IgG labeled with Alexa Fluor 488 diluted 1:100 in PBS-1% Triton with 400 U/ml RNasin and 50 ng/ml DAPI. Incubate for 5 min.
Rinse 2 times in PBS then dehydrate 30 s in a graded series of RNase free ethanol (75%-75%-95%-95%-100%-100%).
Incubate in two washes of Xylene for 1 min then 5 min.
Remove slides from Xylene immediately prior to use for LCM and allow to air dry.
3. Tissue Preparation for Laser Capture – Tyrosine Hydroxylase Immunohistochemistry for Indirect Laser Capture of Immunoreactive Regions
Process one slide with sections adjacent to sections mounted on Silane-prep and/or PEN membrane slides for immunohistochemistry. NOTE: Procedures used here are similar to those previously reported 5. For the demonstration images, the diaminobenzidine (DAB) chromagen reaction was used to visualize the tyrosine hydroxylase neurons of interest.
For slides used for LCM, fix for 5 min in 100% acetone at 4 °C.
Dehydrate tissue in a graded series of RNase Free ethanol (75%-75%-95%-95%-100%-100%) for 1 min each.
Incubate in two washes of Xylene for 1 min then 5 min.
Remove slides from Xylene immediately prior to use for LCM and allow to air dry.
4. Using the Laser Capture Microdissection System
NOTE: The LCM application program has 10 Tool bars and 2 windows to control the microdissection procedure. Tool Bars: 1. Microscope, 2. Capture Laser, 3. Cutting Laser, 4. Study, 5. Navigation, 6. Materials, 7. Capture Groups, 8. Microdissection, 9. Font, 10. Annotation. Windows: 1. Live Video and 2. Road Map.
Open the door by selecting the “Open door” tab in the Materials tool bar in LCM system software.
Load slides on the three available slots. For this demonstration, load one slide with tyrosine hydroxylase immunohistochemistry visualized with DAB, one Silane-prep slide and one PEN membrane slide each labeled for rapid fluorescent tyrosine hydroxylase immunohistochemistry. In a typical experiment, use either unlabeled acetone fixed sections with a reference slide labeled for tyrosine hydroxylase using DAB or slides directly labeled with fluorescent tyrosine hydroxylase for laser capture.
Load the LCM caps into the available slot. NOTE: Macro and HS LCM caps are available for use. The type of LCM cap to use depends on the number of cells or amount of tissue to be collected. Macro slides are listed to collect up to several thousand cells while HS caps can collect several hundred. The Macro slides allow for more tissue collection but require 50 µl of lysis buffer. The HS cap, when used with the adaptors, requires only 10 µl of lysis buffer which allows for greater proportional recovery of RNA.
Close the door.
Load the slide to activate the Road Map window which provides a working view of the entire slide, this allows for selection of the region of interest (Figure 3). View the Road Map on Live video window to navigate to the area of interest.
In the Materials tool bar, identify the PEN membrane slides by checking the box above the slide slot. To supply cap properties, right click on the caps in the Materials tools bar and select “Cap supply properties”. Check the boxes indicating the position of the caps and select either Macro or HS caps.
Select the “Fluorescence” tab in the Microscope tool bar to turn on the fluorescent lamp and allow the fluorescent lamp to warm up. Use the Blue and UV filters to visualize the Alex Fluor 488 and DAPI, respectively. Use the “Adjust Camera” tab to increase the sensitivity of the image in order to see the fluorescently labeled cells.
In the Materials tool bar, right click on a cap and move it to the slide of interest. Double click on a location in the Road Map to select the region that appears in the Live Video window. Move the cap around on the slide by selecting an area on the Road Map (double left click) and then right clicking and selecting “Place cap at center region.”
Before using the IR capture laser, aim and adjust it for strength. Place the cap in a region of the slide away from tissue. On the Capture Laser tool bar, select “Enable.” Adjust the Power (3-100 mW), Pulse (100-1000000 µsec) and # Hits of the laser. Fire the IR laser when the cap is over a portion of the slide without tissue and check to see if the laser sufficiently melts the LCM cap membrane. NOTE: This is called wetting which is melting the polymer membrane on the cap so that it contacts the glass slide. Sufficient wetting is visible as a dark ring fused to the slide with the center of the ring clear (See Figure 4 for examples of sufficient and insufficient melting). If wetting is not sufficient, adjust the power and pulse, and test fire again until this is achieved. Additionally, multiple fires of the laser can also be used.
When the IR laser intensity is adjusted, right click and select “Capture laser is here ” to aim the laser. NOTE: This step tells the computer where the IR capture laser is aimed at the tissue. This step of aiming the laser is critical and needs to be repeated every time the cap is moved or the objective is changed.
5. Laser Capture Microdissection of Individual Dopamine Neurons off of Silane-prep Slides
Move to the region of interest to capture cells. NOTE: The example of isolating dopamine neurons is shown here.
To capture individual cells off of a Silane-prep slide, select the “LCM” tab then the “single point” icon in the Microdissection tool bar.
In the Microscope tool bar, use the “Shutter” tab to switch between fluorescence and bright field with fluorescent filters tabs to choose the appropriate filters and use the objectives tabs to change the magnification. Once the cells of interest are visible in the Live video window, adjust the spot size used to mark cells of interest to the approximate size of the cells. Do this by clicking the Spot tab on the Capture Laser tool bar then clicking on one side of a stained cell, followed by clicking on the opposite side. NOTE: This calculates the diameter of the spot and displays the value.
Mark the cells of interest by clicking on them while the “Single point” icon is selected. Do this to place a spot marker on each cell. Here, the Blue filter and the 10X objective are used to visualize the dopamine neurons. Retest and aim the IR laser on the cap where there is no tissue beneath it as described in steps 4.9 and 4.10.
Once the cells are marked, select the “Go - cut and capture” tab on the Microdissection tool bar and the IR laser will automatically fire at all marked cells selected. Additionally, if needed, fire the IR laser manually at any point of interest (Figure 5D).
Move the cap away from the section and onto an open region of the slide to check if the cells were captured onto the LCM cap. Ensure that the cells of interest are removed from the slide (Figure 5B, E) and attached to the LCM cap (Figure 5C, F). Document this process by clicking on the “Capture image” tab in Microscope tool bar.
When finished collecting tissue, remove the cap by right clicking on the cap and select “Move to” then “Unload.”
6. Laser Capture of Individual Dopamine Neurons off of PEN Membrane Slides
To capture individual cells off of PEN membrane slides using both the IR and UV lasers, select “Cut and Capture” tab in the Microdissection tool bar. Select the “single point” tab to mark the cells of interest. Use the “Cut line” tab to outline each cell. Be sure to test the IR laser as described in step 4.9 and 4.10 and test the UV laser to determine the minimum intensity necessary to cut thought the PEN membrane and tissue (Figure 4 and 5)
Select the “Capture and Cut” tab on the Microdissection tool bar. When collecting individual cells using this technique, be sure to fire the IR laser first followed by the UV laser as these small pieces can be lost if they are cut out prior to being affixed to the LCM cap. NOTE: This process can also be done manually by firing the IR laser at a point of interest and the cells individually outlined with the UV laser.
After collecting the cells of interest, move the LCM cap as described above in step 5.7 to determine if the cells were removed and attached to the cap (Figure 6).
When finished collecting tissue, remove the cap by right clicking on the cap and selecting “Move to” then “Unload.”
7. Capturing the Ventral Tegmental Area from Silane-prep Slides
To identify the region of interest, use the slide processed for immunohistochemistry. NOTE: For this demonstration the ventral tegmental area is isolated (Figure 7A).
Load a LCM cap and test the lasers as in steps 4.9 and 4.10. NOTE: Typically, a Macro LCM cap is used but an HS cap can also be used (See Figure 8).
To isolate a region of tissue (i.e., the ventral tegmental area) from the Silane-prep slide, select the “LCM” tab in the Microdissection tool bar, then select the “polygon-exterior” tab. Outline the region of interest in the Live Video window. Depending of the size of the area for collection, outline the region of interest at low magnification (2X Objective). For collection, however, the instrument uses the 10X objective so adjust and aim the IR laser under the 10X objective prior to capturing.
Select the “Go - capture and cut” tab in the Microdissection tool bar. NOTE: The computer will automatically fire the IR laser throughout the entire selected area (Figure 7F,I). If the LCM membrane has not been melted sufficiently throughout the area, the laser can be fired manually or the entire process repeated.
Move the LCM cap off of the tissue to determine how well the tissue was collected. NOTE: One pass using this method typically leaves some tissue behind on the slide (See Figure7G). If this occurs, repeat the selection and capture step above. Repeating this step can increase the amount of tissue collected (Figure 7J).
When finished collecting tissue, unload the cap by right clicking on the cap and selecting “Move to” then “Unload.”
8. Capturing the Ventral Tegmental Area from PEN Membrane Slides
Use the slide processed for immunohistochemistry as a guide to select the region of interest from the tissue on the PEN membrane slide as above.
Select the “Cut and Capture” tab in the Microdissection tool bar. Select the “Cut Line” tab and outline the region of interest using the immunohistochemistry labeled slide as a guide. NOTE: The program will automatically add spots for the IR laser to attach the tissue to the cap. Use the “single point” tab to add additional spots to better secure the tissue to the cap.
Under the 10X objective, test fire and aim the IR and UV laser as described in 4.9 and 4.10. Test that the UV laser can penetrate the membrane on the slide and that this cut corresponds to the location on the computer screen. Use a UV laser strength that is just sufficient to cut the membrane and tissue but does not damage the tissue (Figure 5).
Select the “Go - capture and cut” tab in the Microdissection tool bar. NOTE: The computer will automatically fire the IR laser at all labeled spots within the tissue to attach it to the cap then fire the UV laser to cut the slide PEN membrane and tissue (Figure 7C). Additional IR melted spots may be necessary to adequately attach the tissue to the LCM cap, which can also be added manually.
Move the LCM cap off of the tissue to determine if the tissue was removed from the section (Figure 7C) and attached to the cap (Figure 7C).
When finished collecting tissue, to remove the cap, right click on the cap and select “Move to” then “Unload.”
9. Isolation of RNA
To collect RNA from the captured cells or tissue, incubate the LCM cap membrane in RNA lysis buffer for 30 min at 37 °C. NOTE: For HS caps, an adaptor is available that allows for the use of only 10 µl of lysis buffer on the cap and attaches to a 0.5 ml microcentrifuge tube. The Macro caps require 50 µl of lysis buffer and attaches directly to a 0.5 ml microcentrifuge tube.
After incubation, spin the lysis buffer down into the 0.5 ml microcentrifuge tube.
To ensure complete lysis of all the cells or tissue, peel off the LCM cap membrane of the cap and place in the lysis buffer.
To test for RNA integrity, use RNA isolated from the tissue remaining on the slide after LCM to measure RNA integrity. NOTE: Due to the limited sample RNA obtained with LCM, it is typically not practical to test RNA integrity on LCM isolated RNA, however, RNA from the remaining tissue provides a good indicator of any RNA degradation due to fixation, immunohistochemistry and time during the LCM procedure.
Representative Results
The micrograph in Figures 6 and 7 demonstrate the capabilities for isolation of individual dopamine neurons. The current anatomical resolution is around 5-10 µm as the smallest spot size that can be consistently produced on the LCM caps to isolate a cell. The only limitation for isolation of specific cell types is the ability to visualize the cells. Although LCM is capable of isolating individual dopamine neurons, contamination of parts of adjacent unlabeled cells most likely occurs. Therefore, the final sample is a concentrated collection of dopamine neurons, not a pure population of dopamine neurons. This technology is also capable of isolating small areas of tissue such as discrete nuclei or regions within the brain as shown with the isolation of the ventral tegmental area. Previous publications have demonstrated this use in brain tissue as well as to isolate pathological tissue from normal tissue5,11.
Because the most common use of cells and tissues isolated using LCM is RNA analysis, the procedures presented were optimized to preserve RNA integrity. In order to determine the quality of RNA that remained following LCM, RNA was isolated, using silica based spin columns, from tissue on the slides following LCM, either after the rapid tyrosine hydroxylase fluorescent immunohistochemistry procedure or fixation in acetone. RNA quality was measured and compared to values for RNA from commercially available whole brain RNA (Figure 9). RNA quality was reduced in the RNA samples from tissue used for LCM with lower integrity after immunohistochemistry (RIN not available) followed by acetone fixation (RIN 2.6) as compared to whole brain RNA (RIN 8.2) as can be seen by a relative reduction in height of the rRNA S28 peak compared to the S18 peak. However, the RNA maintained the 18S and 28S bands and gene expression could be measured using Q-PCR.
To measure the quantity of RNA obtained from neurons acquired via LCM, dopamine neurons (50, 100 and 200 dopamine neurons) were isolated from the ventral tegmental area off of Silane-prep slides. RNA was isolated using silica based spin columns and the quantity of RNA was measured. Based on the standard curve generated, a total of 17.3, 24.8 pg and 50.9 pg/µl was isolated from 50, 100 and 200 dopamine neurons, respectively (Figure 10A). To measure RNA quantity with Q-PCR, a range of concentrations of whole brain RNA and RNA from the dopamine neurons were reverse transcribed and the β-actin gene was measured using Q-PCR as previously described 5. Based on these measurements, a concentration of 3.55, 6.82 and 20.58 pg/µl was calculated from 50, 100 and 200 dopamine neurons, respectively (Figure 10B).
To demonstrate how isolation of dopamine neurons compares to isolation of the entire ventral tegmental area with LCM, dopamine neuron specific genes (Nurr1, tyrosine hydroxylase, and dopamine transporter) were measured with Q-PCR in these two different types of samples and compared to expression of β-actin (Figure 11). Since lower Ct values are generated with a higher concentration of the gene of interest, a decrease in the ΔCt indicates an increased expression of the dopamine neuron genes relative to β-actin. This demonstrates how the isolation of individual dopamine neurons concentrates the expression of dopamine neuron specific genes. In the ventral tegmental area samples, dopamine neuron specific genes are diluted out as other cells (non-dopaminergic neurons and glial cells) will also be collected that express β-actin but do not express dopamine neuron genes.
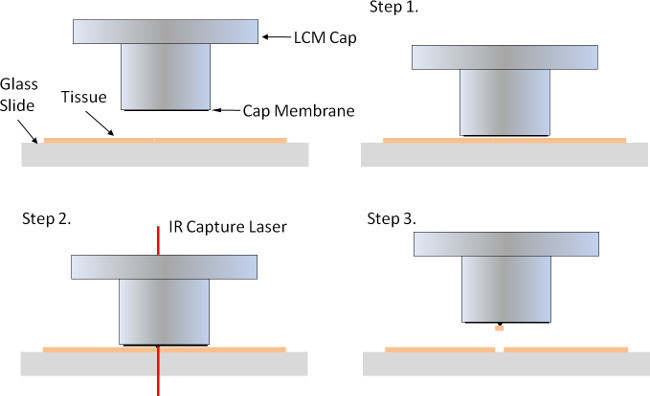 Figure 1. Laser capture using the infrared (IR) capture laser. The small plastic LCM cap has a membrane on the bottom that is placed on top of tissue mounted onto a glass slide (Step 1). When the tissue or cell of interest is identified, an IR capture laser is fired through the LCM cap to melt a small dimple in the membrane onto the underlying tissue/cell (Step 2). When the LCM cap is removed, the tissue is removed from the microscope slide and remains attached to the LCM cap.
Figure 1. Laser capture using the infrared (IR) capture laser. The small plastic LCM cap has a membrane on the bottom that is placed on top of tissue mounted onto a glass slide (Step 1). When the tissue or cell of interest is identified, an IR capture laser is fired through the LCM cap to melt a small dimple in the membrane onto the underlying tissue/cell (Step 2). When the LCM cap is removed, the tissue is removed from the microscope slide and remains attached to the LCM cap.
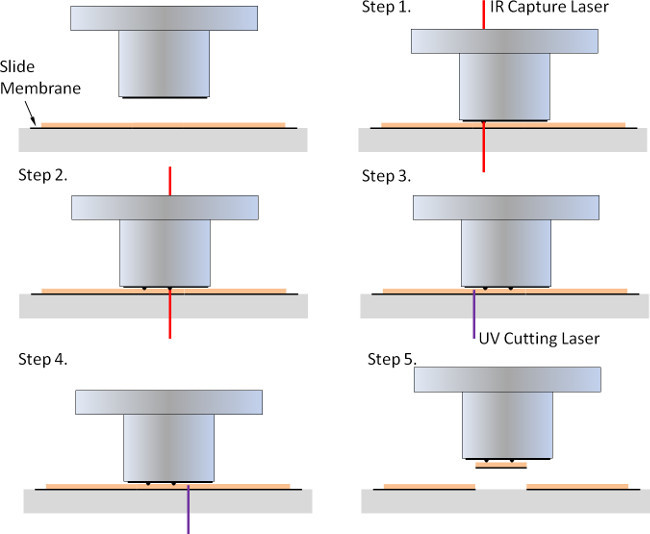 Figure 2. Laser capture using the infrared (IR) capture laser and the ultraviolet (UV) cutting laser. In order to acquire larger areas of tissue or facilitate the recovery of cells using the UV laser, tissue is mounted on a slide with a membrane connected to the slide along the outside corner (PEN membrane slides). The LCM cap is placed on the tissue and the IR capture laser is used to melt the cap membrane and attach the tissue to the LCM cap (Step 1 and 2). The UV cutting laser is then used to cut through the slide membrane and tissue (Step 3 and 4) so that when the cap is removed the tissue is removed from the slide and remains on the LCM cap (Step 5).
Figure 2. Laser capture using the infrared (IR) capture laser and the ultraviolet (UV) cutting laser. In order to acquire larger areas of tissue or facilitate the recovery of cells using the UV laser, tissue is mounted on a slide with a membrane connected to the slide along the outside corner (PEN membrane slides). The LCM cap is placed on the tissue and the IR capture laser is used to melt the cap membrane and attach the tissue to the LCM cap (Step 1 and 2). The UV cutting laser is then used to cut through the slide membrane and tissue (Step 3 and 4) so that when the cap is removed the tissue is removed from the slide and remains on the LCM cap (Step 5).
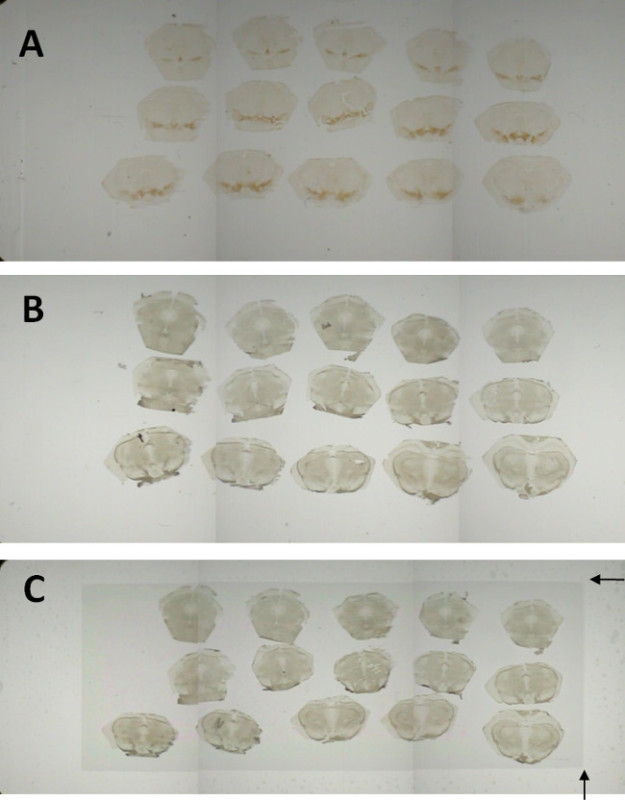 Figure 3. The Laser Capture Microdissection SystemRoad Map. This Laser Capture Microdissection System has space for 3 slides at a time. When slides are loaded into the microscope, low magnification Road Map images for each slide are generated. Selecting an area on the roadmap image moves the Live Image window to that region. For demonstration purposes, the mouse mesencephalon was sectioned into 10 µm section onto 3 slides. The first slide was labeled for tyrosine hydroxylase immunoreactivity using diaminobenzidine as a chromagen (A). Sections were mounted on either Silane-prep slides (B) or PEN membrane slides (C). Both of these slides were labeled using the rapid fluorescent tyrosine hydroxylase immunohistochemistry. Arrows in (C) indicate the attachment of the PEN membrane to the glass slide. No tissue outside this border can be collected with LCM.
Figure 3. The Laser Capture Microdissection SystemRoad Map. This Laser Capture Microdissection System has space for 3 slides at a time. When slides are loaded into the microscope, low magnification Road Map images for each slide are generated. Selecting an area on the roadmap image moves the Live Image window to that region. For demonstration purposes, the mouse mesencephalon was sectioned into 10 µm section onto 3 slides. The first slide was labeled for tyrosine hydroxylase immunoreactivity using diaminobenzidine as a chromagen (A). Sections were mounted on either Silane-prep slides (B) or PEN membrane slides (C). Both of these slides were labeled using the rapid fluorescent tyrosine hydroxylase immunohistochemistry. Arrows in (C) indicate the attachment of the PEN membrane to the glass slide. No tissue outside this border can be collected with LCM.
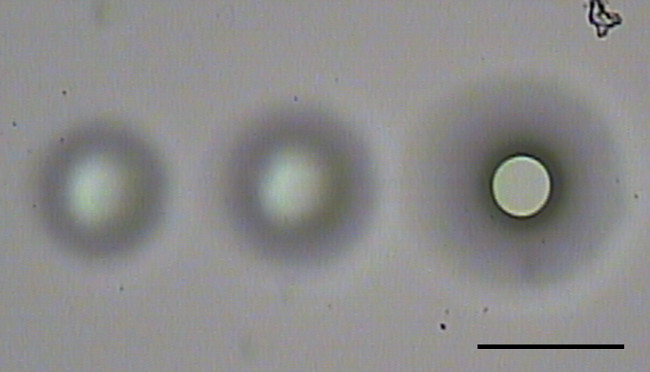 Figure 4. Using the IR capture laser. The IR capture laser is used to melt the LCM cap membrane to attach the tissue to the LCM cap and remove it from the rest of the tissue. The IR capture laser must be at sufficient strength to melt the LCM cap membrane sufficiently to contact the underlying tissue and glass slide. The image above demonstrates when the laser was insufficient to melt the membrane to the slide (left two spots). When the melted membrane touches the glass slide, a thick black outline can be observed (right spot). The intensity of the laser can be adjusted by changing the Power (3-100 mW), Pulse (100-1,000,000 µsec) and # Hits of the laser. Additionally, repeated firing of the IR laser at the same spot can also further melt the membrane. The aim of the IR laser needs to be adjusted anytime the LCM cap is moved or when the objective is changed. Scale bar = 100 µm.
Figure 4. Using the IR capture laser. The IR capture laser is used to melt the LCM cap membrane to attach the tissue to the LCM cap and remove it from the rest of the tissue. The IR capture laser must be at sufficient strength to melt the LCM cap membrane sufficiently to contact the underlying tissue and glass slide. The image above demonstrates when the laser was insufficient to melt the membrane to the slide (left two spots). When the melted membrane touches the glass slide, a thick black outline can be observed (right spot). The intensity of the laser can be adjusted by changing the Power (3-100 mW), Pulse (100-1,000,000 µsec) and # Hits of the laser. Additionally, repeated firing of the IR laser at the same spot can also further melt the membrane. The aim of the IR laser needs to be adjusted anytime the LCM cap is moved or when the objective is changed. Scale bar = 100 µm.
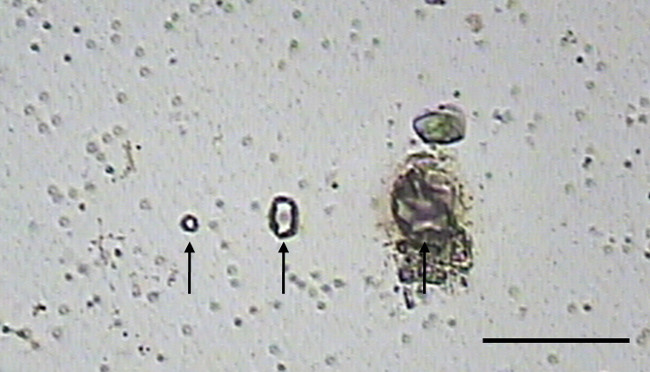 Figure 5. Using the UV cutting laser. The UV cutting laser is used to cut through the PEN membrane and tissue. Prior to use the minimum intensity needed to cut through the membrane and tissue needs to be determine since the UV laser can damage tissue. Above, the three spots (Arrows) of different UV laser intensities are shown with the left spot size sufficient for 10 µm sections, the middle spot for thicker sections and the right spot demonstrating a UV laser intensity that is too high. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Figure 5. Using the UV cutting laser. The UV cutting laser is used to cut through the PEN membrane and tissue. Prior to use the minimum intensity needed to cut through the membrane and tissue needs to be determine since the UV laser can damage tissue. Above, the three spots (Arrows) of different UV laser intensities are shown with the left spot size sufficient for 10 µm sections, the middle spot for thicker sections and the right spot demonstrating a UV laser intensity that is too high. Scale bar = 100 µm. Please click here to view a larger version of this figure.
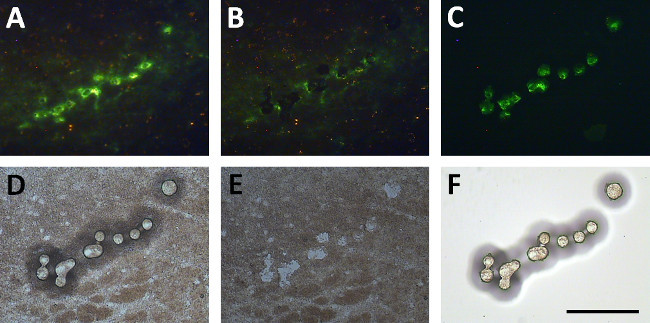 Figure 6. Direct isolation of tyrosine hydroxylase immunoreactive neurons using the IR laser. Dopamine neurons are shown after a rapid fluorescent labeling for tyrosine hydroxylase (A). When the IR laser is fired melting the LCM cap membrane over these neurons (D), removal of the cap picks these cells off of the slide (B, E). These neurons can then be visualized as attached to the LCM cap (C, F). Scale bar = 250 µm. Please click here to view a larger version of this figure.
Figure 6. Direct isolation of tyrosine hydroxylase immunoreactive neurons using the IR laser. Dopamine neurons are shown after a rapid fluorescent labeling for tyrosine hydroxylase (A). When the IR laser is fired melting the LCM cap membrane over these neurons (D), removal of the cap picks these cells off of the slide (B, E). These neurons can then be visualized as attached to the LCM cap (C, F). Scale bar = 250 µm. Please click here to view a larger version of this figure.
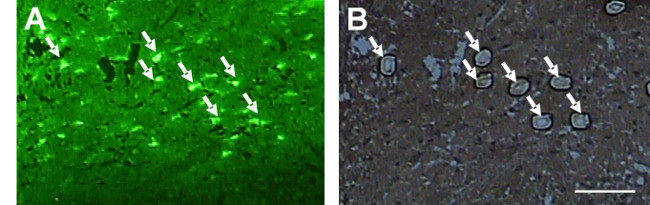 Figure 7. Direct isolation of tyrosine hydroxylase immunoreactive neurons from PEN membrane slides using the IR and UV lasers. Tyrosine hydroxylase labeled neurons are shown above (A). These neurons can be attached to the LCM cap using the IR laser and cut from the PEN membrane slide using the UV laser (B). Scale bar = 250 µm.Please click here to view a larger version of this figure.
Figure 7. Direct isolation of tyrosine hydroxylase immunoreactive neurons from PEN membrane slides using the IR and UV lasers. Tyrosine hydroxylase labeled neurons are shown above (A). These neurons can be attached to the LCM cap using the IR laser and cut from the PEN membrane slide using the UV laser (B). Scale bar = 250 µm.Please click here to view a larger version of this figure.
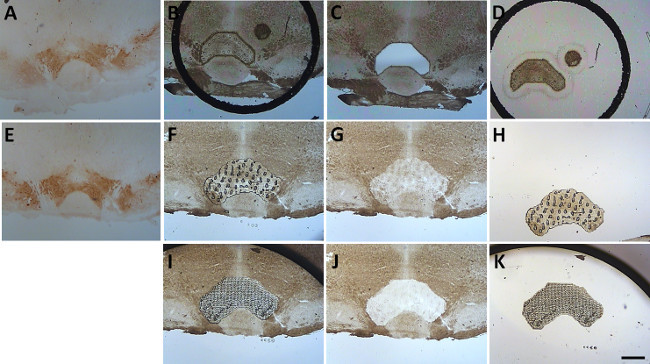 Figure 8. Indirect isolation of the ventral tegmental area using tyrosine hydroxylase immunoreactivity. The location of dopamine neurons in the ventral tegmental area was visualized in a section using standard immunohistochemistry techniques (A and E). This immunohistochemistry labeled section was used as a template to locate the ventral tegmental area in unstained adjacent sections (B and F). Using the UV laser, the ventral tegmental area was attached to the LCM cap with the IR laser and cut from the slide with the UV laser (C and D) and is shown attached to an HS LCM cap (D). The ventral tegmental region isolated from Silane-prep slides using only the IR laser is also shown attached to a Macro cap (F-K). Note that more than one attempt was needed to collect most of the tissue from this region (Compare G and J). Scale bar = 500 µm. Please click here to view a larger version of this figure.
Figure 8. Indirect isolation of the ventral tegmental area using tyrosine hydroxylase immunoreactivity. The location of dopamine neurons in the ventral tegmental area was visualized in a section using standard immunohistochemistry techniques (A and E). This immunohistochemistry labeled section was used as a template to locate the ventral tegmental area in unstained adjacent sections (B and F). Using the UV laser, the ventral tegmental area was attached to the LCM cap with the IR laser and cut from the slide with the UV laser (C and D) and is shown attached to an HS LCM cap (D). The ventral tegmental region isolated from Silane-prep slides using only the IR laser is also shown attached to a Macro cap (F-K). Note that more than one attempt was needed to collect most of the tissue from this region (Compare G and J). Scale bar = 500 µm. Please click here to view a larger version of this figure.
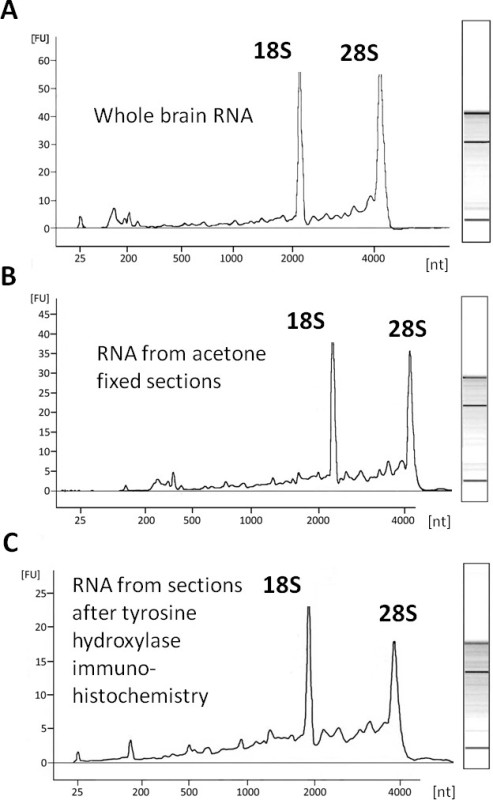 Figure 9. RNA quality. Representative electropherograms and associated gel images of commercially available whole brain RNA (A) and RNA isolated from sections after acetone fixation and LCM (B) or rapid fluorescent tyrosine hydroxylase immunohistochemistry and LCM (C). Although RNA quality was reduced in sections processed for immunohistochemistry, compared to whole brain RNA, the electropherograms demonstrate mostly intact RNA based on the presence of the rRNA S18 and S28 peaks. Whole brain RNA concentration was 6.487 pg/µl with a RIN of 8.2. Acetone fixed tissue RNA concentration was 5.036 pg/µl with a RIN of 2.6. RNA obtained after immunohistochemistry had a concentration of 4.474 pg/µl and RIN was not available.
Figure 9. RNA quality. Representative electropherograms and associated gel images of commercially available whole brain RNA (A) and RNA isolated from sections after acetone fixation and LCM (B) or rapid fluorescent tyrosine hydroxylase immunohistochemistry and LCM (C). Although RNA quality was reduced in sections processed for immunohistochemistry, compared to whole brain RNA, the electropherograms demonstrate mostly intact RNA based on the presence of the rRNA S18 and S28 peaks. Whole brain RNA concentration was 6.487 pg/µl with a RIN of 8.2. Acetone fixed tissue RNA concentration was 5.036 pg/µl with a RIN of 2.6. RNA obtained after immunohistochemistry had a concentration of 4.474 pg/µl and RIN was not available.
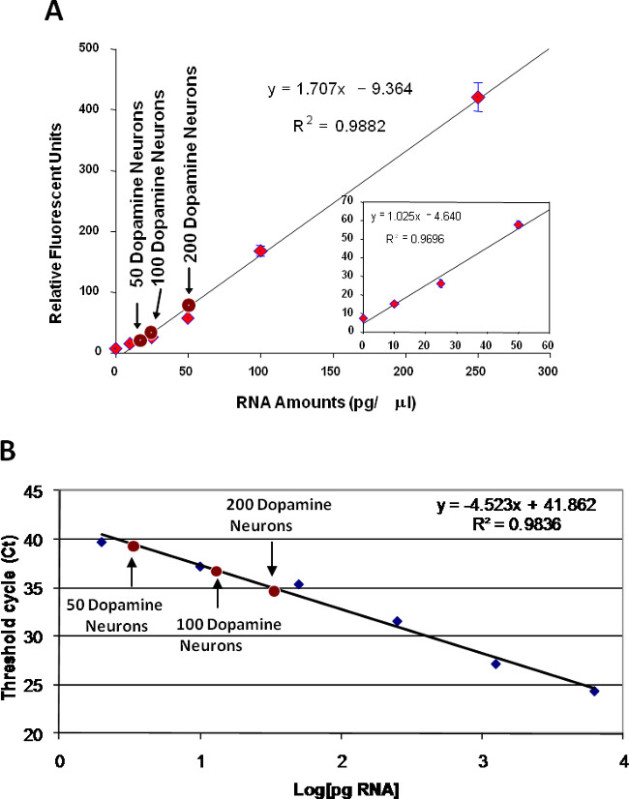 Figure 10. RNA quantity. To determine the quantity of RNA in neurons obtained with LCM, a standard curve of RNA concentrations was produced using a fluorospectrometer and Q-PCR. For the fluorospectrometer measurements (A), the large graph shows the higher range of RNA amounts and the small graph shows the sensitivity of this assay down to 10 pg/µl. Concentrations of RNA obtained from 50, 100 and 200 dopamine neurons was 17.3, 24.8 and 50.9 pg/µl, respectively. Using Q-PCR to measure RNA amounts (B), the concentrations of RNA obtained from 50, 100 and 200 dopamine neurons was 3.55, 6.82 and 20.58 pg/µl, respectively.
Figure 10. RNA quantity. To determine the quantity of RNA in neurons obtained with LCM, a standard curve of RNA concentrations was produced using a fluorospectrometer and Q-PCR. For the fluorospectrometer measurements (A), the large graph shows the higher range of RNA amounts and the small graph shows the sensitivity of this assay down to 10 pg/µl. Concentrations of RNA obtained from 50, 100 and 200 dopamine neurons was 17.3, 24.8 and 50.9 pg/µl, respectively. Using Q-PCR to measure RNA amounts (B), the concentrations of RNA obtained from 50, 100 and 200 dopamine neurons was 3.55, 6.82 and 20.58 pg/µl, respectively.
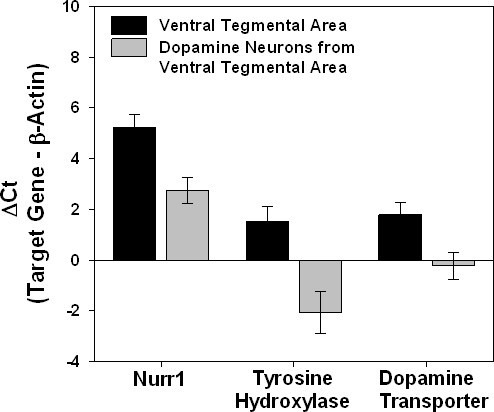 Figure 11. Concentration of dopamine neuron specific genes with the isolation of individual dopamine neurons. The difference in Ct (ΔCt) between dopamine neuron specific genes (Nurr1, tyrosine hydroxylase, and dopamine transporter) and β-actin in samples of dopamine neurons in the ventral tegmental area isolated with LCM as compared to expression in dissections of the entire ventral tegmental region are shown. Since lower Ct values are generated with a higher concentration of the gene of interest, a decrease in the ΔCt indicates an increased expression of the dopamine neuron genes relative to β-actin as other cells (non-dopaminergic neurons and glial cells) will be collected in the ventral tegmental area samples that express β-actin but do not express dopamine neuron genes.
Figure 11. Concentration of dopamine neuron specific genes with the isolation of individual dopamine neurons. The difference in Ct (ΔCt) between dopamine neuron specific genes (Nurr1, tyrosine hydroxylase, and dopamine transporter) and β-actin in samples of dopamine neurons in the ventral tegmental area isolated with LCM as compared to expression in dissections of the entire ventral tegmental region are shown. Since lower Ct values are generated with a higher concentration of the gene of interest, a decrease in the ΔCt indicates an increased expression of the dopamine neuron genes relative to β-actin as other cells (non-dopaminergic neurons and glial cells) will be collected in the ventral tegmental area samples that express β-actin but do not express dopamine neuron genes.
Discussion
LCM is a powerful technique for the dissection of discrete populations of cells or regions of tissue from tissue sections on a microscope slide and the subsequent isolation of RNA, microRNA, DNA and proteins. The use of LCM in combination with analysis using high-throughput technologies such as microarray, next generation RNA sequencing, epigenetic analysis of DNA and proteomics is becoming more and more common as the sensitivity of these techniques improves and the amount of starting material needed is reduced 8-12. With LCM, a better anatomical resolution is achieved for a sample, and the understanding from downstream measures of these samples is greatly enhanced. One caveat with LCM is that it provides a concentrated sample of the cells of interest but not necessarily a pure population of cells. The limits of the precision mean that there will be some contamination from adjacent tissue. However, this technique does concentrate the cells of interest to minimize effects associated with surrounding tissue and cells and maximize the experimental effects on the cells of interest.
Direct versus Indirect Immunohistochemistry
Since LCM is currently utilized primarily for the isolation and measurement of RNA, keeping RNA intact is a very important criterion. Maintaining RNA integrity is the main rationale behind the use of indirect immunohistochemistry to guide tissue dissection which removes the necessity to stain the tissue directly which can degrade the RNA (Figure 10). When the experimental question, however, requires isolation of a specific population of cells, such as dopamine neurons, direct fluorescent immunohistochemistry is required. Using a rapid immunohistochemistry labeling protocol can minimize the degradation of RNA during the procedure. The short incubation times are due to the use of high concentrations of primary and secondary antibodies, concentrations that are around 10-20 fold higher than what is necessary for conventional immunohistochemistry with a 2 h incubation. If however, longer incubation times are required, Brown and Smith used high salt buffers to inhibit RNA degradation and obtain good quality RNA with long incubation times (up to 20 h) 13. This approach can also be used if there is substantial time required between labeling of the tissue and getting access to a LCM system.
Choice of slides - PEN membrane, Silane-prep and uncoated glass slides
The use of uncoated glass slides for LCM have been reported 14. In our laboratory, Silane-prep slides have been found to be an optimal choice, as this coating sufficiently attaches the tissue to the slide for immunohistochemistry without loss of tissue but still allows for attachment of the tissue to the LCM caps and removal from the slide. Uncoated glass slides have showed some tissue loss with the immunohistochemistry procedure. If indirect immunohistochemistry is used, however, tissue retention becomes less of an issue. With proper dehydration, capturing cells or tissue off of Silane-prep slides has not been an issue. Each of the various approaches to isolating neurons or brain regions described in this paper has advantages and disadvantages. For isolation of individual dopamine neurons, the use of Silane-prep slides has the advantage of better optics and is less expensive than the PEN membrane slides. The disadvantage is that sometimes capturing cells can be difficult if there is insufficient dehydration (see below) and some tissue may be left on the slide. The advantage of the PEN membranes slides is that using the combination of the IR laser and UV laser ensures that the dopamine neurons will be collected. Failure to collect cells from a PEN membrane slides almost never occurs, as it is less dependent on dehydration than the glass slides. An additional advantage is that the UV laser can be used to more closely approximate the shape of the neurons of interest, as compared to a circle for capturing neurons off of glass slides with the IR laser. For isolation of regions of tissue, the PEN membrane slides are far superior and justify the added cost. This approach results in complete removal of all the tissue cut out on the membrane, is much faster than capturing a large area of tissue using the IR laser alone and thicker tissue sections can be collected. Using only the IR laser requires that the LCM cap membrane is completely melted over the entire tissue area. Additionally, incomplete collection of the tissue is typical and usually requires multiple attempts. Although this takes longer than isolating tissue from the PEN membrane slide, if the available tissue is already on a glass slide or a UV laser is not available, this is a viable option as most of the tissue can be collected (Figure 8J).
Critical steps
The 100% ethanol incubations are one of the most important steps. In order to collect cells from Silane-prep slides complete dehydration of the tissue is required. Therefore, any water not removed, which is associated mostly with the 100% ethanol incubation, will impact the ability to pick up cells from the slides. In order to address this issue, frequently change the 100% ethanol solutions as absorption of water from the air or transfer from previous washing steps can affect the dehydration. Additionally, molecular sieves are used to absorb any water contamination. The LCM system should ideally be located in a small room with a dehumidifier to maintain low humidity conditions.
Good cryostat sections are critical for proper LCM. Sections need to be flat with folds in the tissue minimized. Folds in the tissue will increase the distance between the LCM cap and prevent it from contacting the tissue. It then becomes difficult to sufficiently melt the membrane cap onto the tissue. For glass slides, typically section thickness needs to be between 6 and 12 µm. Thicker sections can be captured with the PEN membrane slides.
LCM offers a technical capacity to make very precise dissections of tissue and cells from microscope slides. This dramatically increases the resolution of further analysis as compared to gross dissection of tissue pieces. Although, LCM can be time and labor intensive, it does not require extensive training or expertise and, when coupled with other high-throughput technologies, can greatly enhance the understanding of a biological process when discrete cells or anatomical regions are used.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was funded by the National Institute of Mental Health (1R15MH084209 - 01A1), National Institute of Neurological Disorders and Stroke (R21NS058375-01A2) and the National Science Foundation (DBI0723080 07070646).
References
- Emmert-Buck MR, et al. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- Schutze K, Lahr G. Identification of expressed genes by laser-mediated manipulation of single cells. Nature biotechnology. 1998;16:737–742. doi: 10.1038/nbt0898-737. [DOI] [PubMed] [Google Scholar]
- Schutze K, Posl H, Lahr G. Laser micromanipulation systems as universal tools in cellular and molecular biology and in medicine. Cellular and Molecular Biology. 1998;44:735–746. [PubMed] [Google Scholar]
- Vandewoestyne M, Van Nieuwerburgh F, Van Hoofstat D, Deforce D. Evaluation of three DNA extraction protocols for forensic STR typing after laser capture microdissection. Forensic Science International Genetics. 2012;6:258–262. doi: 10.1016/j.fsigen.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Eells JB, Wilcots J, Sisk S, Guo-Ross SX. NR4A gene expression is dynamically regulated in the ventral tegmental area dopamine neurons and is related to expression of dopamine neurotransmission genes. Journal of Molecular Neuroscience. 2012;46:545–553. doi: 10.1007/s12031-011-9642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelan RS, et al. Epigenetic analysis of laser capture microdissected fetal epithelia. Analytical Biochemistry. 2013;442:68–74. doi: 10.1016/j.ab.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, et al. miR-126 contributes to Parkinson's disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiology of Aging. 2014;35:1712–1721. doi: 10.1016/j.neurobiolaging.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm C, et al. Effects of antidepressant treatment on gene expression profile in mouse brain: cell type-specific transcription profiling using laser microdissection and microarray analysis. Journal of Neurochemistry. 2006;97(1):44–49. doi: 10.1111/j.1471-4159.2006.03750.x. [DOI] [PubMed] [Google Scholar]
- Kadkhodaei B, et al. Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2360–2365. doi: 10.1073/pnas.1221077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, et al. Quantitative Proteomics in Laser Capture Microdissected Sleep Nuclei From Rat Brain. Journal of Neurogenetics. 2014. [DOI] [PMC free article] [PubMed]
- Roberts E, et al. Application of laser capture microdissection and protein microarray technologies in the molecular analysis of airway injury following pollution particle exposure. Journal of Toxicology and Environmental Health. A. 2004;67:851–861. doi: 10.1080/15287390490425768. [DOI] [PubMed] [Google Scholar]
- Yuferov V, Nielsen D, Butelman E, Kreek MJ. Microarray studies of psychostimulant-induced changes in gene expression. Addiction Biology. 2005;10:101–118. doi: 10.1080/13556210412331308976. [DOI] [PubMed] [Google Scholar]
- Brown AL, Smith DW. Improved RNA preservation for immunolabeling and laser microdissection. RNA. 2009;15:2364–2374. doi: 10.1261/rna.1733509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran S, McKay JA, McLeod HL, Murray GI. Laser capture microscopy. Molecular Pathology. 2000;53:64–68. doi: 10.1136/mp.53.2.64. [DOI] [PMC free article] [PubMed] [Google Scholar]