

Abstract
Adoptive T cell immunotherapy offers a promising strategy for specifically targeting and eliminating malignant gliomas. T cells can be engineered ex vivo to express chimeric antigen receptors specific for glioma antigens (CAR T cells). The expansion and function of adoptively transferred CAR T cells can be potentiated by the lymphodepletive and tumoricidal effects of standard of care chemotherapy and radiotherapy. We describe a method for generating CAR T cells targeting EGFRvIII, a glioma-specific antigen, and evaluating their efficacy when combined with a murine model of glioblastoma standard of care. T cells are engineered by transduction with a retroviral vector containing the anti-EGFRvIII CAR gene. Tumor-bearing animals are subjected to host conditioning by a course of temozolomide and whole brain irradiation at dose regimens designed to model clinical standard of care. CAR T cells are then delivered intravenously to primed hosts. This method can be used to evaluate the antitumor efficacy of CAR T cells in the context of standard of care.
Keywords: Immunology, Issue 96, Tumor immunotherapy, glioblastoma, chimeric antigen receptor, adoptive transfer, temozolomide, radiotherapy
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor and is invariably fatal. Surgical resection coupled with non-specific standard of care chemotherapy and radiotherapy fails to completely eliminate malignant cells, resulting in a dismal prognosis of less than 15 months in patients with this disease1. In contrast, immunotherapy offers a precise approach for specifically targeting tumor cells, and thus has the potential to serve as a highly effective treatment platform with reduced risk of collateral toxicity2-4. T cells engineered ex vivo to express chimeric antigen receptors (CARs) offer a versatile strategy for tumor immunotherapy. CARs are generated by fusing the extracellular variable region of an antibody with one or more intracellular T cell signaling molecule(s), in lieu of a full-length major histocompatibility complex (MHC)-restricted T cell receptor5. This mode of antibody-like antigen recognition allows for reactive antigen-specific T cells to recognize and respond to tumor antigens in the absence of MHC and can be adapted for a virtually infinite antigen repertoire.
CAR T cells engineered against a variety of tumor antigens have shown preclinical efficacy and outstanding promise in the clinic6-9. Specifically, in the context of GBM, a CAR T cell platform targeting epidermal growth factor receptor variant III (EGFRvIII), a tumor-specific mutation expressed on the cell surface10, was shown to prolong survival in glioma-bearing mice11. Despite their versatility, however, the clinical benefit of CAR adoptive therapy has not been fully realized, due in part to tumor-associated immunosuppression and immune evasion12-16 as well as challenges in establishing and maintaining antigen-specific T cells in vivo. Leveraging standard of care (SOC) with immunotherapy can potentially overcome several of these limitations, resulting in enhanced efficacy in both the preclinical and clinical setting.
SOC for post-resection GBM consists of high-dose temozolomide (TMZ), a DNA alkylating agent17, and whole brain irradiation (WBI)1. These treatments are presumed to synergize with tumor vaccines via upregulation of tumor MHC expression18-20 and the shedding of antigens by dead tumor cells17,19,21,22. Indeed, the addition of TMZ20,23 or WBI18,24 leads to enhanced antitumor efficacy of immune-based treatments in the preclinical setting. Furthermore, like many non-specific cytotoxic chemotherapeutics, TMZ is known to cause systemic lymphopenia25,26, which can be leveraged as a means of host-conditioning for adoptive therapy platforms27-29. TMZ-mediated lymphodepletion has been shown to enhance the frequency and function of antigen-specific T cells, leading to increased efficacy of an adoptive therapy platform against intracranial tumors30. In the context of CAR therapy, lymphodepletion serves as a means of host-conditioning by both reducing the number of endogenous suppressor T cells31, and inducing homeostatic proliferation32 via reduced competition for cytokines33, thus enhancing antitumor activity11,34. Given the synergistic relationship between GBM SOC and immunotherapy platforms, evaluating novel adoptive therapies and vaccine platforms in the context of SOC is critical for drawing meaningful conclusions regarding efficacy.
In this protocol, we outline a method for the generation and intravenous administration of murine EGFRvIII-specific CAR T cells alongside TMZ and WBI in mice bearing EGFRvIII-positive intracranial tumors (see Figure 1 for treatment timeline). Briefly, CAR T cells are made ex vivo by retroviral transduction. Human embryonic kidney (HEK) 293T cells are transfected using a DNA/lipid complex (containing the CAR vector and pCL-Eco plasmids) to produce virus, which is then used to transduce activated murine splenocytes that are harvested and cultured in parallel. During the course of CAR generation, murine hosts bearing EGFRvIII-positive intracranial tumors are administered fractionated whole-brain X-ray irradiation and systemic TMZ treatment at doses comparable to clinical SOC. CAR T cells are then delivered intravenously to lymphodepleted hosts.
The following procedure is described in seven separate phases: (1) Administration of Temozolomide to Tumor-bearing Mice, (2) Whole Brain Irradiation of Tumor-bearing Mice, (3) Transfection, (4) Splenectomy and T cell Preparation, (5) Transduction, (6) CAR T cell Culture and Harvest, and (7) CAR T cell administration to Tumor-bearing Mice. These phases consist of several steps that span 6-7 days and are performed concurrently.
Protocol
This protocol is based on an experimental design where 10 mice are treated with 107 CAR T cells each. This means that 108 CAR T cells will be needed; the yield should be overestimated by 5 x 107-1 x 108 to account for loss in viability. The following protocol is scaled to generate approximately 200 x 106 cells. The cells are then administered intravenously to female C57BL/6 mice with 9 day established syngeneic EGFRvIII-positive intracranial tumors, developed from the existing KR158B astrocytoma or B16 melanoma cell lines. Concomitantly with CAR T cell generation, tumor-bearing mice are administered clinically relevant doses of lymphodepletive TMZ (60 mg/kg) and WBI (16.5 Gy).
Mice were maintained and bred under pathogen-free conditions at Duke University Medical Center (DUMC). All animal experiments were performed according to protocols approved by the Duke University Institutional Animal Care and Use Committee (IACUC).
1. Administration of Temozolomide to Tumor-bearing Mice
Days 0 through 4:
Calculate the total number of mice to be injected and multiply this by 0.5 to determine the volume of TMZ solution needed (e.g., 30 mice x 0.5 ml = 15 ml TMZ) and add 1.5 ml to this volume to account for spillover (16.5 ml TMZ).
Multiply this total volume by 3 mg/ml to determine the weight of lyophilized TMZ needed (e.g., 16.5 x 3 = 49.5 mg TMZ). Weigh this into a 50 ml conical. CAUTION: TMZ is toxic by inhalation, ingestion, and contact with eyes, skin, and mucosal membranes. Consult with institution’s occupational safety and/or environmental health and wellness department for recommendations on reducing risk of exposure.
Multiply the total volume by 0.15 to determine the volume of dimethyl sulfoxide (DMSO) needed (e.g., 16.5 x 0.15 = 2.475 ml DMSO). Filter sterilize this volume of DMSO (plus an additional 2 ml to account for spillover) through a 0.2 µm filter into a sterile tube. Add 2.475 mL of sterile DMSO to the TMZ powder.
Place the TMZ/DMSO solution in a beaker of water over a hot plate at 65 °C for 10-15 min. Once TMZ is fully dissolved, the solution should become pale yellow in color; if the solution becomes pink, then the TMZ is degraded, and a new solution should be prepared with a fresh lot of TMZ.
Calculate the appropriate volume of sterile saline needed by multiplying 0.85 by the total volume (0.85 x 16.5 ml = 14.025 ml saline). Add 15 ml of sterile saline to a 50 ml conical. While TMZ is dissolving in the DMSO, heat saline to 65 °C.
Vortex the TMZ/DMSO solution to ensure that the TMZ powder is fully dissolved and slowly add warmed saline to the TMZ/DMSO solution.
Immediately after preparation, fully load 15 x 1 ml tuberculin syringes with TMZ solution for administration to 4 day established tumor bearing mice.
Repeat this procedure on days 1 through 4 for a total of five TMZ administrations.
2. Whole Brain Irradiation of Tumor-bearing Mice
Days 2 through 4:
Power on the X-ray irradiator and allow it to warm-up to full voltage. Ensure that the appropriate filter is in place and input the proper voltage and current settings (Figure 2A). NOTE: The dosimetry of the irradiator should be established beforehand to determine the appropriate voltage settings and grid layout (Figure 2A,B).
Calculate the length of time that is necessary to result in 5.5 Gy X-ray irradiation. For example, if X-rays are delivered at a rate of 2 Gy/min, then 2.75 min is necessary to deliver a total of 5.5 Gy. Input the appropriate time duration. NOTE: Table 2 provides mouse WBI doses and their clinical equivalents in humans. In the context of high-grade gliomas, 60 Gy fractionated WBI (2 Gy fractions, 5 days/week for 6 weeks) is the clinical standard of care, and the murine equivalent used here is 16.5 Gy (5.5 Gy x 3).
Prepare a fresh solution of ketamine/xylazine for systemic anesthesia by adding 2 ml ketamine and 1 ml xylazine to 17 ml saline such that the final solution is 10 mg/ml ketamine and 1 mg/ml xylazine.
Weigh mice and administer 10 μl of ketamine/xylazine intraperitoneally per gram of body weight such that animals receive a dose of ketamine at 100 mg/kg and 10 mg/kg xylazine. Mice should be fully sedated and visibly breathing within 2 min of ketamine/xylazine administration.
To maintain ophthalmic moisture in sedated animals, gently rub a small amount of artificial tears ointment on each eye.
Place sedated mice on the grid such that heads are positioned in the area that receives the highest X-ray intensity (Figure 2B,C). Shield bodies from the neck down with appropriately sized lead tubing to block systemic X-ray delivery (Figure 2D).
Place positioned mice under the X-ray beam such that the laser denoting the focal point of the X-ray beam is at the coordinate (0,0) on the grid layout. Begin X-ray delivery.
When X-ray delivery is complete, remove animals from the irradiator and place on a warm heating pad. Do not house sedated animals with conscious animals until animals have regained sufficient consciousness to maintain sternal recumbence. Once animals can maintain sternal recumbence, place conscious animals back in their appropriate cages.
Repeat this procedure on days 3 and 4 for a total of three fractionated doses of radiation.
3. Transfection
Day -1:
Prepare D10 media by adding 50 ml fetal bovine serum (FBS) to 500 ml of Dulbecco’s Modified Eagle Medium (DMEM).
Prepare T cell media (TCM) by adding 5.5 ml L-Glutamine, 5.5 ml sodium pyruvate, 5.5 ml non-essential amino acids, and 5.5 ml pencillin/streptomycin (pen/strep) to 500 ml RPMI-1640 media. Next, add 550 μl of 2-mercaptoethanol and 550 μl gentamicin. Finally, add 50 ml FBS.
Harvest in vitro cultured HEK293T cells and bring to a concentration of 7.5 x 106 cells/ml in D10 media.
Plate 10 ml D10 media and add 1 ml of HEK293T cell suspension in each of 16 x 10 cm poly-D-lysine (PDL) coated plates.
Incubate overnight at 37 °C with 5% CO2.
Day 0:
Replace media with 10 ml fresh D10 at least 30 min before transfecting cells.
Determine the amount of vector plasmid, pCL-Eco vector, and liposomal transfection reagent required for transfection by multiplying the total number of plates + 1 (16 + 1 = 17) by 14.1 μg vector plasmid (14.1 x 17 = 239.7 μg), 9.9 μg pCL-Eco vector (9.9 x 17 = 168.3 μg), and 60 μl liposomal transfection reagent (60 x 17 = 1,020 μl). All reagents should be at room temperature before preparing solutions.
Label two tubes A and B. In tube A, add 239.7 μg vector plasmid and 168.3 μg pCL-Eco vector to (1.5 x 17) ml reduced-serum modified eagle’s medium (RS-MEM). In tube B, add 1020 μl liposomal transfection reagent to (1.5 x 17) ml RS-MEM.
Incubate A and B separately for 5 min at room temperature.
Mix A and B together gently (vortex for 1-2 sec or invert several times) and incubate for 20 min at room temperature to form lipid/DNA complex.
Add lipid/DNA complex drop wise to HEK293T cells in 10 cm dish.
Incubate at 37 °C for 6-8 hr or overnight (do not exceed 24 hr).
After 6-8 hr incubation, replace medium with 12 ml of fresh TCM for viral production. This plated media will be used on Day 2 as viral supernatant for T cell transduction.
4. Splenectomy and T Cell Preparation
Day 0:
Pour 10 ml of TCM into a 50 ml conical and place on ice for spleen collection.
Sacrifice the appropriate number of animals by CO2 asphyxiation and secondary decapitation: Place animals in a cage receiving CO2 at a flow rate of 10-30% cage volume/minute, per American Veterinary Medical Association guidelines (no more than 5 animals can be sacrificed simultaneously) until respiration terminates and for two minutes thereafter. Remove animals from the CO2 chamber and decapitate. NOTE: One spleen of a 6-12 week old female C57BL/6 mouse will yield approximately 4.5-5 x 107 splenocytes. Here, 4 spleens will be harvested for approximately 200 x 106 cells.
Lay the mouse such that its right side is facing up, and spray with 70% ethanol. With the forceps, grab a thin fold of skin below the left ribcage and cut a slight incision with the scissors. Peel back the skin, carefully grab a thin fold of the peritoneum with forceps, and cut a small cavity.
The spleen is a small, elongated, dark red organ that resembles a flattened bean; delicately grab the spleen with the forceps and excise by cutting away the surrounding connective tissue. Place the excised spleens into the conical containing 10 ml of TCM on ice.
Pour spleens over a 70 μm mesh cell strainer and disaggregate by mashing with the blunt end of the inside of a 5 ml syringe to generate a single-cell suspension. A maximum of two spleens should be disaggregated per mesh strainer.
Use a small volume of TCM to carefully wash the strainer following disaggregation to collect any remaining splenocytes. Pool all disaggregated spleens into a single-cell suspension. Bring final volume to 50 ml with TCM for one wash and spin at 300 x g for 10 min.
Prepare a solution of 1x lysis buffer by adding 5 ml 10x lysis buffer to 45 ml sterile water. To eliminate red blood cells, resuspend pellet in 5 ml of 1x lysis buffer per spleen in a 50 ml conical, mixing well by gently pipetting up and down. If exceeding 5 spleens, use a 250 ml centrifuge tube. Place conical or centrifuge tube in a 37 °C water bath for 5 min.
Remove the lysis reaction from the water bath and add TCM at a 1:1 ratio with lysis buffer to neutralize the reaction. Wash by spinning at 300 x g for 10 min.
Aspirate supernatant and fully resuspend pellet in TCM (2 ml/spleen, 8 ml total for 4 spleens) by pipetting up and down.
Count cells by adding 10 μl of cell suspension to 190 μl trypan blue (1:20 dilution). Multiply the number obtained in one of the four gridded squares by 20 x 104 x total volume (8 ml) to obtain the total cell number (e.g., 125 cells x 20 x 104 x 8 ml = 200 x 106 splenocytes).
Dilute cells to a concentration of 2 x 106 cells/ml in TCM, supplemented with 2 μg/ml concanavalin A (ConA) and 50 IU/ml recombinant human interleukin-2 (rhIL-2). Thus, for 200 x 106 splenocytes, add 92 ml media to 8 ml cells, 200 μg ConA, and 5,000 IU rhIL-2.
Add 2 mL cells to each well of 24-well tissue-culture treated plates, such that 4 x 106 cells are in each well (e.g., 100 ml of cells will require approximately 4 plates).
Incubate overnight at 37 °C with 5% CO2.
5. Transduction
Day 1:
Calculate the number of non-tissue culture treated 24-well plates needed for transduction by multiplying the number of spleens harvested by 2 (8 plates are needed for 4 spleens).
Next, calculate the required volume of recombinant human fibronectin fragment (RHFF) solution needed to coat plates by multiplying (total number of wells + 3) x 0.5 ml (8 plates x 24 wells = 192 + 3 = 195) x 0.5 ml = 97.5 ml.
Prepare 97.5 ml phosphate-buffered saline (PBS) containing RHFF at a concentration of 25 μg/ml by multiplying the total volume by 25 μg (97.5 x 25 = 2437.5 μg). Add this amount of RHFF to 97.5 ml PBS.
Coat non-tissue culture treated 24-well plates by adding 0.5 ml PBS/RHFF solution per well.
Incubate overnight at 4 °C.
Day 2:
Dump PBS/RHFF solution from non-tissue culture treated 24-well plates.
Add 1 ml/well 2% bovine serum albumin (BSA) in PBS and incubate at room temperature for 30 min.
Remove BSA by firmly upending plate. Wash by adding 2 ml PBS.
Collect viral supernatant by transferring media from each HEK293T PDL plate into a 250 ml centrifuge tube and spin 10 min at 500 x g.
Carefully transfer viral supernatant into a fresh 250 ml centrifuge tube, being sure not to disturb the cell pellet that may have formed.
Add fresh TCM to the viral supernatant such that the final volume is 3 ml more than the amount needed for RHFF-coated wells. For example, for 192 RHFF-coated wells, bring the volume of viral supernatant to 192 + 3 ml = 195 ml.
Remove cultured splenocytes from the incubator and resuspend by gently pipetting up and down 2 - 3 times in each well. Transfer to a 250 ml conical. If using a multichannel pipette, using a sterile reservoir prior to transfer will help expedite this step. Count as previously described and spin at 300 x g for 10 min.
Add rhIL-2 to viral supernatant at a concentration of 50 IU/ml. For example, add 50 x 195 = 9,750 IU rhIL-2 to 195 ml of viral supernatant.
Resuspend splenocytes in viral supernatant at 1 x 106 cells/ml
Add 1 ml/well of splenocyte suspension to RHFF-coated 24-well plates.
Spin for 90 min according to the following settings: 770 x g, acceleration = 4, brake/deceleration = 0, 32 °C.
Prepare a TCM solution with 50 IU/ml rhIL-2. For example, prepare a 200 TCM solution by adding 10,000 IU rhIL-2. Add 1 ml of rhIL-2/TCM to each well after centrifugation.
Culture overnight at 37 °C in 5% CO2.
6. CAR T cell Culture and Harvest
Days 3 and 4:
If T cells achieve >80% confluence, cells may be split (this usually occurs by day 3 or day 4). To split cells, gently pipet up and down in each well 2-3 times, and move 1 ml from each well into new wells of a fresh 24-well tissue-culture treated plate. Then, add 1 ml of fresh TCM with 50 IU/ml IL-2 to each well such that final volume is 2 ml in all wells.
If cells do not reach >80% confluence, perform a half media change by slowly pipetting off 1 ml of media from the top of each well. Avoid disturbing cells settled on the bottom while removing media. Add 1 ml of fresh TCM containing 50 IU/ml rhIL-2.
Day 5:
Resuspend CAR T cells by gently pipetting up and down 3 times in each well and transfer to a 250 ml centrifuge tube. Spin cells at 300 x g for 10 min.
Completely aspirate supernatant without disturbing pellet. Wash once with PBS, count cells as previously described, and wash with PBS a second time. A typical CAR T cell yield is approximately 1 x 106 cells per well (8 plates x 24 wells = 192 x 106 CAR T cells).
Resuspend cells in PBS at a concentration of 5 x 107/ml for an injection of 1 x 107 in a volume of 200 μg (e.g., for 192 x 106 CAR T cells, resuspend washed pellet in 3.84 ml PBS).
7. CAR T cell Administration to Tumor-bearing Mice
Day 5:
Transport cells on ice to animal facility for intravenous injection. Load 500-1,000 μl into insulin syringe with 27 - 31 G needle, ensuring that all bubbles are expelled.
Grab a mouse at the base of the tail and place in tube restrainer.
Pull the tail taut with the vein facing upward, and glide the needle approximately 1-2 mm under the skin into the vein by inserting it parallel to the tail vein. Slowly expel 200 μl into the vein. If the needle is correctly inserted into the vein, the volume will easily flow in; otherwise there will be resistance and the needle will need to be removed from the tail and re-inserted correctly.
Representative Results
CAR T cells are generated by transduction with the EGFRvIII CAR retroviral vector11. This vector, MSGV1, was developed from the SFGtcLuc_ITE4 vector35, which contains the murine stem cell virus (MSCV) long terminal repeats, the extended gag region and envelope splice site (splice donor, sd, and splice acceptor, sa), and viral packaging signal (ψ). The EGFRvIII CAR containing the human anti-EGFRvIII single-chain variable fragment (scFv) 139, in tandem with murine CD8TM, CD28, 4-1BB, and CD3ζ intracellular regions, was cloned into the retroviral vector downstream the NcoI site (Figure 3).
Following transduction, CAR T cells can be quantified and phenotyped by flow cytometry. EGFRvIII CAR T cells can be visualized with a 2-color panel comprised of a streptavidin-phycoerythrin (SA/PE)-conjugated biotynlated EGFRvIII-derived multimer and anti-CD3 FITC. Using the culture and transduction protocols described here, we routinely observe CAR expression among 55-70% of murine splenocytes (Figure 4A)11. Alternatively, CARs can also be stained for expression using goat-anti-human F(ab’)2-biotin primary and SA/PE secondary antibodies which has been previously described36. CAR T cells can be further phenotyped by the addition of other fluorescently labeled antibodies to the two-color CAR panel. For example, staining for CD8 and CD4 shows that 70% of transduced CARs are CD8+ T cells, while 20% are CD4+ T cells (Figure 4B). CAR T cells with this expression profile have been shown to readily traffic to the brain and treat intracranial tumors.
| Body weight | Volume |
| 25 g | 0.50 ml |
| 24 g | 0.48 ml |
| 23 g | 0.46 ml |
| 22 g | 0.44 ml |
| 21 g | 0.42 ml |
| 20 g | 0.40 ml |
| 19 g | 0.38 ml |
| 18 g | 0.36 ml |
| 17 g | 0.34 ml |
| 16 g | 0.32 ml |
| 15 g | 0.30 ml |
| 14 g | 0.28 ml |
| 13 g | 0.26 ml |
| 12 g | 0.24 ml |
| 11 g | 0.22 ml |
Table 1: Temozolomide dose based on animal weight. Temozolomide doses were calculated based on a total dose of 60 mg/kg.
| D | d | # Fractions | BED | SOC |
| 16.5 | 5.5 | 3 | 62 | GBM |
| 14 | 7 | 2 | 63 | GBM |
| 20 | 4 | 5 | 60 | GBM |
| 12 | 6 | 2 | 48 | LGA |
| 16 | 4 | 4 | 48 | LGA |
| 21 | 3 | 7 | 52.5 | LGA |
| 12 | 3 | 4 | 30 | Met |
| 16 | 2 | 8 | 32 | Met |
Table 2: Clinically relevant biologically equivalent radiation doses. Radiation doses were calculated according to BED = D[1+d/(α/β)], where D = total dose, d = fractionated dose, and α/β = 2). The total dose administered as WBI to the murine host is shown in terms of the fractional doses delivered and the human dose that is modeled by those dose fractions. For model purposes, the malignancy for which the BED is standard of care is also shown.
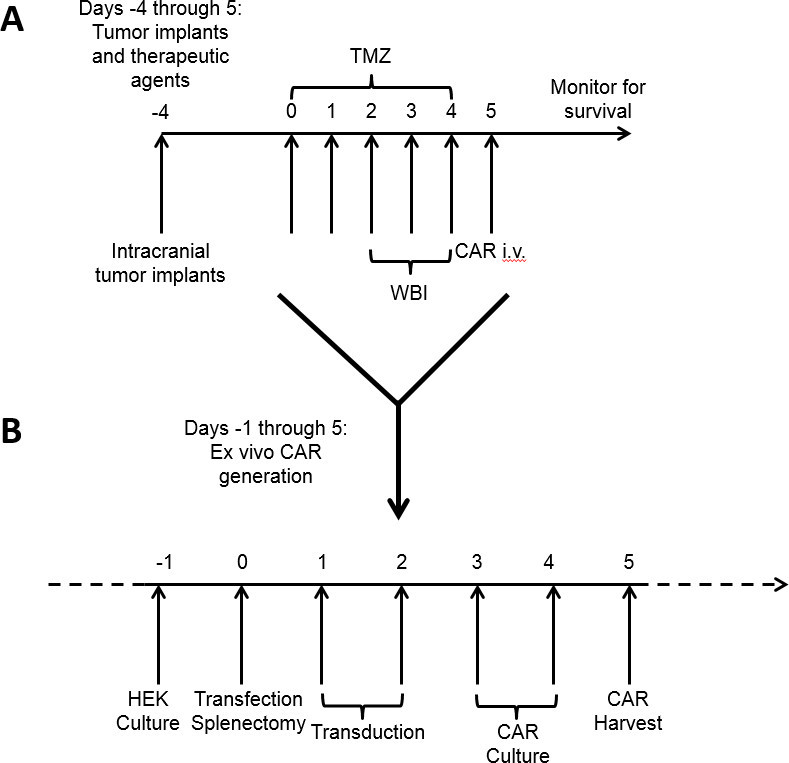 Figure 1: Timeline for treatment of tumor-bearing mice and concurrent ex vivo CAR generation. Standard of care chemotherapy and whole brain irradiation begins 4 days after tumor implantation (A). During this time interval, CAR T cells are generated and cultured ex vivo (B) and delivered after a full course of host conditioning.
Figure 1: Timeline for treatment of tumor-bearing mice and concurrent ex vivo CAR generation. Standard of care chemotherapy and whole brain irradiation begins 4 days after tumor implantation (A). During this time interval, CAR T cells are generated and cultured ex vivo (B) and delivered after a full course of host conditioning.
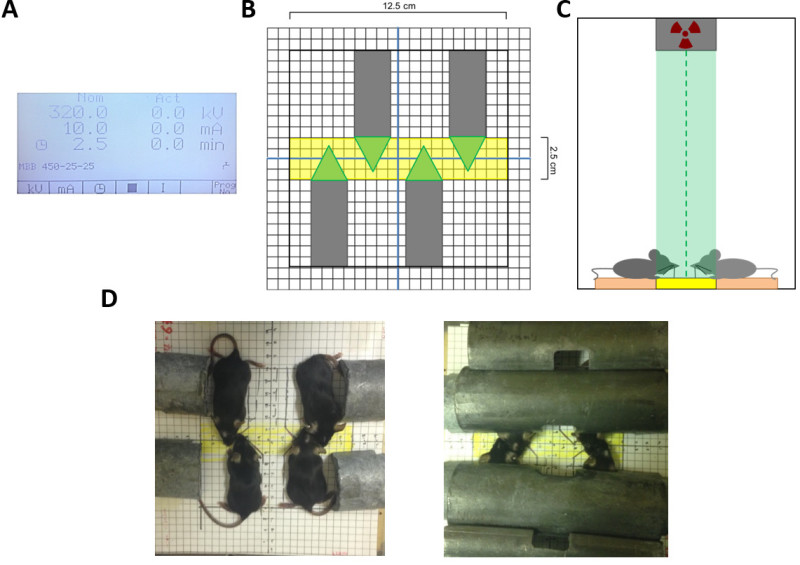 Figure 2: Layout and settings for radiation delivery. X-rays are delivered according to a dosimetry of 320 kV and 10 mA, with a variable duration chosen according to the desired dose (A). The focal field of X-ray radiation is a narrow 2.5 cm area. Sedated animals are arranged according to the grid (B) such that their heads lie in the area of highest X-ray intensity; (C) shows the view from one end of the X-ray beam. Approximately 4 animals can be under the irradiator during one course of X-ray administration according to this grid layout (B, D). Lead tubing is used to shield the body, leaving only their heads exposed for WBI (D).
Figure 2: Layout and settings for radiation delivery. X-rays are delivered according to a dosimetry of 320 kV and 10 mA, with a variable duration chosen according to the desired dose (A). The focal field of X-ray radiation is a narrow 2.5 cm area. Sedated animals are arranged according to the grid (B) such that their heads lie in the area of highest X-ray intensity; (C) shows the view from one end of the X-ray beam. Approximately 4 animals can be under the irradiator during one course of X-ray administration according to this grid layout (B, D). Lead tubing is used to shield the body, leaving only their heads exposed for WBI (D).
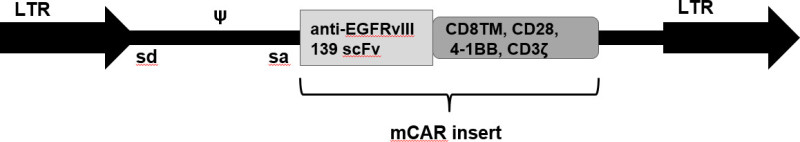 Figure 3: The modified SFGtcLuc_ITE4 retroviral vector, MSGV1, was used for transduction and generation of CAR T cells. The EGFRvIII CAR insert containing the human anti-EGFRvIII single chain variable fragment (scFv) 139, in tandem with murine CD8TM, CD28, 4-1BB, and CD3ζ intracellular regions, is flanked by murine stem cell virus (MSCV) long terminal repeats (LTR). Upstream of the CAR insert are the extended gag region and envelope splice site (splice donor, sd, and splice acceptor, sa), and the viral packaging signal (ψ).
Figure 3: The modified SFGtcLuc_ITE4 retroviral vector, MSGV1, was used for transduction and generation of CAR T cells. The EGFRvIII CAR insert containing the human anti-EGFRvIII single chain variable fragment (scFv) 139, in tandem with murine CD8TM, CD28, 4-1BB, and CD3ζ intracellular regions, is flanked by murine stem cell virus (MSCV) long terminal repeats (LTR). Upstream of the CAR insert are the extended gag region and envelope splice site (splice donor, sd, and splice acceptor, sa), and the viral packaging signal (ψ).
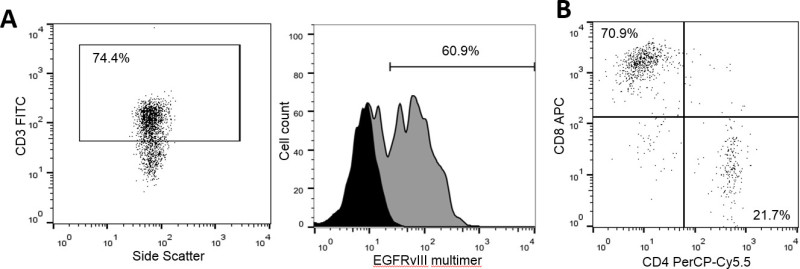 Figure 4: EGFRvIII CARs are expressed on the surface of T cells. 48 hr following transduction, T cells were stained for surface expression of EGFRvIII CARs using a multimer composed of LEEKKGNYVVTDHC-K(biotin)-NH2/streptavidin-PE. Untransduced T cells from the same donor were also stained as a negative control. Data shows the number of CAR T cells (y-axis) positive for staining by the PE-conjugated EGFRvIII multimer (x-axis), gated on CD3+ cells as a T cell marker, where expression was found to be 60.9% (A). CAR T cells stained for CD4-PerCP-Cy5.5 and CD8-APC show an expression profile of 21.7% and 70.9%, respectively (B).
Figure 4: EGFRvIII CARs are expressed on the surface of T cells. 48 hr following transduction, T cells were stained for surface expression of EGFRvIII CARs using a multimer composed of LEEKKGNYVVTDHC-K(biotin)-NH2/streptavidin-PE. Untransduced T cells from the same donor were also stained as a negative control. Data shows the number of CAR T cells (y-axis) positive for staining by the PE-conjugated EGFRvIII multimer (x-axis), gated on CD3+ cells as a T cell marker, where expression was found to be 60.9% (A). CAR T cells stained for CD4-PerCP-Cy5.5 and CD8-APC show an expression profile of 21.7% and 70.9%, respectively (B).
Discussion
The treatment timeline described here was designed to model clinical standard of care and leverage its effects for CAR adoptive therapy. CAR T cell doses, TMZ regimens, and radiotherapy administration can be modified to enhance in vivo T cell activity, lymphodepletion, and tumor killing. TMZ regimens can be increased to yield host myeloablation and increased expansion of adoptively transferred cells30. Furthermore, the lymphodepletive effects of TMZ can be recapitulated by low-dose (4 - 6 Gy) single-fraction total body irradiation (TBI), thus circumventing the tumoricidal properties of SOC. The radiation regimen applied here mimics a biologically equivalent dose of 60 Gy; however, the number of fractions and fractional doses can be tuned to mimic other clinical doses (Table 2). Importantly, our model of SOC utilizes biologically equivalent doses of WBI, rather than external beam radiation delivered focally to the tumor and margins, as is often utilized in the clinic, and thus may lead to a lesser dose of radiation delivered to the tumor and an increased risk of toxicities to healthy mouse brain. Despite these limitations, however, WBI is an accepted technique for modeling clinical radiotherapy in a preclinical setting.
CAR T cell doses can be increased or decreased to observe dose effects on tumor killing and overall survival11. Additionally, the timing of treatment and route of delivery can be modified to see differences in efficacy. For instance, CAR T cells can be delivered intracranially to ensure activity at the tumor site. The yield of CAR-positive T cells can vary depending on the efficiency of transduction. One way of ensuring a successful transduction is by carrying a green fluorescent protein (GFP)-control throughout the procedure. To do this, 1 HEK293T 10 cm PDL plate can be allotted for GFP transfection and transduction of a single well of T cells to screen for GFP on days 3-5. A critical factor in transduction efficiency is the extent of T cell activation and proliferation upon addition of viral supernatant. We note an important discrepancy between our method of T cell activation, which is achieved by the addition of ConA to the culture medium, and that used in the clinical setting, achieved by CD3 and CD28 agonist monoclonal antibodies. Although the latter is routinely used for generating patient retroviral transduced CAR T cells and has also been successful in transduction of murine T cells, we previously determined that ConA stimulation led to better and more consistent transductions37. We have also observed that overall CAR T cell yield can vary based on when step 5.18 is performed. If you experience poor CAR T cell yield, we recommend performing step 5.18 immediately after step 5.17 instead of waiting for the 5-6 hr incubation. Although this may impact the efficiency of transduction, we have not observed significant differences in CAR surface expression in our studies.
Following transduction, CAR T cells can be quantified and phenotyped by flow cytometry; we stain our EGFRvIII CAR T cells with a 2-color panel comprised of a streptavidin-phycoerythrin (SA/PE)-conjugated biotynlated EGFRvIII-derived multimer and anti-CD3. Alternatively, CARs can also be stained for expression using goat-anti-human F(ab’)2-biotin primary and SA/PE secondary antibodies which has been previously described36. We routinely observe CAR expression among 55-70% of murine splenocytes11. We have previously shown that delivery of T cells with this CAR expression profile can readily traffic to the brain and treat tumors. TMZ- or TBI-induced lymphopenia enhances clonal expansion of adoptively transferred cells, increasing the frequency of CAR-positive cells and their overall antitumor response. CAR T cells can be tracked in vivo by staining peripheral blood with the appropriate tetramer; this is a useful technique for understanding CAR T cell survival over time and the effects of host-conditioning on CAR T cell function and persistence. Alternatively, if no tetramer is available for the antigen receptor, a fluorescent reporter can be included in the CAR vector construct, or CAR T cells can be labeled ex vivo with carboxyfluorescein succinimidyl ester (CFSE) prior to administration for in vivo tracking.
Depending on the tumor model, immunotherapy platform, and SOC regimen, clinical SOC administered alongside adoptive therapy or vaccines can lead to toxicities. TMZ is a DNA alkylating agent that leads to non-specific cell killing, and intensified doses can result in weight loss and morbidity in murine hosts. High-dose and prolonged fractions of WBI can result in hydrocephalus. Furthermore, inflammatory effects from strong vaccine and/or adoptively transferred T cell responses can lead to morbidity due to toxic cytokine storms. It is also important to note the effects of systemic anesthesia on morbid animals. If TMZ and WBI lead to significant toxicities, then ketamine/xylazine doses can be decreased by 20-25% to reduce the risk of mortality, as animals only need to be sedated and immobilized for 10 min for delivery of small radiation doses (2-8 Gy).
Host conditioning is critical for all immunotherapy platforms that include adoptive transfer. As immunotherapy trials are often evaluated within the context of SOC, preclinical evaluation of immunotherapies should be done within this setting to recapitulate the clinical scenario. We present here one example of leveraging the lymphodepletive and tumoricidal effects of SOC with adoptive therapy using CAR T cells. This regimen is a powerful tool that can be applied to other immunotherapy platforms to evaluate the effects of combined SOC.
Disclosures
The authors have no conflicts of interests to declare.
Acknowledgments
The authors would like to acknowledge Dr. Laura Johnson and Dr. Richard Morgan for providing the CAR retroviral construct. The authors also thank Giao Ngyuen for her assistance with dosimetry for whole brain irradiation. This work was supported by an NIH NCI grant 1R01CA177476-01.
References
- Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Kantoff P, Higano C, Shore N, Berger E, Small E. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010. pp. 411–422. [DOI] [PubMed]
- Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber DJ, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc. 1989;21(1 Pt 1):127–130. [PubMed] [Google Scholar]
- Pule MA, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrand CJ, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995;55(14):3140–3148. [PubMed] [Google Scholar]
- Sampson JH, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20(4):972–984. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppner MC, Hamou MF, Sawamura Y, Bodmer S, de Tribolet N. Inhibition of lymphocyte function by glioblastoma-derived transforming growth factor beta 2. J Neurosurg. 1989;71(2):211–217. doi: 10.3171/jns.1989.71.2.0211. [DOI] [PubMed] [Google Scholar]
- Wintterle S, et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63(21):7462–7467. [PubMed] [Google Scholar]
- Fecci PE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66(6):3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- Wilmotte R, et al. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport. 2005;16(10):1081–1085. doi: 10.1097/00001756-200507130-00010. [DOI] [PubMed] [Google Scholar]
- Yang BC, et al. Mediation of enhanced transcription of the IL-10 gene in T cells, upon contact with human glioma cells, by fas signaling through a protein kinase A-independent pathway. Journal of Immunology. 2003;171(8):3947–3954. doi: 10.4049/jimmunol.171.8.3947. [DOI] [PubMed] [Google Scholar]
- Reilly SM, et al. Temozolomide: a new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumours. Eur J Cancer. 1993;29A(7):940–942. doi: 10.1016/s0959-8049(05)80198-4. [DOI] [PubMed] [Google Scholar]
- Newcomb E, et al. The Combination of Ionizing Radiation and Peripheral Vaccination Produces Long-term Survival of Mice Bearing Established Invasive GL261Gliomas. Clin Cancer Res. 2006;12:4730–4737. doi: 10.1158/1078-0432.CCR-06-0593. [DOI] [PubMed] [Google Scholar]
- Park B, Yee C, Lee KM. The effect of radiation on the immune response to cancers. Int J Mol Sci. 2014;15(1):927–943. doi: 10.3390/ijms15010927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzell S, et al. Intratumoral temozolomide synergizes with immunotherapy in a T cell-dependent fashion. Cancer Immunol Immunother. 2013;62(9):1463–1474. doi: 10.1007/s00262-013-1449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SD, et al. Cross-priming by temozolomide enhances antitumor immunity of dendritic cell vaccination in murine brain tumor model. Vaccine. 2007;25(17):3485–3491. doi: 10.1016/j.vaccine.2006.12.060. [DOI] [PubMed] [Google Scholar]
- Emens LA, Jaffee EM. Leveraging the activity of tumor vaccines with cytotoxic chemotherapy. Cancer Res. 2005;65(18):8059–8064. doi: 10.1158/0008-5472.CAN-05-1797. [DOI] [PubMed] [Google Scholar]
- Murphy KA, et al. An in vivo immunotherapy screen of costimulatory molecules identifies Fc-OX40L as a potent reagent for the treatment of established murine gliomas. Clin Cancer Res. 2012;18(17):4657–4668. doi: 10.1158/1078-0432.CCR-12-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb EW, et al. Radiotherapy enhances antitumor effect of anti-CD137 therapy in a mouse Glioma model. Radiat Res. 2010;173(4):426–432. doi: 10.1667/RR1904.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YB, Krown SE, Livingston PO, Wolchok JD, Chapman PB. How lymphotoxic is dose-intensified temozolomide? The glioblastoma experience. J Clin Oncol. 2005;23(18):4235–4236. doi: 10.1200/JCO.2004.00.8417. [DOI] [PubMed] [Google Scholar]
- Neyns B, Tosoni A, Hwu WJ, Reardon DA. Dose-dense temozolomide regimens: antitumor activity, toxicity, and immunomodulatory effects. Cancer. 2010;116(12):2868–2877. doi: 10.1002/cncr.25035. [DOI] [PubMed] [Google Scholar]
- Muranski P, et al. Increased intensity lymphodepletion and adoptive immunotherapy-how far can we go. Nat Clin Pract Oncol. 2007;3(12):668–681. doi: 10.1038/ncponc0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Current Opinion in Immunology. 2005;17(2):195–201. doi: 10.1016/j.coi.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Perez LA, et al. Myeloablative Temozolomide Enhances CD8(+) T-Cell Responses to Vaccine and Is Required for Efficacy against Brain Tumors in Mice. Plos One. 2013;8(3) doi: 10.1371/journal.pone.0059082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YB, et al. Selective CD4+ lymphopenia in melanoma patients treated with temozolomide: a toxicity with therapeutic implications. J Clin Oncol. 1200;22(4):610–616. doi: 10.1200/JCO.2004.07.060. [DOI] [PubMed] [Google Scholar]
- Dummer W, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110(2):185–192. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzesinski C, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. 2010;33(1):1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MS, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16(4):457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23(10):1043–1053. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar SP, et al. Genetic engineering of murine CD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies. J Immunother. 2011;34(4):343–352. doi: 10.1097/CJI.0b013e3182187600. [DOI] [PMC free article] [PubMed] [Google Scholar]