

Abstract
Cigarette smoke (CS) has a major impact on lung biology and may result in the development of lung diseases such as chronic obstructive pulmonary disease or lung cancer. To understand the underlying mechanisms of disease development, it would be important to examine the impact of CS exposure directly on lung tissues. However, this approach is difficult to implement in epidemiological studies because lung tissue sampling is complex and invasive. Alternatively, tissue culture models can facilitate the assessment of exposure impacts on the lung tissue. Submerged 2D cell cultures, such as normal human bronchial epithelial (NHBE) cell cultures, have traditionally been used for this purpose. However, they cannot be exposed directly to smoke in a similar manner to the in vivo exposure situation. Recently developed 3D tissue culture models better reflect the in vivo situation because they can be cultured at the air-liquid interface (ALI). Their basal sides are immersed in the culture medium; whereas, their apical sides are exposed to air. Moreover, organotypic tissue cultures that contain different type of cells, better represent the physiology of the tissue in vivo. In this work, the utilization of an in vitro exposure system to expose human organotypic bronchial and nasal tissue models to mainstream CS is demonstrated. Ciliary beating frequency and the activity of cytochrome P450s (CYP) 1A1/1B1 were measured to assess functional impacts of CS on the tissues. Furthermore, to examine CS-induced alterations at the molecular level, gene expression profiles were generated from the tissues following exposure. A slight increase in CYP1A1/1B1 activity was observed in CS-exposed tissues compared with air-exposed tissues. A network-and transcriptomics-based systems biology approach was sufficiently robust to demonstrate CS-induced alterations of xenobiotic metabolism that were similar to those observed in the bronchial and nasal epithelial cells obtained from smokers.
Keywords: Bioengineering, Issue 96, human organotypic bronchial epithelial, 3D culture, in vitro exposure system, cigarette smoke, cilia beating, xenobiotic metabolism, network models, systems toxicology
Introduction
Lungs are directly and constantly exposed to inhaled air that may contain diverse toxicants such as pollutants and constituents of cigarette smoke (CS). Studying the impact of exposure to those toxicants on respiratory tissues is most informative when done in a manner that resembles in vivo exposure. Compared with the classical 2D immersed cell cultures (e.g., normal human bronchial epithelial cells (NHBE)), 3D organotypic tissue models better recapitulate the morphological, physiological, and molecular aspects of the human airway epithelium in vivo1,2: the 3D tissue models contain the diversity of the cell types observed in vivo, including differentiated epithelial cells, ciliated and non-ciliated cells, goblet cells, and basal cells. They have functional tight junctions and exhibit a mucociliary phenotype 1-3. Moreover, the cultures can be grown on a permeable porous membrane, in an air-liquid interface, allowing a direct exposure to aerosol at the apical side (whereas the basolateral side is immersed in culture medium) 3-5. Dvorak and colleagues reported that gene expression profiles of bronchial tissue models were similar to those obtained from human bronchial brushings 3. In addition, Mathis and colleagues showed that the responses of these tissue models to CS were similar to the differences observed between bronchial epithelial cells obtained from smokers and cells obtained from non-smokers 6. Finally, because the bronchial tissue models could be cultured for up to several months 4,5, they could potentially be used to examine the effects of long-term exposure of test items.
Cytotoxicity assessments are common parameters measured following chemical insults or to assess the toxicity of specific compounds or mixtures. For instance, membrane integrity can be measured by a luminescent assay and allows the measurement of a dose-dependent cytotoxic effect on the cell culture 7. However, to assess pathophysiological effects of compounds at subtoxic concentrations, other parameters should be measured. For example, tissue integrity determined using the transepithelial electrical resistance (TEER) assay ensures the functionality of tight junctions and monitors the disruption of the epithelial layer 8,9. Ciliary beating frequency also allows the measurement of CS-related effects on respiratory tissues. A normal beating frequency for the cilia lining bordering the upper and lower respiratory tract is important to protect against airway infections 10. Each of the ciliated columnar epithelial cells of the respiratory epithelium has 200-300 cilia beating at a particular frequency to eliminate infectious agents or inhaled particulate matter trapped in the mucus released by interspersed goblet cells 11. CS contains chemicals that may inhibit ciliary beating 12, leading to a reduced protection of the respiratory tract. This work shows that ciliary beating can be measured in organotypic tissue models. This approach allows assessment of whether epithelial cells exhibit their normal function in the organotypic tissue culture. CS also activates xenobiotic metabolism responses in the respiratory tract to metabolize tobacco smoke constituents 13. The activity of the phase I xenobiotic metabolism enzymes, CYP1A1 and CYP1B1, of the tissue models can be measured. Additionally, as previously reported, global gene expression can be measured in the organotypic bronchial tissue models 6,14,15. A transcriptomic data and network-based systems biology approach is leveraged to assess the impact of CS on xenobiotic metabolism 15.
The methodologies used to expose organotypic 3D bronchial and nasal tissue models to mainstream CS using an in vitro exposure system and to measure the tissue responses to this exposure compared to fresh air exposure (control) are detailed here.
Protocol
1. Culture of the Organotypic Tissue Bronchial Culture Model
Note: Reconstituted tissue models were purchased as inserts that are ready to use. Alternatively, the culture could be developed from primary cells as described for example by Karp and colleagues16. Following receipt of tissues in sterile packaging, always handle the human organotypic tissues under sterile conditions.
Add 0.7 ml of pre-warmed (37 °C) assay medium to each well of a new sterile 24-well plate under the hood.
Remove the packaging of the tissues under the hood, and transfer the tissue culture inserts to the new prepared 24-well plate (described in step 1.1. above).
Maintain and culture the tissues in an incubator at 37 °C (5% CO2, 90% humidity).
Replace the medium every 2 days.
During a medium change, examine the tissues under a light microscope to ensure they they are contamination-free and that there is no leakage of medium.
2. Cigarette Smoke (CS) Exposure using an In Vitro Exposure System
Three days prior to exposure experiments, wash the apical side of the tissue culture with 200 µl of culture medium.
On the day of exposure, if the measurement of ciliary beating is planned before exposure, handle the tissues for ciliary beating measurement as described in section 3.
Pre-warm the climatic chamber of the in vitro exposure system and the cultivation base module to 37 °C. Fill the cultivation base module with 17.5 ml of medium per row.
Remove the tissues from the incubator and place them under the hood to transfer the tissue culture inserts from the culture plate to the cultivation base module of the in vitro exposure system. Additionally, cover the cultivation base module with a glass lid.
Transfer the tissues in the climatic chamber of the in vitro exposure system for exposure to mainstream CS.
- If the in vitro exposure system is equipped with a microbalance, set-up the Quartz crystal microbalance before running the exposure to measure the real-time particle deposition of the smoke on a quartz crystal using the microbalance software.
- Replace the crystals in the microbalance modules connected to each row of the dilution/distribution system.
- Connect the microbalance module to the microbalance software. Set the scale of the measurement to zero in the software.
- Expose the tissues according to the experimental procedure (e.g., as in Figure 1) Note: Prior exposure, reference cigarettes (3R4F) are conditioned between 7 and 21 days under controlled conditions at 22 ± 1 °C and relative humidity of 60 ± 3% according to ISO standard 3402 17.
- Generate cigarette smoke (e.g., from 3R4F cigarettes) using a 30-port carousel smoking machine according to the Health Canada Intense regime: smoke the cigarettes to a standard butt length, i.e., approximately 35 mm at 55 ml puff over 2 sec, twice a minute and 8 sec pump exhaust time. Note: If necessary, dilute the mainstream CS so that the dose is not too toxic. In results reported here, smoke was diluted to 16% (vol/vol).
- Use 60% humidified-air for the control samples (air-exposed).
- Expose the tissues for 6-7 min (i.e., 1 cigarette or air-exposed) in the in vitro exposure system
- Put the tissues back in the incubator at 37 °C (5% CO2, 90% humidity) for 1 hr.
- Repeat steps 2.7.3 and 2.7.4 three more times.
At the end of the exposure experiment, stop the microbalance software. The software will generate a file containing all the measurements along with a graphical representation of the particle deposition.
Transfer back the tissues from the cultivation base module to the culture plate. Place the tissues back in the incubator at 37 °C (5% CO2, 90% humidity) until the measurements for the different endpoints at the specific post-exposure time-points.
3. Measurement of Ciliary Beating
Note: As per experimental plan (Figure 1), record ciliary beating prior to the exposure and directly after the exposure.
Remove the tissue culture plates from the incubator and leave them under the hood at RT for 30 min to stabilize the cilia beating.
Place the tissues under the light microscope connected to the CiliaMetrix Software.
Record the movie and frequency (in Hertz) of the ciliary beating. Measure the frequency every 3 sec for 1 minute.
Return the tissues back to the incubator at 37 °C (5% CO2, 90% humidity).
4. Transepithelial Electrical Resistance (TEER) Measurement
Note: The measurement of TEER is conducted 3 days before and 48 hr after exposure under the hood under sterile condition using Chopstick Electrode (STX-2) connected to a voltohmmeter.
Add 200 µl of culture media to the apical surface of the human bronchial tissues.
Turn on the EVOMX machine; wash the electrode with a 70% ethanol solution.
Wash the electrode with the culture medium.
Measure the resistance (Ω) by inserting the electrode into the medium on the apical side of the tissue culture without touching the tissue.
Repeat step 4.4 five times to obtain quintuplicate measurements.
After the measurements, gently remove the medium from the apical side of the tissue cultures using an aspirator.
Return the tissue cultures to the incubator at at 37 °C (5% CO2, 90% humidity) for culturing.
5. Cytochrome P450 (CYP) 1A1/1B1 Activity
Before the start of the study, prepare the luciferin detection reagent by transferring the entire contents of the bottle of the appropriate reconstitution buffer to the amber bottle containing the luciferin detection reagent (according to manufacturer’s instructions). Store aliquots of 2 ml at -20 °C for a maximum of 1 month.
For the 48 hr post-exposure time, incubate the tissues for 48 hr following CS exposure at 37 °C.
Warm the Luciferin detection reagent to RT.
Incubate tissue inserts for 3 hr or O/N with the Luciferin-CEE substrate (diluted at 1:50) in the basal culture medium.
Transfer 50 µl of culture medium from each tissue insert to a 96-well opaque white luminometer plate at RT.
Add 50 µl of luciferin detection reagent to each well to initiate a luminescent reaction.
Incubate the plate at RT for 20 min.
Read the luminescence using a luminometer. Note: For the luminometer, use an integration time of 0.25–1 sec per well as a guideline.
6. RNA Extraction.
- Before collecting 3D organotypic tissue inserts, prepare 1) a box with dry ice, 2) sufficient amount of blades (one per 3D insert), 3) cold PBS without calcium chloride and magnesium chloride, 4) one 25 ml glass pipet, and 5) sterile glass needles for aspiration.
- Label tubes filled with ceramic beads for homogenizing solid cellular samples and add 700 µl of Qiazol.
- Turn on aspiration machine.
- Collect the plate of the 3D organotypic tissue inserts from the incubator.
- Wash each insert on the plate 3 times with cold PBS. Between the washes, aspirate PBS with the glass needle. After the third wash, aspirate with PBS and cover plate to prevent drying of the insert.
- For each insert from the plate, cut out the insert membrane (on which 3D tissue insert resides) until the membrane lies flat down on the blade.
- Transfer the tissue (along with the insert membrane) from the blade to the homogenizing tube appropriately labelled and screw the lid on the tube, shake it and vortex it.
- Freeze the sample in dry ice and store at -80 °C or continue to the extract the RNA. Note: Prior to the extraction of RNA, label all tubes and columns according to the randomization design (if applicable). Prepare all reagents according to manufacturer’s recommendations and take out samples from -80 °C freezer and thaw them on ice until they are completely defrosted.
- Homogenization
- Grind samples with homogenizing instrument on 6000 Hz for 45 sec. Repeat if the samples are not properly homogenized and leave samples for 5 min at RT.
- Add 140 µl chloroform to the sample and vortex for 10-15 sec.
- Transfer the supernatant from the homogenizing tube to a Phase lock tube and shake on a thermoshaker for 2 min at 1400 rpm and RT.
- Further incubate samples for 2 min at RT and centrifuge samples at 12,000 x g for 15 min at 4 °C.
- RNA precipitation, wash, suspension and purification in QIAcube.
- Transfer the upper aqueous phase in a new 2 ml tube.
- Load all necessary reagents in the extraction robot according to the the manufacturer protocol sheet, select the appropriate protocol, set the elution volume (30 µl) and run the extraction robot.
- When the run is finished, remove the rotor adapters and keep elution tubes immediately at 4 °C for further analysis or at -80 °C for long term storage.
- Quality control of the isolated RNA
- Determine the quantity of the isolated RNA using an UV reader according to manufacturer’s instructions.
- Check the quality of RNA and the RNA integrity using microfluidic-gel-based lab-on-a-chip and an UV reader, according to the manufacturer instructions.
7. Microarray Workflow
Note: The process described below focuses on the method for Affymetrix GeneChip High throughput 3’IVT Express Kit, which is used for target synthesis for hybridization on Affymetrix 3’ gene expression cartridges starting from the preparation of the RNA to the use of the a complex liquid handling system (e.g., pipetting, diluting, dispensing, or integrating).
- Preparation
- Randomize samples to avoid batch effects. The number of samples processed by batches is from 1 to 96, including one positive (Commercial Human universal reference RNA) and one negative (RNase free water) control per batch.
- Start the target synthesis using 100 ng of total RNA in 3 µl of RNAse-free water. For 100 ng of total RNA, use a 16 h incubation time.
- RNA Amplification
- Prepare the Poly-A RNA spike-in control solution by a serial dilution of the Poly-A RNA Stock with Poly-A Control Dilution Buffer:
- For dilution 1, prepare 2 µl polyA control stock in 36 µl dilution buffer.
- For dilution 2, prepare 2 µl of dilution 1 in 98 µl of dilution buffer.
- For dilution 3, prepare 4 µl of dilution 2 in 196 µl of dilution buffer.
- For dilution 4, prepare 20 µl of dilution 3 in 180 µl of dilution buffer.
- Dilute the RNA in RNAse free water to get a concentration of 33.3 ng/µl.
- Add 2 µl of the polyA control solution to the 3 µl of total RNA sample directly in a 96 well plate. This step is performed by the liquid handling system.
- Prepare the liquid handling system and target synthesis. Place the plates on the robot. Place all the required reagents and labware on the deck. The robot will start the procedure by preparing the appropriate mastermixes and incubate the samples in the automated remote controlled PCR block.
- First Quality Control Check: Control Of The Amplification Process
- If the quality of aRNA is acceptable, perform the fragmentation step by adding to each sample 8 µl of fragmentation 5X buffer (This step is performed by the liquid handling system). Otherwise, perform the previous step again starting from the RNA. Use fragmented aRNA immediately or store undiluted and fragmented aRNA at -20 °C (or -70 °C for longer-term storage).
- Perform a second Quality Control analysis by picking up randomly 12 samples in the plate and transfer 1 µl on the microfluidics gel based system to check the efficiency of the fragmentation.
- Hybridization procedure
- Scan the chip barcodes and register each sample in the manufacturer’s software according to the instructions.
- Prepare the hybridization mix as follows for a single reaction and add it to 33 µl of the fragmented and labelled aRNA: 4.2 µl of control oligonucleotide B2 (3nM), 12.5 µl of 20x eukaryotic hybridization controls, 25 µl of 100% DMSO, 125 µl of 2x hybridization buffer and 50 µl of H2O for a final volume of 216.7 µl Note: Hybridize, wash and scan positive and negative controls before processing the samples.
- Pre-wet the array with 200 µl of pre-hybridization mix contained in the Hybridization Wash and Stain kit.
- Place the chip in the hybridization oven set at 45 °C and 60 RPM for 10 min.
- In the meantime, denature the plate (containing the final cocktail hybridization mix) in a PCR block at 99 °C for 5 min and 45 °C for 5 min.
- Remove the pre-hybridization mix from the array and replace it with 200 µl of the hybridization cocktail target (one sample per chip).
- Place the array into the hybridization oven set at 45 °C for 16 hr (O/N) with a rotation speed of 60 RPM.
- Washing and staining
- Run “Fluidics” from the software menu bar. In the Fluidics dialog box, select the module of interest (1 - 4), then select the Prime_450 program to all modules. Place the tubing for Wash Buffer A in a bottle (400 ml) and for Wash Buffer B in a bottle (200 ml), as well as for milliQ Water in a bottle (500 ml).
- Subsequently, follow the LCD screen instructions. Lift up the needles and place 600 µl stain cocktail 1 (SAPE Solution Mix) and 600 µl stain cocktail 2 (Antibody Solution Mix) containing microcentrifuge tubes at positions #1 and #2, and 900 µl Array Holding buffer solution at position #3.
- After the O/N incubation in the oven, aspirate the cocktail from the chip and fill the microarray with 250 µl Wash Buffer A. Assign the right chip to each module, select the FS450-001 protocol and run each module, following instructions on screen. This process lasts 90 min.
- Once the LCD screen indicates “Eject Cartridge” message, remove the chip and inspect if there are any bubbles. If bubbles are present, fill the array entirely with the Array holding buffer. Apply stickers onto the septa to avoid any leaks in the scanner and clean the microarray window prior to loading in the Scanner.
- Scanning.
- Warm up the scanner. Load the chip into the autoloader of the scanner and start scanning. Note: After ~12 min per chip, a .CEL file per chip is generated.
- Check the image and align the grid (if necessary) to the spot to identify the probe cells. Note: The dataset has been submitted to Arrayexpress (Accession code = E-MTAB-1721).
8. Network-based Systems Biology Analysis for an Impact Assessment
- Microarray data processing. Note: Data processing and scoring methods were implemented using the R statistical environment version 2.14.
- Open an R 18 session and load the affy 19, gcrma, and affyPLM packages installed by running the commands: library(affy) > library(gcrma) > library(affyPLM)
- Read raw data files running the command: data.affybatch<-ReadAffy(“path to the folder where are stored the CEL files”)
- Substract the background correction and quantile normale to generate probe set expression values using the gcrma package, by running the command: eset.norm<-gcrma(data.affybatch)
- Quality control.
- Generate RNA degradation plots with the command: deg<-AffyRNAdeg(data.affybatch)
- Extract the coefficient of the RNA degradation slope running the command: slope=deg$slope
- Plot the slope coefficient and identify possible outliers.
- Generate NUSE and RLE plots running the following commands: Pset<-fitPLM(data.affybatch) > RLE(Pset) > NUSE(Pset).
- Identify if some of the boxplots generated are outliers: an array is considered outlier if at least 2 of the 3 QC metrics defined below deviate from the other arrays: - deg$slope is different from the average deg$slope - NUSE plot: an array is considered outlier if the upper quartile falls below 0.95 or the lower quartile above 1.05, i.e., if the boxplot is entirely above 1.05 or entirely below 0.95. - RLE plot: an array is considered outlier if the median of the RLE distribution is greater than 0.1 or smaller than -0.1.
- If an array is identified as an outlier, then discard, and start again from step 8.1.2.
- Fit an overall linear model to the data for the specific contrasts of interest (i.e., the comparisons of “treated” and “control” conditions) generating raw P values for each probe set on the microarray, which can be further adjusted using the Benjamini-Hochberg procedure of the Limma package. Select a probeset per gene to keep as representative of the gene for further analyses as described in 20. Note: A blocking factor (the exposure plate) from the experiment design was accounted in the model for data processing.
- Network-based analysis. Note: The method as implemented here is described in detail in Martin et al. (in revision in BMC bioinformatics).
- For each pairwise comparison of interest, start from the computed (log2-) fold changes (treatment vs. control) for each gene under the network (from step 8.2).
- Compute the signed Laplacian matrix L of the network defined by L(i,j)=- sign(i~j)w(i,j) if there is an edge of weight w(i,j) between node i and j, L(i,j)=deg(i) is the weighted degree of i and L(i,j)=0 else. The weight w(i,j) are equal to 1 if i and j are in the backbone and w(i,j)=1/n if i is a backbone node and j is one of its n neighbors in the transcript layer.
- Compute scores for the backbone by f=L3-1L2Tx where L3 is the sub-matrix of L to the backbone nodes and L2 is the sub-matrix of L whose rows correspond to the transcriptional layer nodes and column to the backbone nodes
- Compute the signed Laplacian, Q, of the network defined by the backbone network where all the edge signs are reversed.
- Compute the NPA score by NPA=fTQf.
- Compute the confidence interval, the backbone edge permutation p-value and the transcript layer permutation p-value. Note: In addition to the confidence intervals of the NPA scores, which account for the experimental error (e.g., the biological variation between samples in an experimental group), companion statistics were derived to describe the specificity of the NPA score to the biology described in the network. Because NPA is a quadratic form of the fold changes, its variance can be computed based on the fold-change estimated variances. A confidence interval is subsequently derived using the central limit theorem. Two permutation tests were implemented 21 whereby first, to assesses if the results were specific to the underlying evidence (i.e., gene fold-changes) in the model, leading to a permutation P-value (denoted by *O in the figures when P-value < 0.05). Second, to assess whether the “cause-and-effect” layer of the network significantly contributed to the amplitude of the network perturbation (denoted by K* in the figures when P-value < 0.05).
- Consider the network as impacted by the treatment if the confidence interval does not contain zero AND the two permutation p-values < 0.05.
- Compute the leading nodes which are defined as the key nodes in the backbone contributing up to 80% of the NPA score.
Representative Results
In results described below, the organotypic tissues were primary human epithelial cells isolated from healthy, non-smoking, Caucasian donors and were reconstituted using fibroblasts 15.
3D bronchial and nasal tissue modelsIn vitro bronchial and nasal organotypic models resemble at the cellular level the human respiratory tract epithelium (Figure 2).
CS exposure Organotypic bronchial and nasal tissue models can be repeatedly exposed to mainstream CS at the air-liquid interface. The smoke exposure experimental procedure used for the results illustrated here is depicted in Figure 1.
Recording of ciliary beating Cilia beating was recorded in the air-exposed tissues as illustrated in the videos. CS exposure was associated with a reduction of the ciliary beating frequency: the stronger peak observed around 4 Hz in the sham exposed and before exposure is not observed any longer after CS exposure (Figure 3). This reduced frequency would make the cilia beating less efficient to remove mucus and infectious agents 22.
Measurement of TEER and CYP1A1/1B1 activity TEER was measured 48 hr after the last exposure to ensure the tissue is still functional. CYP1A1/1B1 activity was also measured 48 h after end of exposure as illustrated in Figure 1. The TEER values in the CS-exposed tissues were not significantly different as compared to the air-exposed tissues confirming that there is no major cytotoxic effect of smoke at this concentration. At the 48 h post-exposure, CYP1A1/1B1 activity of the tissues was slightly increased, indicating a modest increase of tissue defense mechanism following the exposure (Figure 4).
Gene expression profiles and network-based systems biology approach to study the impact of CS exposure on the alteration of xenobiotic metabolism Tissues were collected at 48 h post-exposure time points. Subsequently, microarray analysis was conducted to generate gene expression profiles of the CS- and air-exposed tissues. Impact of the CS exposure on xenobiotic metabolism was investigated using a network-based systems biology approach leveraged from the gene-expression profiles and as reported earlier 15. Using a network-based systems biology approach shows that CS exposure impacts at the level of backbone nodes in the Xenobiotic Metabolism network model were nicely correlated between the in vitro and in vivo datasets (Figure 5). In contrast, at the level of each individual gene-expression changes, poor correlations were observed between the in vitro (CS-exposed vs. air-exposed) and in vivo (smokers vs. nonsmokers).
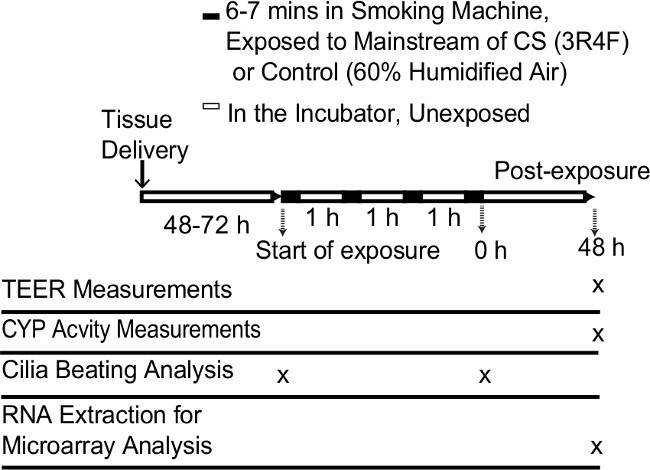 Figure 1. Schematic procedure of the repeated exposure of CS to the organotypic bronchial tissue models. Abbreviations: CYP, cytochrome P450s; CS, cigarette smoke; TEER, transepithelial electrical resistance.
Figure 1. Schematic procedure of the repeated exposure of CS to the organotypic bronchial tissue models. Abbreviations: CYP, cytochrome P450s; CS, cigarette smoke; TEER, transepithelial electrical resistance.
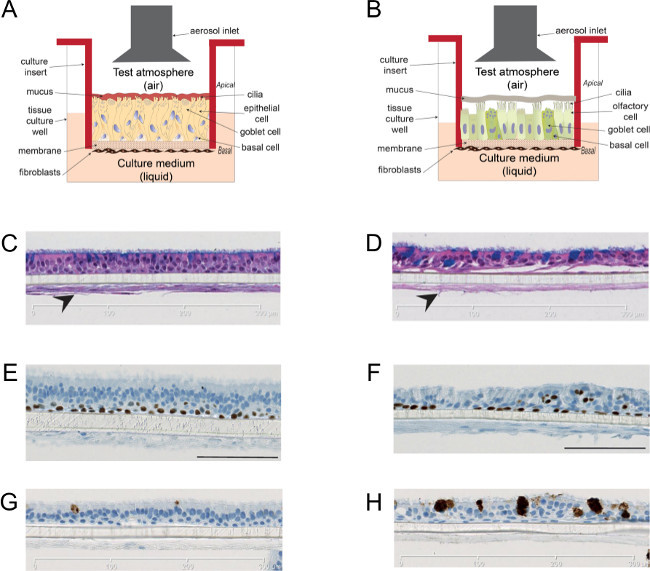 Figure 2.Organotypic bronchial and nasal epithelial tissue culture models. Cartoon illustrating the bronchial (A) and nasal (B) tissue culture insert at the air-liquid interface. The in vitro models contained ciliated cells shown in the apical layer of the Hematoxylin & Eosin stained cells (C for bronchial, D for nasal) as reported previously 15. The models were co-cultured with fibroblasts that are important for the growth and differentiation of epithelial cells (indicated by arrows). Staining of airway lineage markers: p63 (E for bronchial, F for nasal) and Muc5AC (G for bronchial, H for nasal) are shown as reported previously 15. Please click here to view a larger version of this figure.
Figure 2.Organotypic bronchial and nasal epithelial tissue culture models. Cartoon illustrating the bronchial (A) and nasal (B) tissue culture insert at the air-liquid interface. The in vitro models contained ciliated cells shown in the apical layer of the Hematoxylin & Eosin stained cells (C for bronchial, D for nasal) as reported previously 15. The models were co-cultured with fibroblasts that are important for the growth and differentiation of epithelial cells (indicated by arrows). Staining of airway lineage markers: p63 (E for bronchial, F for nasal) and Muc5AC (G for bronchial, H for nasal) are shown as reported previously 15. Please click here to view a larger version of this figure.
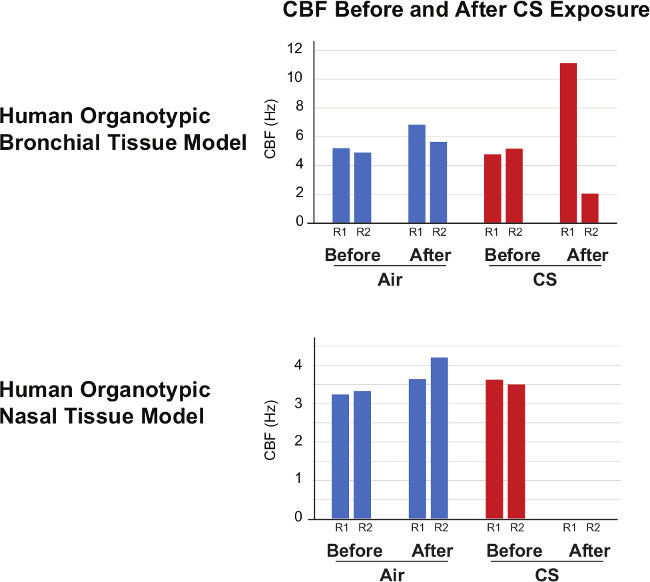 Figure 3. Cilia Beating Frequency. Cilia beating frequency (CBF) was measured in air-exposed and in the culture before and after exposure. Decreased CBF was seen after CS exposure (representative results from two insert are shown as replicate 1 (R1) and replicate 2 (R2)).
Figure 3. Cilia Beating Frequency. Cilia beating frequency (CBF) was measured in air-exposed and in the culture before and after exposure. Decreased CBF was seen after CS exposure (representative results from two insert are shown as replicate 1 (R1) and replicate 2 (R2)).
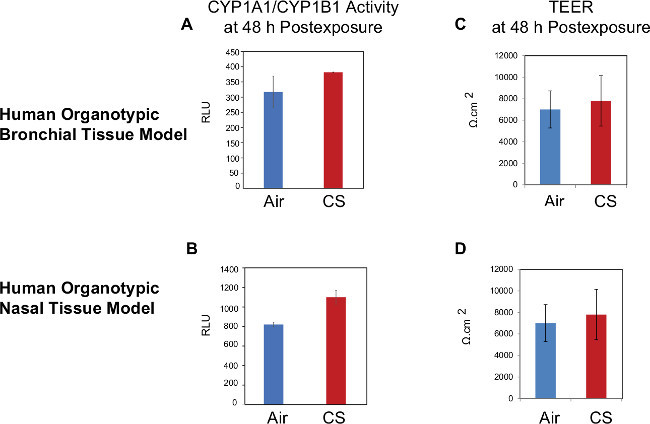 Figure 4.Measurement of CYP1A1/CYP1B1 activity and TEER. Left pannels indicate the CYP1A1/CYP1B1 activity in the bronchial (A) and nasal (B) tissue models. Right panels indicate the TEER measurement in the bronchial (C) and nasal (D) tissue models. Means ± SD are shown. Abbreviations: CS, cigarette smoke; CYP, cytochrome P450; TEER, transepithelial electrical resistance; RLU, relative luminescense unit.
Figure 4.Measurement of CYP1A1/CYP1B1 activity and TEER. Left pannels indicate the CYP1A1/CYP1B1 activity in the bronchial (A) and nasal (B) tissue models. Right panels indicate the TEER measurement in the bronchial (C) and nasal (D) tissue models. Means ± SD are shown. Abbreviations: CS, cigarette smoke; CYP, cytochrome P450; TEER, transepithelial electrical resistance; RLU, relative luminescense unit.
 Figure 5.Network-based systems biology approach for the impact assessment of CS exposure in the context of xenobiotic metabolism. The changes of gene expressions levels were used to compute the activation of backbone node of the Xenobiotic Metabolism network model (A) as reported in a previous publication 15. The figure on the right illustrates the concept of the quantification of the backbone nodes, also known as “differential network backbone value,” using the NPA approach. The blue ovals represent the activity of the backbone nodes (i.e., the functional layer) and the green balls represent the expression of genes (i.e., the transcriptional layer). Impacts of CS exposure in organotypic bronchial tissues (in vitro, CS-exposed vs. air-exposed) at the 48 h of postexposure were compared to those of smoking on human bronchial epithelial collected by bronchoscopy and nasal epithelial by burshing the inferior turbinate (GSE16008) (in vivo, smokers vs. nonsmokers) (B). Insets, correlation of the gene expression changes between the in vitro and in vivo datasets. Abbreviations: CS, cigarette smoke; NPA, network perturbation amplitude. Please click here to view a larger version of this figure.
Figure 5.Network-based systems biology approach for the impact assessment of CS exposure in the context of xenobiotic metabolism. The changes of gene expressions levels were used to compute the activation of backbone node of the Xenobiotic Metabolism network model (A) as reported in a previous publication 15. The figure on the right illustrates the concept of the quantification of the backbone nodes, also known as “differential network backbone value,” using the NPA approach. The blue ovals represent the activity of the backbone nodes (i.e., the functional layer) and the green balls represent the expression of genes (i.e., the transcriptional layer). Impacts of CS exposure in organotypic bronchial tissues (in vitro, CS-exposed vs. air-exposed) at the 48 h of postexposure were compared to those of smoking on human bronchial epithelial collected by bronchoscopy and nasal epithelial by burshing the inferior turbinate (GSE16008) (in vivo, smokers vs. nonsmokers) (B). Insets, correlation of the gene expression changes between the in vitro and in vivo datasets. Abbreviations: CS, cigarette smoke; NPA, network perturbation amplitude. Please click here to view a larger version of this figure.
Discussion
Here, we have demonstrated the applicability of human organotypic bronchial and nasal tissue models to assess the impact of repeated CS exposure. As an alternative to animal testing, a number of exposure systems were developed for toxicological assessments of aerosol exposure in vitro (e.g., Vitrocell, Cultex, ALICE, etc.). These exposure modules can also be utilized for a toxicological assessment of airborne pollutants, airborne particles, nanoparticles, etc. In this study, we used the Vitrocell system that can accomodate up to 48 different samples simultaneously, allowing for larger scale experiments and lower variability between treatments. For every aerosol exposure in vitro, the risk of tissue culture contamination remains a major risk whose mitigation demands a careful handling of the tissue cultures throughout the experiments.
Measuring particle deposition in real time on the microbalance during the exposure experiment allows to monitor that the CS doses generated from the exposure system are conforming to the expectations. To ensure the accuracy of the measured particle deposition, setting-up the online measurement correctly before the exposure is critial, e.g., setting up the scale to 0. Additionally, because of the difference between the areas of the microbalance (cm2) and the tissue culture insert (0.33 cm2), we adjusted the final calculation to the area of the culture insert: only 33% of the deposition on the microbalance reflect the actual deposition in the culture insert.
Measurement of TEER to ascertain tight-junction barrier functionality and to assess disruption of the epithelial layer is a relatively easy procedure to implement, which we reported here. However, because the bronchial and nasal tissue models contain mucus-producing interspersed goblet cells, apical washing needs to be performed before the TEER measurement. The apical washing is critical because the presence of the mucus layer and the variability of its thickness can bias the measurement of TEER, interferring with the impact of CS exposure. This notion is in agreement with what was reported by Hilgendorf and colleagues, in which the permeability of Caco-2 cells was affected by the co-culturing with the mucus-producing goblet cell line HT29 23. The mucus needs to be washed off prior to TEER measurement, because apical washing right before the exposure may interfere with the tissue responses to CS. Therefore, the measurement is performed three days before exposure, and not right before exposure.
We showed that CYP1A1/CYP1B1 activity could be measured from the organotypic culture models following CS exposure although the activity was only modestly increased by CS. This weak signal can be amplified by a longer incubation of the CYP1A1/1B1 substrate (i.e., luciferin-CEE), for example for 24 hr (data not shown). One of the limitations of the CYP activity measurement in the present work is the absence of normalization to the CYP protein level or to the cell count, which could be taken into consideration for future studies to ensure that the alteration on the enzyme activity is not influenced by the alteration of either the protein level or the cell count.
We reported that CS exposure inhibited the ciliary beating in both the nasal and bronchial tissue models. Similar observations were done in diverse mammalian and non-mammalian models 12. For ciliary beating measurement, ensuring that the tissues are handled and treated in a similar manner is critical, for example if medium change is implemented, it should be applied for all samples. Sutto and colleagues reported that pH affected the mammalian ciliary beating frequency 24. Thus, when comparing beating frequencies between cells treated with different compunds/mixtures, pH adjustment should be considered to minimize the variability of ciliary beating measurement. Moreover, the temperature at which the measurement is conducted is also critical as the frequency of ciliary beating drops with decreasing temperature. To minimize the variability due to these changes, a stage-top incubator, equiped with temperature, CO2, and humidity control was used in this study. Despite this, we observed that the ciliary beating frequencies in the bronchial tissues after CS exposure were highly variable (i.e., increase in one insert and decrease in the other insert), suggesting that the ciliary beating is highly disturbed right after exposure. In contrast, we observed the absence of measurable ciliary beating frequencies in the nasal tissue after CS exposure, suggesting that the response of the nasal tissue is more sensitive and consistent. This is in agreement with a previous publication showing that the nasal tissue has a lower capacity to detoxify as compared with the bronchus 25.
Finally, we showed that gene expression profiling from the organotypic bronchial and nasal tissue models exposed to CS could demonstrate an impact of CS on xenobiotic metabolism. Interestingly, the observed alteration in xenobiotic metabolism in the organotypic bronchial and nasal in vitro models exposed to CS resemble that of the in vivo situation in smokers as discussed in greater detail in a previous publication 15. For gene expression analyses, using the robotic instrument makes a high-throughput analysis possible. Moreover, automatic robotic handling further increases the consistency and accuracy of the gene expression results. Nonetheless, fast collection of the tissue samples was critical to avoid RNA degradation during the RNA extraction. The RNA processing and transcriptomic approaches described here can also be applied to in vivo tissue samples.
Disclosures
This study was funded by Philip Morris International.
Acknowledgments
The authors would like to thank Maurice Smith and Marja Talikka for their review of the manuscript.
References
- Pezzulo AA, et al. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. American Journal of Physiology - Lung Cellular and Molecular Physiolog. 2011;300:L25–L31. doi: 10.1152/ajplung.00256.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nature reviews Molecular cell biolog. 2007;8:839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- Dvorak A, Tilley AE, Shaykhiev R, Wang R, Crystal RG. Do airway epithelium air–liquid cultures represent the in vivo airway epithelium transcriptome. American journal of respiratory cell and molecular biolog. 2011;44:465. doi: 10.1165/rcmb.2009-0453OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiszniewski L, et al. Long-term cultures of polarized airway epithelial cells from patients with cystic fibrosis. American Journal of Respiratory Cell and Molecular Biolog. 2006;34:39–48. doi: 10.1165/rcmb.2005-0161OC. [DOI] [PubMed] [Google Scholar]
- Balharry D, Sexton K, BéruBé KA. An in vitro approach to assess the toxicity of inhaled tobacco smoke components: nicotine, cadmium, formaldehyde and urethane. Toxicolog. 2008;244:66–76. doi: 10.1016/j.tox.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Mathis C, et al. Human bronchial epithelial cells exposed in vitro to cigarette smoke at the air-liquid interface resemble bronchial epithelium from human smokers. Am J Physiol Lung Cell Mol Physio. 2013;304:L489–L503. doi: 10.1152/ajplung.00181.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho M-H, et al. A bioluminescent cytotoxicity assay for assessment of membrane integrity using a proteolytic biomarker. Toxicology in Vitr. 2008;22:1099–1106. doi: 10.1016/j.tiv.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CE, Torr EE, Mohd Jamili NH, Bosquillon C, Sayers I. Evaluation of Differentiated Human Bronchial Epithelial Cell Culture Systems for Asthma Research. Journal of Allerg. 2012;2012:11. doi: 10.1155/2012/943982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume LF, Denker M, Gieseler F, Kunze T. Temperature corrected transepithelial electrical resistance (TEER) measurement to quantify rapid changes in paracellular permeability. Pharmazi. 2010. pp. 19–24. [PubMed]
- Chhin B, et al. Ciliary beating recovery in deficient human airway epithelial cells after lentivirus ex vivo gene therapy. PLoS genetic. 2009;5:e1000422. doi: 10.1371/journal.pgen.1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Meng N, Wang H, Zhang L. Comparison of human nasal epithelial cells grown as explant outgrowth cultures or dissociated tissue cultures in vitro. Front Me. 2013;7:486–491. doi: 10.1007/s11684-013-0287-x. [DOI] [PubMed] [Google Scholar]
- Talbot P. In vitro assessment of reproductive toxicity of tobacco smoke and its constituents. Birth defects research. Part C, Embryo today : review. 2008;84:61–72. doi: 10.1002/bdrc.20120. [DOI] [PubMed] [Google Scholar]
- Port JL, et al. Tobacco smoke induces CYP1B1 in the aerodigestive tract. Carcinogenesi. 2004;25:2275–2281. doi: 10.1093/carcin/bgh243. [DOI] [PubMed] [Google Scholar]
- Hoeng J, et al. A network-based approach to quantifying the impact of biologically active substances. Drug Discov Toda. 2012;17:413–418. doi: 10.1016/j.drudis.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Iskandar AR, et al. Systems approaches evaluating the perturbation of xenobiotic metabolism in response to cigarette smoke exposure in nasal and bronchial tissues. Biomed Res In. 2013;2013:512086. doi: 10.1155/2013/512086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PH, et al. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods in molecular biolog. 2002;188:115–137. doi: 10.1385/1-59259-185-X:115. [DOI] [PubMed] [Google Scholar]
- ISO 3402: Tobacco and tobacco products -- Atmosphere for conditioning and testing. International Organization for Standardization; 1999. [Google Scholar]
- R: A Language and Environment for Statistical Computing. The R Core Team; 2011. [Google Scholar]
- Gautier L, Moller M, Friis-Hansen L, Knudsen S. Alternative mapping of probes to genes for Affymetrix chips. BMC Bioinformatic. 2004;5:111. doi: 10.1186/1471-2105-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermida L, et al. Confero: an integrated contrast data and gene set platform for computational analysis and biological interpretation of omics data. BMC genomic. 2013;14:514. doi: 10.1186/1471-2164-14-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson TM, et al. Quantitative assessment of biological impact using transcriptomic data and mechanistic network models. Toxicology and applied pharmacolog. 2013;272:863–878. doi: 10.1016/j.taap.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Teff Z, Priel Z, Gheber LA. The forces applied by cilia depend linearly on their frequency due to constant geometry of the effective stroke. Biophysical journa. 2008;94:298–305. doi: 10.1529/biophysj.107.111724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf C, et al. Caco-2 versus Caco-2/HT29-MTX co-cultured cell lines: permeabilities via diffusion, inside- and outside-directed carrier-mediated transport. J Pharm Sc. 2000;89:63–75. doi: 10.1002/(SICI)1520-6017(200001)89:1<63::AID-JPS7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Sutto Z, Conner GE, Salathe M. Regulation of human airway ciliary beat frequency by intracellular pH. The Journal of physiolog. 2004;560:519–532. doi: 10.1113/jphysiol.2004.068171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, et al. Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiological genomic. 2010;41:1–8. doi: 10.1152/physiolgenomics.00167.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]