

Abstract
Here we describe a protocol to generate a co-culture consisting of 2 different neuronal populations. Induced pluripotent stem cells (iPSCs) are reprogrammed from human fibroblasts using episomal vectors. Colonies of iPSCs can be observed 30 days after initiation of fibroblast reprogramming. Pluripotent colonies are manually picked and grown in neural induction medium to permit differentiation into neural progenitor cells (NPCs). iPSCs rapidly convert into neuroepithelial cells within 1 week and retain the capability to self-renew when maintained at a high culture density. Primary mouse NPCs are differentiated into astrocytes by exposure to a serum-containing medium for 7 days and form a monolayer upon which embryonic day 18 (E18) rat cortical neurons (transfected with channelrhodopsin-2 (ChR2)) are added. Human NPCs tagged with the fluorescent protein, tandem dimer Tomato (tdTomato), are then seeded onto the astrocyte/cortical neuron culture the following day and allowed to differentiate for 28 to 35 days. We demonstrate that this system forms synaptic connections between iPSC-derived neurons and cortical neurons, evident from an increase in the frequency of synaptic currents upon photostimulation of the cortical neurons. This co-culture system provides a novel platform for evaluating the ability of iPSC-derived neurons to create synaptic connections with other neuronal populations.
Keywords: Developmental Biology, Issue 96, Neuroscience, Channelrhodopsin-2, Co-culture, Neurons, Astrocytes, induced Pluripotent Stem Cells, Neural progenitors, Differentiation, Cell culture, Cortex
Introduction
In recent years, our understanding of neuron and neuronal circuit function have been revolutionized by several optogenetic tools that control neuronal excitability. One such tool is the light-activated cation channel, channelrhodopsin-2 (ChR2): neurons expressing ChR2 fire action potentials in response to light stimulation1-3. Because neurons are not ordinarily sensitive to light, this remarkable ability opens up new avenues for the precise temporal and spatial control of the activity of genetically-defined populations of neurons4 and has greatly accelerated studies of brain function. ChR2 has been applied to stem cell research for different purposes, from monitoring neuronal development5 to regulating activity of neural networks6.
Here we used ChR2 to increase the utility of a neuronal co-culture system. Co-culture systems enable the assessment of interactions between different cell types. Co-cultures of neurons and glia, the main cell types of the nervous system, are commonly used to investigate signaling between these different cell types, as well as to evaluate the significance of this signaling for physiological responses, propagation of damage and to examine the molecular mechanisms that are potentially involved in this signaling7. A previous study revealed that human iPSCs differentiate very quickly into neurons in the presence of rat cortical primary culture containing neurons, astrocytes, oligodendrocytes, and microglia8.
In this protocol, we demonstrate a method that utilizes ChR2 in a co-culture system to interrogate synaptic connections between rat cortical neurons and neurons derived from human induced pluripotent stem cells (iPSCs). We describe steps to dissect and harvest brain tissues9 as well as to maintain and differentiate mouse neural progenitor cells (NPCs) into astrocytes and human NPCs into neurons to create the co-culture system. To establish this co-culture system, we used the integration-free reprogramming method described by Okita et al.10 to generate human iPSCs. These iPSCs were then efficiently differentiated into NPCs in chemically-defined conditions using small-molecule inhibitors as described by Li et al.11
In the co-cultures, the different populations of neurons can be identified via detection of ChR2 expression in cortical neurons and tagging of iPSC-derived neurons via the florescent protein, tandem dimer Tomato (tdTomato). Combining electrophysiological recordings from iPSC-derived neurons with optogenetic photostimulation of the cortical neurons allows assessment of the ability of these two types of neurons to form circuits with each other. Specifically, photostimulation of ChR2-expressing cortical neurons evoke synaptic responses in iPSC-derived neurons that have formed synaptic circuits with the photostimulated cortical neuron network. We demonstrate that increased postsynaptic currents are indeed detected in iPSC-derived neurons under such conditions. This protocol allows the assessment of synaptic circuitry under a variety of experimental conditions, including recapitulation of different neurological disease models by using iPSCs derived from fibroblasts from disease patients.
Protocol
NOTE: All experiments are performed following protocols approved by the Institutional Animal Care and Use Committee at SingHealth.
1. Harvesting Early Postnatal Mouse Brains
Before dissection, prepare the hippocampal tissue digestion solution: 10 µl papain per 1 ml 1x Earle’s Balanced Salt Solution (EBSS) (+ 1% HEPES) with Deoxyribonuclease I (DNase I) (250 U/ml) and Dispase II (1 U/ml). Make approximately 2.5 ml of the tissue digestion solution and filter it using a 0.20 µm filter unit. Keep the solution warm at 37 °C until use.
Anesthetize the postnatal day 5 (P5) mice pups by putting them in ice for 5 min. Remove from ice and test for pain reflex via toe or tail pinch. Spray the pups with 70% ethanol. Decapitate the pups and place the heads in a petri dish over ice.
Make an incision in the skin along the midline, from the base of the skull to the nose, with a pair of small scissors or fine dissecting forceps. Peel open the skin and make a similar incision through the skull. Make two additional horizontal cuts on each side of the skull, one rostral (at the bregma) and one caudal (at the lambda).
Peel open the skull along the incised lines to expose the underlying brain. Use a pair of forceps to remove the skull overlying the olfactory bulbs, and any midline bone between them. Do this while holding the head stable with a pair of forceps in the non-dissecting hand.
- Ease the flat part of a spatula between the ventral part of the brain and the base of the skull at the caudal end. Keeping the spatula parallel to the base of the skull under the brain, move it towards the rostral end of the skull to sever any adhesions between the brain and the base of the skull.
- Scoop the brain out with the spatula and place it in a Petri dish containing ice-cold 1x EBSS (+ 1% HEPES). Keep tissues submerged at all times.
For all micro-dissection steps, work under a dissecting microscope and use a pair of fine dissecting forceps in each hand, one for cutting tissue, and the other for holding the tissue stable.
Make a cut just posterior to the cerebral cortex to separate it from the hindbrain. Cut along the midline to separate the cerebral cortex into its two hemispheres. Remove the meninges from the cerebral hemispheres.
- Place a hemisphere medial side up and insert the forceps into the lateral ventricle underneath the hippocampus. Cut away the midbrain.
- Make cuts at the superior and inferior ends of the hippocampus, and near the dentate/entorhinal cortex junction to isolate the hippocampus from the cortex. Collect the hippocampi in a dish containing ice-cold 1x EBSS (+ 1% HEPES).
2. Hippocampal Tissue Dissociation
Transfer the hippocampi to a dish containing the pre-warmed hippocampal tissue digestion solution. Mince the hippocampal tissues to as fine as possible and incubate for 30 min at 37 °C.
Gently dissociate tissues into single cells by mechanical pipetting and transfer cells into a 15 ml centrifuge tube.
Centrifuge at 200 x g for 5 min and remove the supernatant.
Resuspend cell pellet in 20 ml 0.5x Hank’s Balanced Salt Solution (HBSS) containing sucrose (0.9 M) and centrifuge at 200 x g for 10 min.
Remove the supernatant and resuspend cell pellet in 2 ml of NPC maintenance medium (Table 1), placed on top of 10 ml of 4% Bovine Serum Albumin (BSA) in 1X EBSS solution and centrifuge at 200 x g for 5 min.
Remove supernatant and add mouse NPC maintenance medium (Table 1) to the cell pellet. Gently pipette up and down 5-10 times to ensure cell pellet is broken up into single cells.
Transfer medium containing cells into pre-coated Matrigel plates and add epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF2) (both 20 ng/ml) in heparin (5 mg/ml). Incubate plates at 37 °C and 5% CO2.
3. Maintenance of Adherent Mouse Hippocampal NPCs
Prepare the coated plates/flasks at least an hour prior to passaging mouse hippocampal NPCs. Add enough Matrigel to cover the surface area of each plate or flask and incubate at room temperature for 1 hr.
Remove media from 90% confluent mouse NPC cultures and wash once with 1x Phosphate Buffered Saline (PBS) then aspirate PBS off.
Add enough cell detachment solution to cover all wells/flasks and incubate for 5 min at 37 °C.
Ensure that all cells have detached by looking under an inverted bright field microscope at 10X magnification. If there are still cells attached, incubate culture vessel for a further 1-2 min and check cells again. Add twice the amount of Dulbecco’s Modified Eagle Medium/Ham’s F-12 Nutrient Mixture (DMEM/F-12) as that of cell detachment solution to the wells/flasks once all cells have detached.
Transfer DMEM/F-12 containing cells into a 15 ml centrifuge tube and centrifuge at 200 x g for 5 min.
Remove supernatant and resuspend cell pellet in 5 ml of mouse NPC maintenance medium (Table 1). Gently pipette up and down to break up cell pellet into single cells.
Plate a third of total cells into new culture wells/flasks and top up sufficient culture medium supplemented with EGF and FGF2. Incubate culture vessels at 37 °C and 5% CO2. Replenish medium every second day and split cells 1:3 every 3 to 4 days. Avoid overgrowth of mouse NPC cultures to prevent aggregation and detachment.
4. Differentiation of Mouse NPCs into Astrocytes
- Prepare poly-L-Lysine (PLL)/laminin coated 12 mm cover glasses at least a day in advance before initiating differentiation of mouse NPCs into astrocytes. Add coverslips into a 24-well plate and cover entire surface area of wells with enough PLL (1 mg/ml in borate buffer).
- Incubate the plate overnight at 4 °C. The following day, remove PLL and wash once with 1x PBS.
Add laminin (1 mg/ml in ice cold DMEM/F-12), again covering the entire surface area of each well, then incubate for at least 4 hr at 37 °C.
Remove media from mouse NPC cultures and wash once with 1x PBS then aspirate PBS off.
Add enough cell detachment solution to cover all wells/flasks and incubate for 5 min at 37 °C.
Ensure that all cells have detached then add twice the amount of DMEM/F-12 as that of cell detachment solution to the wells/flasks.
Transfer DMEM/F-12 containing cells into a 15 ml centrifuge tube and centrifuge at 200 x g for 5 min.
Remove supernatant and resuspend cells in 5 ml astrocyte differentiation medium (Table 1). Gently pipette cell suspension up and down to break up cell pellet into single cells. Count the number of single cells with a hemocytometer.
Plate cells at 5 x 104 cells/cm2 in 500 µl of medium for each well of a 24-well plate. Incubate plates at 37 °C and 5% CO2. Replace half of the medium with fresh medium every second day. Mouse NPCs rapidly differentiate into astrocytes within 2 days and are differentiated for a week before commencing further experiments.
5. Harvesting Embryonic Rat Brains and Cortical Tissue Dissociation
Remove astrocyte differentiation medium (Table 1) from astrocyte cultures and replace with primary neuron medium (Table 1) at least 4 hr prior to commencing harvest of embryonic rat brains.
Before dissection, prepare the cortical tissue digestion solution: 10 µl papain per 1 ml 1x EBSS (+ 1% HEPES) with 12.1 mg/ml L-cysteine. Make approximately 2.5 ml of the tissue digestion solution and filter it using a 0.20 µm filter unit. Keep the solution warm at 37 °C until use.
Euthanize the pregnant dam by carbon dioxide asphyxiation using compressed gas. Test for pain reflex via toe or tail pinch.
Lay the animal ventral side up on an absorbent pad and soak its abdominal area with 70% ethanol. Pinch the skin with a pair of forceps and make a lateral incision with a pair of surgical scissors, wide enough to expose the abdomen. Grasp the peritoneum with the forceps and cut open the abdominal cavity.
- Lift up the uterine horn with the forceps and cut it free along the mesometrium. Place the dissected uterus in a Petri dish over ice. Separate each embryo by cutting the uterine tissue between them.
- Make an incision in the embryonic sac. Gently shell the fetus out through the opening. Decapitate the fetus and place the head in a Petri dish over ice.
- To harvest embryonic rat brains, follow the description from steps 1.3 to 1.5.
- To dissect cortical tissues, place a brain hemisphere lateral side up. Cut the hemisphere in two laterally from the middle. Discard the half closer to the base of the brain. Trim the excised tissue to remove the midbrain.
Transfer the cortical tissues to a dish containing the pre-warmed cortical tissue digestion solution. Mince the cortical tissues to as fine as possible and incubate for 30 min at 37 °C.
Transfer the cortical tissues from the tissue digestion solution to a 50 ml centrifuge tube containing 10 ml pre-warmed primary neuron medium (Table 1). Dissociate the cortical tissues by repeatedly pipetting them up and down in a serological pipette, until a homogenous solution with no tissue pieces is obtained.
Pass the tissue solution through a 70 µm cell strainer to filter out remaining tissue clumps. Aliquot the tissue solution into 15 ml centrifuge tubes, adding approximately 3 ml in each tube.
Add 800 µl 7.5% BSA in 1x PBS to the bottom of each tube, forming two layers with the tissue solution on top and BSA at the bottom.
Centrifuge at 200 x g for 5 min. Remove the supernatant, aspirating from the interface of the two layers. Avoid disturbing the cell pellet at the bottom of the tube.
Resuspend each cell pellet in 1 ml primary neuron medium (Table 1) and combine them in a 15 ml centrifuge tube. Determine the cell density using a hemocytometer.
6. Electroporation and Plating of Primary Cortical Neurons
NOTE: Gene transfer can be achieved using several methods, including electroporation, calcium phosphate transfection and viral transduction. In this protocol, we describe electroporation to deliver ChR2 construct into primary cortical neurons.
Centrifuge 6 x 106 rat cortical neurons at 200 x g for 5 min and remove the supernatant. At least 6 x 106 cortical neurons are required for electroporation to ensure the formation of a neuronal network by surviving neurons.
Add 100 µl of electroporation solution to cell pellet and resuspend gently. Combine 100 µl of the cell suspension with 6 µg of pLenti-Synapsin-hChR2(H134R)-EYFP-WPRE4 plasmid and transfer into a certified cuvette. Avoid producing air bubbles during the transfer.
Select and apply appropriate program for electroporation according to manufacturer’s instructions.
After electroporation, add 500 µl of MEM to cell/DNA suspension to the cuvette. Transfer the sample into 11.5 ml primary neuron medium (Table 1) and resuspend gently. The total amount of media is sufficient for an entire 24-well plate. Aliquot 500 µl of medium for every well of a 24-well plate. Incubate plate at 37 °C and 5% CO2.
7. Generation of Induced Pluripotent Stem Cells
NOTE: This method has been adapted from Okita et al.10
Culture human fibroblasts in DMEM supplemented with 10% fetal bovine serum (FBS) in 6-well plates.
Prior to commencing reprogramming of fibroblasts, coat 6-wells with Matrigel and incubate for at least an hour at room temperature. At 80% confluence, remove media from fibroblast cultures and wash once with 1x PBS.
Add sufficient 0.25% trypsin-EDTA to cover entire plate and incubate at 37 °C for 10 min.
Once cells have dislodged from wells, add twice the amount of DMEM with 10% FBS as that of trypsin to quench trypsin digestion. Gently pipette cell suspension up and down to break up cells. Count the number of single cells with a hemocytometer.
Transfer 7 x 105 cells into a 15 ml centrifuge tube and centrifuge the cell suspension at 200 x g for 5 min.
Remove supernatant and resuspend cell pellet in electroporation solution. Electroporate the fibroblasts according to manufacturer’s instructions. Resuspend cells in 100 µl of electroporation buffer and add 1 µg of each episomal vector. Electroporate cells with settings of 1,650 V, 10 ms and 3 time pulses.
Transfer cell suspension into DMEM supplemented with 10% FBS and plate 5 x 104 cells per well of a 6-well plate. Incubate plates at 37 °C and 5% CO2.
After 48 hr, remove media from transfected fibroblast cultures and replace with mTeSR medium. Replace culture media daily until induced pluripotent stem cell (iPSC) colonies are ready to be isolated. iPSC colonies can be observed after 28-35 days.
When ready for isolation, mechanically cut an iPSC colony and reseed in mTeSR medium with 10 µM Rho-associated protein kinase (ROCK) inhibitor (Y-27632) onto a Matrigel coated 6-well plate12. Replace culture medium daily.
8. Neural Induction and Maintenance of Human NPCs
NOTE: This method has been described by Li et al.11
When iPSC cultures are around 20% confluence, remove mTeSR medium and replace with neural induction medium (Table 1). Replenish medium every second day.
After 7 days, remove neural induction medium and replace with human NPC maintenance medium (Table 1). Replenish medium every second day up to 7 days before passaging cultures.
Prior to passaging cultures, coat plates/flasks with Matrigel at room temperature for at least an hr.
Remove media from confluent human NPC cultures and wash once with 1x PBS then aspirate PBS off.
Add enough cell detachment solution to cover all wells then incubate for 5 min at 37 °C.
Ensure that all cells have detached. Add twice the amount of DMEM/F-12 as that of cell detachment solution to the wells/flasks.
Transfer DMEM/F-12 containing cells into a 15 ml centrifuge tube and centrifuge at 200 x g for 5 min.
Remove supernatant and resuspend cells in 5 ml human NPC maintenance medium (Table 1). Gently pipette cell suspension up and down to break up cells.
- Plate a third of total cells into Matrigel coated culture wells/flasks and top up sufficient medium supplemented with 10 µM Y-27632. Incubate culture vessels at 37 °C and 5% CO2.
- Split cultures 1:3 every 3 to 4 days and replenish medium every other day. Maintain human NPC cultures at high density to prevent differentiation.
9. Differentiation of Human NPCs into Neurons in Co-cultures
Add the lentiviral vector Synapsin1-tdTomato to human NPC culture before initiating differentiation into neurons. Prepare astrocyte/cortical neuron co-cultures a day in advance before differentiating human NPCs.
Wash human NPC cultures once with 1x PBS and add enough cell detachment solution to cover all wells/flasks then incubate for 5 min at 37 °C.
Ensure that all cells have detached. Add twice the amount of DMEM/F-12 as that of cell detachment solution to the wells/flasks. Transfer into a 15 ml centrifuge tube. Centrifuge at 200 x g for 5 min.
Remove supernatant and resuspend cells in 5 ml neuronal differentiation medium (Table 1). Gently pipette cell suspension up and down to break up cell pellet into single cells. Count the number of single cells with a hemocytometer.
Carefully aspirate medium from astrocyte/cortical neuron cultures. Plate human NPCs at 5 x 103 cells/cm2 in 500 µl of neuronal differentiation medium (Table 1) supplemented with 10 µM Y-27632 for each 24-well. Gently add medium containing human NPCs into each well to avoid disturbing the astrocytes and cortical neurons.
Incubate plates at 37 °C and 5% CO2. Replace half of the medium with fresh medium every two to three days for at least 28 days.
10. Electrophysiological Recordings of iPSC-derived Neurons
- Confirm whether human iPSCs behave like mature neurons.
- Find cells differentiated from human iPSCs by visualization of tdTomato using an upright confocal microscope equipped with a 60X (0.9 NA) water immersion lens.
- Pull recording pipette to a resistance of 4 to 6 M. Perfuse cells at room temperature in an external solution (Table 1) bubbled constantly with 95% O2/ 5% CO2. Fill the recording micropipette with internal solution (Table 1).
- Patch tdTomato+ cell in whole-cell mode and record action potential firing in response to current injection (1 sec duration, 10 pA increments) in current clamp.
- Confirm if action potential of cortical cells expressing ChR2 is reliably evoked by light stimulation.
- Find cortical cells expressing ChR2 by visualization of GFP under a confocal microscope. Perform whole-cell patch clamp recording on cortical cells by following steps 10.1.2 to 10.1.3.
- Check action potential is reliably evoked by light illumination with mercury arc lamp (100 W). The wavelength of excitation filter is 480/40 nm.
- Perform optogenetic stimulation.
- Patch tdTomato+ cell in whole-cell mode and set voltage clamp at -70mV. Filter current signals at 2 kHz and digitize at 10 kHz. Monitor series resistances ranging from 10 to 20 M for consistency during recordings.
- Stimulate whole field by 100 W mercury lamp with 480/40 nm excitation filter for 30 sec.
- Record postsynaptic currents (PSCs) of patched cell induced by photostimulation of ChR2-expressing presynaptic cortical neurons. Detect and measure PSCs. Set threshold for amplitude and currents area at 5 pA and 20 pC, respectively. Manually inspect these events to discard any non-PSC traces.
Representative Results
Here, a protocol describing the multiple steps involved in generating a co-culture consisting astrocytes, cortical neurons and human neurons is illustrated. Human iPSCs were obtained by reprogramming fibroblasts using episomal vectors10 and specified into a neural lineage with a cocktail of small-molecule inhibitors, making NPCs11 which were maintained at high density (Figure 1A). Several steps are required to generate the co-culture: differentiation of mouse NPCs into astrocytes, plating of rat cortical neurons expressing ChR2, and seeding of human NPCs tagged with tdTomato (Figure 1B). At day 2 of differentiation, Glial Fibrillary Acidic Protein (GFAP)+ astrocytes can readily be observed in our cultures (Figure 1C). Mouse NPCs were allowed to differentiate for at least a week before plating cortical neurons expressing ChR2 onto the astrocyte monolayer (Figure 1D). After 3 to 4 weeks of human NPC differentiation, tdTomato+ neurons were detected in the cultures (Figure 1E).
This co-culture system allows consistent detection of postsynaptic currents from individual iPSC-derived neurons in response to light stimulation (Figure 2A). When stimulated by light, action potentials were generated in cortical neurons expressing ChR2 (Figure 2B). iPSC-derived neurons are also excitable (Figure 2C), showing increased action potential firing as the amplitude of depolarizing current pulses was increased (Figure 2D). Thus, these cells exhibit mature neuronal properties. In the absence of light, iPSC-derived neurons received spontaneous synaptic inputs (Figure 2E). These inward postsynaptic currents were predominantly mediated by AMPA receptors, because currents through GABA receptors would be outward and NMDA receptors do not carry current at the holding potential of -70 mV. At least some of these inputs were from presynaptic cortical neurons expressing ChR2, because light stimulation greatly increased the frequency of postsynaptic currents (Figure 2E). The time course of this increase in synaptic input is shown in Figure 2F: photostimulation of the cortical neurons was sustained throughout the 30 sec long light flash. While the frequency of PSCs was elevated when ChR2-expressing cortical neurons were photostimulated (Figure 2G), their amplitude was unaffected (Figure 2H). In summary, these results indicate that the iPSC-derived neurons exhibited mature neuronal properties and could form synaptic connections with presynaptic cortical neurons.
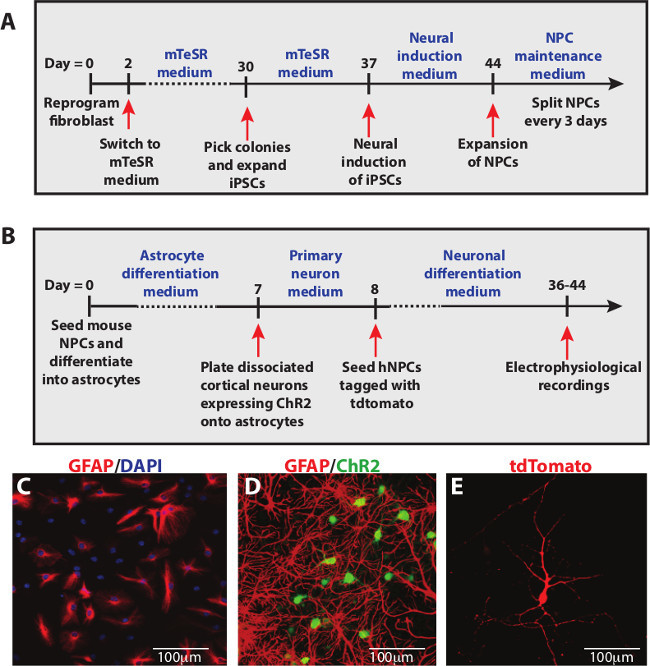 Figure 1: Schematic diagrams for the generation of co-culture system. (A) Timeline for reprogramming human fibroblasts into iPSCs and neural induction to NPCs. (B) Timeline for terminal differentiation of human NPCs into mature neurons on cortical neuron and astrocyte cultures. (C) Mouse NPCs rapidly differentiate into GFAP+ astrocytes after 2 days in vitro. (D) After one week, cortical neurons expressing ChR2 were plated onto GFAP+ astrocytes. (E) At 4 to 5 weeks after differentiation of human NPCs, electrophysiological recordings were performed on tdTomato+ neurons. Scale bars represent 100 µm.
Figure 1: Schematic diagrams for the generation of co-culture system. (A) Timeline for reprogramming human fibroblasts into iPSCs and neural induction to NPCs. (B) Timeline for terminal differentiation of human NPCs into mature neurons on cortical neuron and astrocyte cultures. (C) Mouse NPCs rapidly differentiate into GFAP+ astrocytes after 2 days in vitro. (D) After one week, cortical neurons expressing ChR2 were plated onto GFAP+ astrocytes. (E) At 4 to 5 weeks after differentiation of human NPCs, electrophysiological recordings were performed on tdTomato+ neurons. Scale bars represent 100 µm.
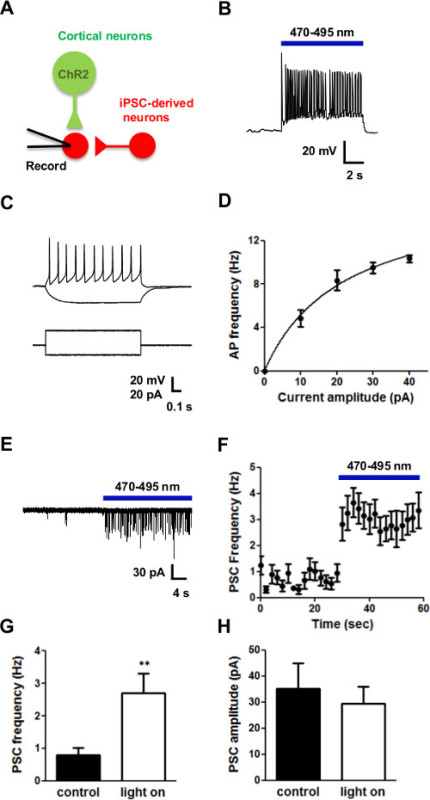 Figure 2: Optogenetic analysis of functional synaptic connectivity. (A) iPSC-derived cells tagged with tdTomato (red) were co-cultured with cortical neurons expressing the light-activated channel, ChR2 (green). Recordings from iPSC-derived neurons revealed postsynaptic current (PSC) responses, which could come from either the cortical neurons or other iPSC-derived neurons. (B) Illumination (blue bar) evoked a series of action potentials in cortical neurons expressing ChR2. (C) iPSC-derived cells are evoked by current injection. (D) Firing vs current curve of iPSC-derived cells. (E) Photostimulation of the ChR2-expressing cortical neurons (blue bar) increased the frequency of PSCs in iPSC-derived neurons. (F) Time course of the increase in PSC frequency produced by photostimulation. Blue bar indicates time of photostimulation. (G, H) While PSC frequency was increased by photostimulation (G), PSC amplitude was unaffected (H). * indicates p <0.05 compared to control with student’s t-test.
Figure 2: Optogenetic analysis of functional synaptic connectivity. (A) iPSC-derived cells tagged with tdTomato (red) were co-cultured with cortical neurons expressing the light-activated channel, ChR2 (green). Recordings from iPSC-derived neurons revealed postsynaptic current (PSC) responses, which could come from either the cortical neurons or other iPSC-derived neurons. (B) Illumination (blue bar) evoked a series of action potentials in cortical neurons expressing ChR2. (C) iPSC-derived cells are evoked by current injection. (D) Firing vs current curve of iPSC-derived cells. (E) Photostimulation of the ChR2-expressing cortical neurons (blue bar) increased the frequency of PSCs in iPSC-derived neurons. (F) Time course of the increase in PSC frequency produced by photostimulation. Blue bar indicates time of photostimulation. (G, H) While PSC frequency was increased by photostimulation (G), PSC amplitude was unaffected (H). * indicates p <0.05 compared to control with student’s t-test.
Discussion
Optogenetics provides temporal and spatial precision for activation of defined populations of neurons13. In our experiments, the entire field of the microscope objective was illuminated for 30 sec to photo-stimulate only cortical neurons expressing ChR2. This allowed us to determine whether synaptic connections were formed between different populations of neurons within the co-culture. Our results revealed that photostimulation increases PSC frequency in iPSC-derived neurons, which demonstrates that these neurons receive synaptic input from presynaptic cortical neurons expressing ChR2. This, in turn, established that the iPSC-derived neurons successfully incorporated into circuits with the cortical neurons.
There are several critical steps for the generation of this co-culture system. It is important to ensure that the mouse NPC-derived astrocyte cultures are healthy, with minimal cell death, before proceeding with plating of other cell types. Cortical neurons and human NPCs attach well onto healthy astrocytes and electrophysiological recordings depend heavily on culture conditions. The other key steps in this protocol are the electroporation and plating of cortical neurons onto astrocyte cultures. As a large number of cells die after electroporation, it is important to ensure that at least 6 million cortical neurons are electroporated for plating onto astrocyte cultures in 24-well plates. We have observed that when fewer than 6 million cells were electroporated, the number of neurons that survive did not form a dense neuronal network, which affected electrophysiological recordings. The number of electroporated cortical neurons should also be consistent for each co-culture experiment to allow comparison between different studies. For all steps involving enzymatic digestion, it is essential not to leave cells in digestive solution for too long as it may lead to cell death.
This approach can be used to screen the capability of neurons derived from patient-specific fibroblasts to form synaptic contacts with other neurons. Neurons derived from fibroblasts from patients suffering from the neurodevelopmental disorder Rett Syndrome (RTT) exhibit synaptic transmission defects: RTT neurons exhibit a significant decrease in PSC frequency and amplitude compared to healthy neurons, presumably due to less synaptic connectivity14. Our co-culture system allows examination of drug effects on neurons displaying developmental defects. This can be carried out via addition of compounds of interest during seeding of diseased human NPCs as well as during continuous exposure to compounds throughout the time of neuronal differentiation.
While this co-culture system can also be used as a model for drug testing, a limitation with this approach is that the process of investigating the effects of each candidate compound can be long and tedious as it is not a high-throughput screening method. Following treatment with candidate compounds, iPSC-derived neurons have to be individually patched to determine the effects of each compound on PSCs. Moreover, there are currently not many available fibroblasts and iPSCs from patients. Therefore, it will be of interest in the near future to have more patient cells and also to adapt this protocol for high throughput screening. For example, by labelling iPSC-derived neurons with an activity indicator, such as genetically-encoded sensors of ions or membrane potential, it would be possible to increase the throughput rate substantially by side-stepping the time-consuming electrophysiological procedure. As the whole process of generating this co-culture system, from reprogramming fibroblasts to performing electrophysiological recordings, requires more than 90 days, it is recommended that either the human iPSCs or NPCs be expanded, after the reprogramming and neural induction steps respectively, and cryopreserved to reduce the time needed for future experiments.
In our experiments, we expressed ChR2 in cortical neurons under the synapsin1 promoter. Because this promoter is pan-neuronal, we presumably were photostimulating both inhibitory and excitatory cortical neurons. If the goal of future experiments is to specifically interrogate either inhibitory or excitatory circuits, more specific promoters could be used. Alternatively, cortical neurons could be obtained from transgenic (or virus-injected mice) where ChR2 expression is specifically targeted to genetically-defined subpopulations of neurons. For example, harvesting cortical tissues from a mouse that has Cre recombinase expressed in parvalbumin-containing interneurons (PV interneurons) that is crossed with a mouse with floxed ChR2 will yield a situation where the only cortical neurons expressing ChR2 will be PV interneurons15. The use of such ChR2-expressing interneurons in our co-culture system will enable the determination of whether a patched iPSC-derived neuron is able to incorporate into a circuit with PV interneurons.
Based on a previous study9, cortical neurons are mature with overlapping dendrites after 14 days in vitro. Thus, if photostimulation does not elicit a response from a patched iPSC-derived neuron, it should indicate that the neuron does not have the ability to make synaptic connections with cortical neurons. An iPSC-derived neuron is able to receive synaptic input either from other iPSC-derived neurons or cortical neurons. If traditional patch clamp recordings were used, it would not be possible to distinguish where the synaptic input is coming from. The advantage of our system is that postsynaptic responses from cortical presynaptic input can be selectively evoked and identified. The procedures outlined here provide a simple approach to evaluate the capability of iPSC-derived neurons to integrate into a defined neuronal network.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank K. Deisseroth and S. Je for the ChR2 and Synapsin1-tdTomato lentiviral constructs respectively, C. Chai for sharing expertise and protocol on iPSC generation, W.Y. Leong for technical support, members of the Goh lab for sharing reagents and expertise. This work was supported by Abbott Nutrition and the Academic Centre of Excellence (ACE) research award from GlaxoSmithKline (GSK) to E.L.G., by the National Research Foundation Singapore under its Competitive Research Program (NRF 2008 NRF-CRP 002-082) to E.L.G. and G.J.A., and by the World Class Institute (WCI) Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology of Korea (MEST) (NRF Grant Number: WCI 2009-003) to G.J.A.
References
- Wang H, et al. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proceedings of the National Academy of Sciences. 2007;104(19):8143–8148. doi: 10.1073/pnas.0700384104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale genetically targeted optical control of neural activity. Nature Neuroscience. 2005;8(9):1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Nagel G, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proceedings of the National Academy of Sciences. 2003;100(24):13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446(7136):633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- Stroh A, et al. Tracking stem cell differentiation in the setting of automated optogenetic stimulation. Stem Cells. 2011;29(1):78–88. doi: 10.1002/stem.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick JP, Liu Y, Zhang S-C. Human embryonic stem cell-derived neurons adopt and regulate the activity of an established neural network. Proceedings of the National Academy of Sciences. 2011;108(50):20189–20194. doi: 10.1073/pnas.1108487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B. Preparation and coculture of neurons and glial cells. Curr Protoc Cell Biol. 2006;32:2.7.1–2.7.2.1. doi: 10.1002/0471143030.cb0207s32. [DOI] [PubMed] [Google Scholar]
- Braun H, Günther-Kern A, Reymann K, Onteniente B. Neuronal differentiation of human iPS-cells in a rat cortical primary culture. Acta Neurobiologiae Experimentalis. 2012;72(30):219–229. doi: 10.55782/ane-2012-1895. [DOI] [PubMed] [Google Scholar]
- Shivaraj MC, et al. Taurine induces proliferation of neural stem cells and synapse development in the developing mouse brain. PLoS ONE. 2012;7:e42935. doi: 10.1371/journal.pone.0042935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nature Methods. 2011;8(5):409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Li W, et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proceedings of the National Academy of Sciences. 2011;108(20):8299–8304. doi: 10.1073/pnas.1014041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, et al. Chemically defined conditions for human iPSC derivation and culture. Nature Methods. 2011;8(50):424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AM, Roska B, Hausser M. Targeting neurons and photons for optogenetics. Nature Neuroscience. 2013;16(7):805–815. doi: 10.1038/nn.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MCN, et al. A Model for neural development and treatment of Rett Syndrome using human induced pluripotent stem cells. Cell. 2010;143(4):527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrican B, et al. Next-generation transgenic mice for optogenetic analysis of neural circuits. Frontiers in Neural Circuits. 2013;7:160. doi: 10.3389/fncir.2013.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]