

Abstract
Degeneration of mesencephalic dopaminergic (mesDA) neurons is the pathological hallmark of Parkinson’s diseae. Study of the biological processes involved in physiological functions and vulnerability and death of these neurons is imparative to understanding the underlying causes and unraveling the cure for this common neurodegenerative disorder. Primary cultures of mesDA neurons provide a tool for investigation of the molecular, biochemical and electrophysiological properties, in order to understand the development, long-term survival and degeneration of these neurons during the course of disease. Here we present a detailed method for the isolation, culturing and maintenance of midbrain dopaminergic neurons from E12.5 mouse (or E14.5 rat) embryos. Optimized cell culture conditions in this protocol result in presence of axonal and dendritic projections, synaptic connections and other neuronal morphological properties, which make the cultures suitable for study of the physiological, cell biological and molecular characteristics of this neuronal population.
Keywords: Neuroscience, Issue 96, Ventral midbrain, Parkinson's disease, Dopaminergic, Primary neuronal culture, Neuronal development, Neurodegeneration
Introduction
Loss of dopaminergic neurons from the substantia nigra pars compacta leads to the cardinal motor symptoms of Parkinson’s Disease (PD), the second most prevalent neurodegenerative disorder. The underlying cause of the demise of this mesencephalic neuronal population is not known. To study the biochemical pathways responsible for the development and modulating neurophysiological properties and survival of mesDA neurons, several cell culture and animal model systems have been used. Immortalized cell lines, including the rat dopaminergic cell line 1RB3AN27(N27), the human dopaminergic neuroblastoma cell line SH-SY5Y, the mouse dopaminergic hybrid cell line MN9D and human mesencephalic LUHMES cells have been used for biochemical and limited mechanistic studies 1-5. For the study of the specific loss of mesDA neurons, several neurotoxin-based and genetic models have been developed 6-8. Primary ventral midbrain cultures, provide an indispensable tool for studying the neuronal and synaptic properties of the dopaminergic neurons and the pathways involved in pathogenesis of this common disorder.
Here we present a detailed protocol for the isolation of mesencephalic dopaminergic neurons, which contains modifications resulting in higher survivability and increased yield of coverslips per embryo. Use of pre-mature E12.5 mouse mesencephalon (E14.5 in rat) enhances survivability. At this age neurons have not developed axons yet, which leaves cells intact during dissection and minimizes the stress thereby significantly increasing viability. In addition, careful dissection of the ventral midbrain, as described in section 2 of this protocol, further enhances survivability. To increase the numbers of coverslips per embryo, an alternative plating method is presented in section 4 of this protocol. This leads to a yield of up to 10 coverslips per embryo as compared to 4 coverslips under standard plating conditions thus reducing the amount of animals per experiment.
Neurons in culture exhibit outgrowth of axons and dentrites, form synaptic connections and reveal the presence of neuronal and synaptic markers making these cultures suitable for live cell imaging, immunocytochemical and electrophysiological studies. Furthermore, the use of neuronal cultures facilitates genetic and pharmacologic manipulation. Outgrowth of neurites from day 2 in vitro allows for developmental studies. Furthermore, the long-term survival of cultures (up to six weeks) makes them suitable for study of the slow, progressive degeneration of these neurons.
Protocol
NOTE: The animals were maintained and handled in compliance with the institutional guidelines and all animal procedures were approved by the Imperial College's Animal Welfare and Ethical Review Body (AWERB) and the Home Office and Harvard University Institutional Animal Care and Use Committee (IACUC), in compliance with federal and state regulations.
1. Reagent and Equipment Setup
Thaw, aliquot and store 1 mg/ml Laminin solution at –80 ºC. Dissolve 2 µl in 1 ml DMEM/F12, resulting in a coating concentration of 1-2 µg/cm2. NOTE: Thaw Laminin slowly on ice and dissolve in cold DMEM/F12 and immediately add it to the plates/coverslips.
Dilute 75 ml of 0.01% Poly-L-ornithine in 425 ml PBS and store at 4 ºC. NOTE: The shelf life of the working solution is up to one month.
Dissolve 25% (wt/vol) BSA in PBS, pH 7.4, and store at 4 ºC for up to a year.
Prepare 50% FBS (in HBSS) deactivation media.
Prepare complete medium by adding 50 U/ml penicillin and streptomycin, 1x N2 supplement, 5% (vol/vol) FBS, 0.36% D-(+)-Glucose (wt/vol), 0.25% BSA (wt/vol) to DMEM/F12 and store at 4 ºC. NOTE: The shelf life is up to 10 days.
Place coverslips in boiling 70% (vol/vol) ethanol for at least 30 min to remove any traces of grease and autoclave.
Place coverslips in 24-well plates and add 500 µl Poly-L-ornithine solution to each well. Incubate 1 hr under the tissue culture hood at RT and then, wash 3 times with 500 µl water. Add 500 µl laminin solution to each plate and incubate O/N in the humidified tissue culture incubator at 37 ºC. NOTE: While no washes are necessary after laminin treatment, washing the poly-L-ornithine less than three times results in death of the cells 24 h after culturing.
Polish the tips of glass pipettes over natural gas or torch flame. NOTE: After this step the diameter of the tip should be about half of its normal size.
Cut the caps of 1.5 ml microcentrifuge tubes, using an autoclaved scissor and autoclave.
2. Dissection of Embryonic Ventral Midbrain
Narcotize timed-pregnant (E12.5) dams by CO2 inhalation and then euthanize by cervical dislocation.
Spray the abdomen with 70% ethanol and make an abdominal incision until the uterine sacs are all exposed. Using forceps cut the vaginal attachment and remove uterus. Place the uterus in ice-cold HBSS, in a 100 mm Petri dish.
Remove embryos from the uterus and amniotic sac, using forceps (Figure 1C). Place embryos in fresh ice-cold HBSS. NOTE: The rectangle in Figure 1D indicates the location of embryonic ventral midbrain.
Place the embryos under a dissection microscope (10x magnification) and dissect the mesencephalic arch by cutting the brain at the isthmus and mesencephalic-diencephalic boundary region, using the bioscissor and forceps (Figure 1E, please refer to Figure 1A for lines of incision).
After taking out the entire midbrain, make a cut in the mediodorsal midbrain (Figure 1F).
Remove meninges, using two forceps (Figure 1H). NOTE: CRITICAL STEP: This step is essential for optimal neuronal yield and survival. Since the culture conditions are optimal for brain cells, most of the cells from the surrounding tissue die after plating and the apoptotic signal from this tissue may damage integrity of the cultures.
Flatten the midbrain tissue on the Petri dish to observe the butterfly shape and cut approximately half of each wing, using a sterilized shaving blade (Figure 1H,I). NOTE: Figure 1J depicts the isolated ventral midbrain.
Transfer the piece of ventral midbrain into ice-cold HBSS in a 15 ml conical tube and proceed with the next embryo. NOTE: Steps 2.1-2.8 should not take more than 1 hr. Keeping the embryos beyond this time frame on ice decreases cell viability significantly.
3. Dissociation of Ventral Midbrain Cells
Transfer the pieces of ventral midbrain under a laminar flow hood. Remove the HBSS and add 1 ml pre-warmed (37 ºC) 0.05% trypsin-EDTA into the conical tube (volumes enough for up to 12 pieces of ventral midbrain). Incubate the tissue at 37 ºC for 5-10 min.
Remove trypsin-EDTA under the hood and add 1 ml de-activation medium (the serum will deactivate trypsin) to the tissue.
Remove the deactivation medium and wash the tissue in 1 ml complete medium twice. NOTE: Avoid removing all the medium from the tube and pipetting the tissue, to prevent discarding the dissociated cells.
Add 1ml complete medium and triturate the tissue with a fire-polished glass pipette. Avoid formation of bubbles and continue triturating until single cell suspension is achieved. NOTE: Usually, 8-10 passes through the restricted tip are required for complete dissociation.
Centrifuge the cells at 400 x g for 5 min at RT and remove the medium.
Re-suspend the pellet in 1 ml complete medium by slowly pipetting up and down for up to four times.
4. Plating Ventral Midbrain Cells
Mix 10 µl of the cell suspension with 90 µl Trypan blue solution and count the number of cells with a hemocytometer. NOTE: Viability of the cells can also be checked at this stage.
Place sterilized microcentrifuge tube caps in 100 mm Petri dishes and transfer the Poly-L-ornithine/Laminin coated coverslips, using forceps, one at a time on top of the caps. NOTE: Do not wash the coverslips and avoid drying the laminin, since this may cause uneven cultures and decrease in survivability of the cells.
Adjust the volume to 1,500 cells per 1 µl by adding complete medium and add 100 µl (total of 150,000 cells) of the cell suspension on top of each coverslip (the volume is optimal to cover the surface of the coverslip without spillage). Close the Petri dishes and incubate them for 1 hr in a humidified tissue culture incubator (37 °C, 5% CO2). NOTE: CRITICAL STEP: This step is required for enhancing the attachment and viability of the cells and for increasing the number of coverslips per embryo. Alternatively, the cells can be transferred directly into the wells (containing coverslips) but the number of cells should be increased to 350,000 per well (instead of 150,000). In other words, using this method, 8-10 coverslips can be acquired, while direct transfer yields about 3-4 coverslips because of the cells, which attach to the plates (beside or underneath the coverslips). The average viability of the cultures, using this method was around 90%.
5. Culture Growth and Maintenance
After 1 hr of incubation, carefully transfer the coverslips including the medium into 24-well plates, containing 400 µl pre-warmed (to 37 ºC) complete medium (total volume: 500 µl). Incubate the cells O/N at 37 ºC.
Within 24 hr after incubation of the wells, gently add 500 µl complete medium into each well. NOTE: The first few hours is the most critical stage for survival of the dopaminergic neurons and the number of surviving cells does not change significantly beyond this point.
Do not add additional media to the cultures for the first two weeks or until the medium becomes yellow. If culturing for more than two weeks or medium color changes to yellow, exchange half of the media (500 µl) with fresh complete pre-warmed media (typically every two weeks). NOTE: Dopaminergic neurons within the cultures would survive for more than 6 weeks under these conditions.
Representative Results
Immunohistochemistry against Tyrosine hydroxylase (Th) shows that between 0.5-1% of the cells in culture are dopaminergic. Neuronal projections appear within 2 hr after plating and by the first day, axons and dendrites are distinguishable (Figure 2), using Tyrosine Hydroxylase (TH) and Microtubule-Associated Protein 2 (Map2) antibodies (Figure 3). The neurons survive for more than six weeks and show extensive outgrowth. The neuron-glia ratio in the cultures is directly related to the concentration of serum in the medium, as shown previously. While eliminating FBS from the cultures may yield relatively pure neuronal cultures, we and others have found that addition of serum increases survivability of dopaminergic neurons in culture 9.
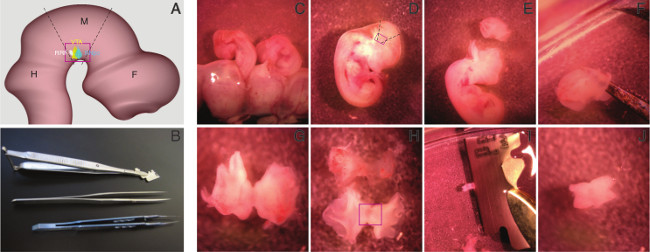 Figure 1. Procedure for isolation of embryonic ventral midbrain. The schematic view of E12.5 mouse brain, midbrain boundaries (dashed lines), and the approximate position of the region of interest, ventral midbrain, within the red rectangle (A) and the tools for dissection of the ventral midbrain, a bioscissor, foreceps, and a blade holder (B) are shown. The embryos are taken out from amniotic sac by rupturing it (C). The region of interest, ventral midbrain, is shown by the rectangle (D). Midbrain is isolated by cutting at the isthmus (left of the cut piece) and the midbrain-forebrain boundary (right; E). The mediodorsal part of the midbrain is cut (F) and it is flattened, to resemble a butterfly shape (G,H). Then, meninges are dissociated from brain (H) and ventral midbrain (rectangle) is isolated from the dorsal part by cutting off half of the wings, using a shaving blade (I). Ventral midbrain (J) is transferred to a 15 ml conical tube and kept on ice, until dissociation. Please click here to view a larger version of this figure.
Figure 1. Procedure for isolation of embryonic ventral midbrain. The schematic view of E12.5 mouse brain, midbrain boundaries (dashed lines), and the approximate position of the region of interest, ventral midbrain, within the red rectangle (A) and the tools for dissection of the ventral midbrain, a bioscissor, foreceps, and a blade holder (B) are shown. The embryos are taken out from amniotic sac by rupturing it (C). The region of interest, ventral midbrain, is shown by the rectangle (D). Midbrain is isolated by cutting at the isthmus (left of the cut piece) and the midbrain-forebrain boundary (right; E). The mediodorsal part of the midbrain is cut (F) and it is flattened, to resemble a butterfly shape (G,H). Then, meninges are dissociated from brain (H) and ventral midbrain (rectangle) is isolated from the dorsal part by cutting off half of the wings, using a shaving blade (I). Ventral midbrain (J) is transferred to a 15 ml conical tube and kept on ice, until dissociation. Please click here to view a larger version of this figure.
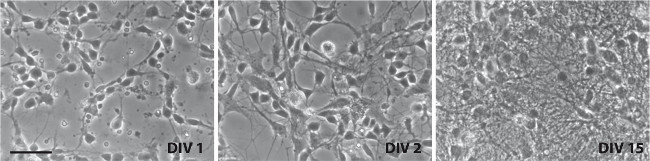 Figure 2. Representative brightfield images of ventral midbrain cultures. Images were taken on DIV1 (left), DIV2 (center), and DIV15 (right). Scale bar: 50 µm. Please click here to view a larger version of this figure.
Figure 2. Representative brightfield images of ventral midbrain cultures. Images were taken on DIV1 (left), DIV2 (center), and DIV15 (right). Scale bar: 50 µm. Please click here to view a larger version of this figure.
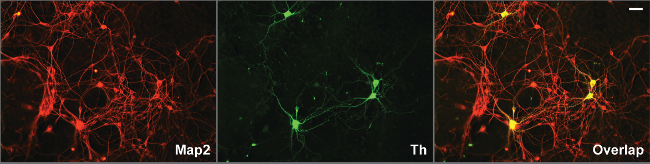 Figure 3. Long term survival, outgrowth of processes and synaptogenesis in two week old ventral midbrain cultures. Midbrain dopaminergic neurons, identified by tyrosine hydroxylase antibody grow extensive axons and dendrites, identified by Map2 antibody, in the ventral midbrain cultures two weeks after dissociation from E12.5 embryos. Dendrites can be distinguished by overlap of Th and Map2-positive processes, where only Th-positive fibers represent axons. Antibodies were used at a dilution of 1:200. Scale bar: 20 µm. Please click here to view a larger version of this figure.
Figure 3. Long term survival, outgrowth of processes and synaptogenesis in two week old ventral midbrain cultures. Midbrain dopaminergic neurons, identified by tyrosine hydroxylase antibody grow extensive axons and dendrites, identified by Map2 antibody, in the ventral midbrain cultures two weeks after dissociation from E12.5 embryos. Dendrites can be distinguished by overlap of Th and Map2-positive processes, where only Th-positive fibers represent axons. Antibodies were used at a dilution of 1:200. Scale bar: 20 µm. Please click here to view a larger version of this figure.
Discussion
Dopaminergic neurons in midbrain are the main source of dopamine in the central nervous system. They are divided into three groups, substantia nigra pars compacta (SNpc), ventral tegmental area (VTA) and retrorubral field (RRF) 10,11. The neurons in SNpc and VTA give rise to major dopaminergic pathways, mesocortical, mesolimbic and nigro-striatal, involved in functions such as control of emotion, motivation and motor behavior. Demise of the neurons in SNpc and functional disruption of the nigrostriatal system is the main pathological characteristic of the second most prominent neurodegenerative disorder, Parkinson’s disease (PD) 12,13. Current therapeutic approaches address symptoms of the disease and do not halt or slow down degeneration and the causes of the slow, progressive cell loss are still unknown 14-16. Therefore, development of in vivo and in vitro techniques for study of the developmental, biochemical, and physiological characteristics of this neuronal population is imperative to understanding the etiology of PD and to development of new therapeutic approaches.
Here we describe an optimized method for dissection, dissociation and culturing of primary neurons from embryonic mouse and rat ventral midbrain, which results in long-term in vitro survival of the dopaminergic neurons from this region, allowing study of the processes such as differentiation, axonal outgrowth and synapse formation. Several advantages of this protocol include the increased number of coverslips per embryo, independence from growth factors and the length of in vitro survival, maximizing development of neurons 4. This method may complement the in vivo studies in allowing for understanding the mechanisms underlying normal physiological functions of the midbrain dopaminergic neurons as well as progressive degeneration of this neuronal population in cytotoxic and genetic models of Parkinson’s disease.
One of the main disadvantages of this protocol is the lack of distinction between SNpc and VTA dopaminergic neurons. This problem exists because at the time of culturing (E12.5 in mice or E14.5 in rats) terminal differentiation of the neurons and their regionalization is not complete and while the use of older embryos is possible, it significantly reduces the survival of the neurons due to axotomy. Distinguishing the two populations would be possible by immunocytochemistry, using antibodies against specific markers for each population such as Girk2 (to identify SNpc neurons) and Calbindin (to identify VTA neurons)17.
The optimized plating described in section 4 decreases the amount of embryos per experiment by more than 50% and therefore contributes to reducing the use of animals in research. The most critical steps for survivability of the cultures are the time between euthanization of the dams and complete dissection of the embryonic midbrains (step 2.1 – 2.8), as well as the complete removal of the meninges (step 2.6, Figure 1H).
Disclosures
No conflict of interest declared.
Acknowledgments
This work was performed using the Division of Brain Sciences, Department of Medicine, Imperial College London startup funds to K.N.A.
References
- Prasad KN, et al. Establishment and characterization of immortalized clonal cell lines from fetal rat mesencephalic tissue. In Vitro Cell Dev Biol Anim. 1994;30A:596–603. doi: 10.1007/BF02631258. [DOI] [PubMed] [Google Scholar]
- Fall CP, Bennett JP. Characterization and time course of MPP+ -induced apoptosis in human SH-SY5Y neuroblastoma cells. J Neurosci Res. 1999;55:620–628. doi: 10.1002/(SICI)1097-4547(19990301)55:5<620::AID-JNR9>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Choi HK, Won L, Roback JD, Wainer BH, Heller A. Specific modulation of dopamine expression in neuronal hybrid cells by primary cells from different brain regions. Proc Natl Acad Sci U S A. 1992;89:8943–8947. doi: 10.1073/pnas.89.19.8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, Sgado P, Alberi L, Subramaniam S, Simon HH. Elevated P75NTR expression causes death of engrailed-deficient midbrain dopaminergic neurons by Erk1/2 suppression. Neural Dev. 2009;4:11. doi: 10.1186/1749-8104-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, et al. The transcription factor orthodenticle homeobox 2 influences axonal projections and vulnerability of midbrain dopaminergic neurons. Brain. 2010;133:2022–2031. doi: 10.1093/brain/awq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson's disease. Bioessays. 2002;24:308–318. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- Schober A. Classic toxin-induced animal models of Parkinson's disease: 6-OHDA and MPTP. Cell Tissue Res. 2004;318:215–224. doi: 10.1007/s00441-004-0938-y. [DOI] [PubMed] [Google Scholar]
- Chesselet MF, Richter F. Modelling of Parkinson's disease in mice. Lancet neurology. 2011;10:1108–1118. doi: 10.1016/S1474-4422(11)70227-7. [DOI] [PubMed] [Google Scholar]
- Takeshima T, Johnston JM, Commissiong JW. Mesencephalic type 1 astrocytes rescue dopaminergic neurons from death induced by serum deprivation. J Neurosci. 1994;14:4769–4779. doi: 10.1523/JNEUROSCI.14-08-04769.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgado P, et al. Slow progressive degeneration of nigral dopaminergic neurons in postnatal Engrailed mutant mice. PNAS. 2006;103:15242–15247. doi: 10.1073/pnas.0602116103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, et al. The lifelong maintenance of mesencephalic dopaminergic neurons by Nurr1 and engrailed. J Biomed Sci. 2014;21:27. doi: 10.1186/1423-0127-21-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HH, Bhatt L, Gherbassi D, Sgado P, Alberi L. Midbrain dopaminergic neurons: determination of their developmental fate by transcription factors. Ann N Y Acad Sci. 2003;991:36–47. [PubMed] [Google Scholar]
- Bjorklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. doi: 10.1016/j.tins.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med. 2013;62:132–144. doi: 10.1016/j.freeradbiomed.2013.01.018. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Aetiopathogenesis of Parkinson's disease. J Neurology. 2011;258:S307–S310. doi: 10.1007/s00415-011-6016-y. [DOI] [PubMed] [Google Scholar]
- Chung CY, et al. Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum Mol Gen. 2005;14:1709–1725. doi: 10.1093/hmg/ddi178. [DOI] [PMC free article] [PubMed] [Google Scholar]