

Abstract
An urgent need exists to test the contribution of new genes to the pathogenesis and progression of human glioblastomas (GBM), the most common primary brain tumor in adults with dismal prognosis. New potential therapies are rapidly emerging from the bench and require systematic testing in experimental models which closely reproduce the salient features of the human disease. Herein we describe in detail a method to induce new models of GBM with transposon-mediated integration of plasmid DNA into cells of the subventricular zone of neonatal mice. We present a simple way to clone new transposons amenable for genomic integration using the Sleeping Beauty transposon system and illustrate how to monitor plasmid uptake and disease progression using bioluminescence, histology and immuno-histochemistry. We also describe a method to create new primary GBM cell lines. Ideally, this report will allow further dissemination of the Sleeping Beauty transposon system among brain tumor researchers, leading to an in depth understanding of GBM pathogenesis and progression and to the timely design and testing of effective therapies for patients.
Keywords: Medicine, Issue 96, Glioblastoma models, Sleeping Beauty transposase, subventricular zone, neonatal mice, cloning of novel transposons, genomic integration, GBM histology, GBM neurospheres.
Introduction
Glioblastoma multiforme (GBM) is the most common (60%) primary brain tumor in adults, with a median survival of 15-21 months when treated with surgery, radiation therapy, and chemotherapy1. Novel therapies for GBM are imperative. Experimental therapies require testing in animal models which adequately reproduce the salient features of the human disease. Strategies to induce GBM in rodents include chemical mutagenesis with alkylating agents, germline or somatic genetic alterations, or transplantation using glioma cell lines2. The most commonly used models employ the implantation of glioma cell lines, either orthotopically, into the brain or subcutaneously using syngeneic cells in animals with identical genotype; or xenogeneic cells, most commonly human GBM cell lines, implanted in immune compromised mice3. Xenografts offer many advantages for the study of intracranial tumors: convenience of reproducibility, standardized growth rates, time of death and tumor localization. However, these models have limitations due to the artificial, invasive surgical approach used for implantations and limited ability to accurately reproduce histological features characteristic of human GBM (WHO grade IV): pseudo-pallisading necrosis, nuclear atypias, diffuse invasion, micro-vascular proliferation and the formation of glomeruloid vascular abnormalities4-7. Induction of GBM by altering the genome of somatic cells with oncogenic DNA, either with viral vectors8-12, or with transposon-mediated integration13, reproduces more closely the etiology of human disease and recapitulates histo-pathological features of human GBM.
Historically, tumors of the CNS have been classified based on the perceived cell of origin, which it was observed, would be a predictive factor for survival14. GBMs are classified into primary and secondary. Emerging evidence today points to the highly heterogeneous nature of primary glioblastoma15 . Secondary GBM (15%), the result of malignant transformation of low grade astrocytomas (WHO grade I) and anaplasic astrocytomas (WHO grade II), are associated with earlier onset of the disease, better prognosis and a “proneural” pattern of gene expression, whereas primary GBM (85%) show a late onset, poor prognosis and glial (classical), neural or mesenchymal expression patterns. Whether these patterns of gene expression correlate with the actual cell of origin of the tumor is still being actively investigated. Accumulating data shows that the combination of genetic mutations associated with GBMs are predictive for survival. For example, loss of heterozygocity (LOH) of chromosomes 1p/19q, IDH1 mutations, PDGFRα amplifications, are associated with secondary GBMs, proneural expression pattern and better prognosis, whereas EGFR overexpression, Notch and Sonic hedgehog pathway activation, Nf1 and PTEN loss and mutations of p53 are correlated with neural, classical, or mesenchymal primary GBM and worse prognosis16,17. The advent of large scale sequencing projects and the accumulation of numerous patient specimens available for testing brings a wealth of new information with respect to genetic mutations and pathways implicated in GBM pathogenesis and progression and the possibility of individualized medicine, where therapies can be specifically tailored to the genetic abnormalities of the patient. Ultimately, to assess the predictive value of these mutations and pathways in a systematic way, and to test possible treatments in each case, requires animal models of GBM with pre-determined genetic alterations. Transposon mediated integration of genomic DNA offers a feasible approach.
The Sleeping Beauty transposon system, member of the Tc1/mariner class of transposons, was “awakened” (constructed) in a multi-step process of site specific mutagenesis from a salmonoid transposase gene, which became dormant more than 10 million years ago18,19. In essence, DNA transposons flanked by specific sequences (inverted repeats/direct repeats: IR/DR) can be integrated into the genome in a “cut and paste” manner by means of the activity of the Sleeping Beauty transposase. The transposase recognizes the ends of the IR sites, excises the transposon and integrates it randomly into another DNA site between the bases T and A, bases which are duplicated at each end of the transposon during transposition (Figure 1a). The Sleeping Beauty transposase is comprised of three domains, a transposon binding domain, a nuclear localization sequence and a catalytic domain. Four transposase molecules are required to bring the two ends of the transposon together and allow for transposition, however, if too many molecules of transposase are present, they can dimerize and tetramerize to inhibit the transposition reaction20. An efficient transposition reaction requires an optimal ratio of transposase to transposons. The DNA encoding the transposase can be delivered on the same plasmid with the transposon (in cis) or on a different plasmid (in trans). To ensure the optimal ratio between the transposase and the transposons, a promoter with adequate activity can be chosen for the expression of the transposase (for the “cis” model) or the ratio of the plasmids in the injection solution can be optimized (for the “trans” model). The Sleeping Beauty transposon system can be used successfully for functional genomics, insertional mutagenesis, transgenesis and somatic gene therapy21. Being a synthetic construct re-engineered to a functional molecule from a dormant salmonoid variant, the Sleeping Beauty transposase does not bind to other transposons in humans or other mammals20. Since its discovery, molecular engineering has enhanced the transposition efficacy of the SB transposon system through changes in the IR sequences and addition of TATA dinucleotides flanking the transposon, resulting in the pT2 transposons. These transposons have optimized binding of the SB to the IR site and increased efficacy of excision. The SB transposase also underwent significant improvement; the transposase used in experiments presented herein is the SB100X, a hyperactive transposase generated by a DNA shuffling strategy followed by a large-scale genetic screen in mammalian cells22.
In this report we present a rapid, versatile and reproducible method to induce intrinsic GBM in mice with non-viral, transposon-mediated integration of plasmid DNA into cells of the sub-ventricular zone of neonatal mice23. We present a simple way to create transposons with novel genes amenable for genomic integration using the Sleeping Beauty transposase and demonstrate how to monitor plasmid uptake and disease progression using bioluminescence. We also characterize histological and immuno-histochemical features of the GBM reproduced with this model. In addition, we present a quick method to generate primary GBM cell lines from these tumors. The Sleeping Beauty model, in which tumors are induced from cells original to the animal, allows the functional assessment of the role of candidate GBM genes in the induction and progression of tumors. This system is also well suited for the testing of novel GBM treatments, including immune therapies in immuno-competent mice, without the need of invasive inflammatory surgical procedures, which may alter the local microenvironment.
Protocol
NOTE: All animal protocols have been approved by the University of Michigan Committee for the Use and Care of Animals (UCUCA).
1. Cloning of the Sequence of Interest into New Sleeping Beauty Transposons
NOTE: To insert genes or inhibitory elements (as shRNA) using the Sleeping Beauty transposase system, clone the sequence of interest into the backbone of a pKT or pT2 plasmid. Direct the cloning such that the regulatory elements, gene of interest and markers remain flanked by the inverted repeats (IR/DR). An example of cloning PDGFβ into a pKT backbone is detailed below (see also Figure 1b).
Digest 5 µg of plasmid vector pKT-IRES-Katushka for 4 hours at 37 °C with 5-10 units of XbaI/XhoI restriction enzymes in 50 µl of reaction volume.
Digest the mPDGFβ cDNA of 5 µg of the pGEM-T- mPDGFβ plasmid for 4 hours at 37 °C with 5-10 units of NcoI/SacI restriction enzymes.
Precipitate the total DNA by adding 2.5 volumes of 100% ethanol, and 1/10 volume of 3 M Na Acetate, pH 5.8 and incubate overnight at -20 °C.
Centrifuge the samples at 13,800 x g for 15 min.
Wash the DNA pellet with 500 µl of 70% ethanol, centrifuge at 13,800 x g for 1 min, repeat once and re-suspend in 19 µl of sterile DNase free water.
Follow the manufacturer’s instructions in the Quick Blunting Kit to blunt the ends of both vector (pKT-IRES-Katushka) and insert (mPDGFβ).
Load the blunted vector and insert on a 1.2% ethidium bromide stained agarose gel and run at 80 V for 40 min. Visualize the bands, excise them with a clean razor-blade and gel purify the DNA using a gel purifying kit according to manufacturer’s protocol.
Ligate the insert and vector DNA purified in step 1.7 by adding them in a tube at a 4:1 molar ratio (insert:vector). Into this tube, pipette 1 µl of T4 ligase and 2 µl of T4 ligase buffer (containing ATP), and adjust the volume to 20 µl with sterile water. Incubate the ligation reaction for 18 hr at 16 °C.
- Transform chemically competent DH5α bacterial cells with the ligated products.
- Thaw the competent cells on ice.
- Add the entire volume of the ligation reaction to 50 µl of competent cells, gently shake the tube and incubate the mix on ice for 30 min. Heat-shock the bacteria for 45 sec at 42 °C and return immediately to ice; incubate on ice for another 2 min.
- Add 950 µl of Luria Broth to the culture and incubate with shaking at 37 °C for 1 hr. Centrifuge bacteria at 2,400 x g for 3 min and re-suspend the pellet in 200 µl of LB.
- Spread the transformed bacteria onto a Petri dish coated with LB-agarose and the corresponding antibiotic. Incubate the plate overnight at 37 °C.
- Finally, collect 5-10 bacterial colonies and culture overnight at 37 °C in 5 ml of LB media with antibiotic.
- Purify plasmid DNA using a plasmid DNA mini kit according to manufacturer’s instructions. Perform a diagnostic restriction digest to verify the proper incorporation of the insert into the vector and submit positive clones for DNA sequencing to ensure the right orientation of the insert.
Select the correct colonies and scale up the DNA production using a high quality, endotoxin free plasmid preparation kit.
Elute the DNA in sterile water or 1 mM Tris-HCl. Store DNA at -20 °C until ready to inject into neonates.
2. Intra-ventricular Neonatal Injections
Three to four weeks prior to experiment, set up a mouse breeding cage with one male and one female mouse, and a plastic colored igloo. This provides an enriched environment and aids the mating process.
When pregnancy is confirmed, remove the male to a new cage. Eighteen days after mating, monitor cage daily for delivery. Perform intraventricular injections on postnatal day 1 (P1).
- Preparation of the injection solution NOTE: The injection solution is made by mixing the plasmid DNA with the transfection reagent to create the particles for transfection. Below is an example on the preparation of an injection solution using a plasmid encoding the Sleeping Beauty transposase and luciferase: pT2/SB100x-Luc and two transposon plasmids: pT/CAGGS-NRASV12 and pT/CMV-SV40-LgT, at a ratio of 1:2:2. All the plasmids have a concentration of 2 µg/µl. Important details on the preparation of the injection solution are presented in the Discussion.
- Prepare the DNA solution by adding: 4 µg (2 µl) of pT2/SB100x-Luc, 8 µg (4 µl) of pT/CAGGS-NRASV12 and 8 µg (4 µl) of pT/CMV-SV40-LgT and 10 µl of 10% glucose to a final volume of 20 µl at a concentration of 1 µg /ml DNA and 5% glucose.
- Prepare the PEI solution by adding 2.8 µl of in vivo jet PEI, 7.2 µl of sterile water, and 10 µl of 10% glucose to a final concentration of 5% glucose.
- Add the PEI solution to the DNA solution, mix and vortex and let sit at room temperature (RT) for 20 min. The solution is now ready for injection. After one hour at RT, keep the solution on ice.
- Fit a 10 µl clean syringe equipped with a 30 gauge hypodermic needle with 12.5° bevel into the micropump (Figure 2 #1).
To verify proper functioning of the injection system, use the automatic injector (Figure 2 #1) to withdraw 10 µl of water into the syringe and then empty the syringe completely
Cool the neonatal stereotaxic stage (Figure 2 #3) with a slurry of dry ice and alcohol to a temperature of 2-8°C.
Induce anesthesia by placing the pups on wet ice for 2 min. Anesthesia will continue on the chilled stereotaxic frame for the remainder of the procedure. Maintaining the temperature of the frame above freezing, between 2-8 °C will prevent thermal burn injuries. As special precaution pups can be wrapped in gauze before placing on wet ice.
Fill the syringe with the DNA/PEI solution.
Immobilize the pup in the stereotaxic frame by placing its head between the gauze covered ear bars (Figure 2b). Ensure that the dorsal side of the skull is horizontal, parallel to the surface of the frame and that the cranial sutures are clearly visible. Wipe the head with 70% ethanol.
Lower the needle of the syringe and adjust stereotaxic coordinates using dials of the stereotaxic frame until the needle touches the lambda (the midline intersection of the parietal and occipital bones) (Figure 2b).
Lift the needle and adjust stereotaxic coordinates to 0.8 mm lateral and 1.5 mm rostral to the lambda (Figure 2b).
Lower the needle until it touches and slightly dimples the skin. Measure the coordinates. Lower the needle another 1.5 mm. The needle will pierce the skin and skull, and pass through the cortex to enter the lateral ventricle (Figure 2c).
Using the automatic injector, inject 0.75 µl of the DNA/PEI solution at a rate of 0.5 µl/min
Keep the pup on the frame for another minute to allow for the solution to disperse into the ventricles. In the meantime, anesthetize another pup, on ice for 2 min in preparation for the next injection.
Gently and slowly lift the needle, and place the pup under a heating lamp. Monitor breathing and activity. If necessary, provide gentle stimulation of the limbs until first breathing ensues.
Return the pup to its mother after it has warmed up, has reached a rosy color, is breathing regularly and is active, normally 5-7 min. If more than one pup is injected at a time, keep all pups together until all the injections are done. If injections take longer than 30 min, return half of the pups after the first 30 min and the rest at the end of the procedure.
Monitor that the dam is responsive and nurturing to her pups. If necessary, add a surrogate mom to the cage.
Ensure that the interval between placing the pup on ice and placing it under the warming lamp is less than 10 min. The quicker the procedure the better the survival. Usually, the time it takes to anesthetize and inject a pup is 4 min.
3. Bioluminescence Monitoring of Tumor Formation and Progression
3.1) Monitoring Plasmid Uptake After Injection
24-72 hr after injection, remove all pups from the mother’s cage and place in a 6 well clean tissue culture dish, one pup per well.
Prepare a 30 mg/ml solution of luciferin dissolved in normal saline or phosphate buffer. Prepare this solution in advance and keep in small aliquots at -20 °C.
Using a 1 ml syringe equipped with a 30 gauge hypodermic needle inject 30 µl of luciferin subcutaneously between the shoulder-blades of the pup. Ensure that the needle does not pierce any organs, by pinching the skin and lifting it slightly before inserting the needle.
Image immediately, using an in vivo bioluminescence imaging system. For the IVIS Spectrum, use the following settings for acquisitions: automatic exposure, large binning, and aperture f = 1.
3.2) Monitoring Tumor Formation and Progression
NOTE: Animals will start forming macroscopic tumors detectable by bioluminescence and histology within 2½-6 weeks, depending on the oncogenic plasmids injected.
Using a 1 ml syringe equipped with a 26 gauge needle inject 100 µl of a 30 mg/ml luciferin solution intra-peritoneally.
Place the animal in the anesthesia chamber. Inject up to five animals at the same time.
Wait 5 min then start the oxygen/isoflurane flow (1.5-2.5% isoflurane) to anesthetize the animals. After 3-4 min, remove the animals from the anesthesia chamber, start the oxygen /isoflurane flow in the bioluminescence chamber. Place the animals in the respective slots equipped with glass nose cones to allow for anesthesia and unobstructed passage of light.
Typically, acquire a series of 6 images, at 2 min intervals with an automatic exposure time, median binning and an open aperture of f = 1.
Set the region of interest for all the animals imaged, typically an oval over the head region, and measure the luminescence intensity using the calibrated units photons/s/cm2/sr.
Record the maximal value for each animal. Subsequently, recordings can be compared during the progression of the tumor for each animal and/or between animals, for example when different treatments are employed.
4. Histological and Immunohistochemical Analysis of New GBMs
NOTE: When the tumors have reached the desired experimental time-point, animals can be sacrificed, brains perfused, fixed and analyzed. For survival analyses the moribund stage represents the endpoint of the experiment, when animals are humanely sacrificed at the first signs of tumor burden, as defined by the clinical stage when the animal becomes symptomatic showing impaired mobility, hunched posture and scruffy fur. A brief description of the standard histological and immuno-histochemical methods used is presented below.
4.1) Perfusion and Fixation
Anesthetize the mouse with an intra-peritoneal injection of 100 mg/kg of ketamine and 10 mg/kg of xylazine.
Check the pedal reflex by pinching the foot of the mouse. When this reflex is abolished, indicating deep anesthesia, immobilize the mouse on a dissection board with pins, open the abdominal cavity, dissect the diaphragm and open the thoracic cavity to visualize the heart.
Insert a blunt 20 gauge cannula into the left ventricle of the heart. Connect the cannula with plastic tubing passing through a peristaltic pump to a container with oxygenated and heparinized Tyrode’s solution (0.8% NaCl, 0.0264% CaCl2 2H2O, 0.005% NaH2PO4, 0.1% glucose, 0.1%NaHCO3, 0.02% KCl). Start the flow of Tyrode’s solution by adjusting the speed of the peristaltic pump to ensure a slow and constant flow, approximately one drop per second or less if the animals are smaller.
Make a small incision into the right atrium of the heart to permit the drain of blood and Tyrode’s.
Monitor perfusion efficiency by observing blanching of internal organs (most obvious in the liver and kidney) as they become devoid of blood.
When the solution draining from the heart is clear (3-5 min), switch the solutions from Tyrode’s to 4% paraformaldehyde for fixation (4% PFA, pH 7.34 in phosphate buffered saline).
Monitor good fixation of the tissues by the increased rigidity of the neck and tail.
- Decapitate the mouse and carefully dissect the brain from the skull.
- Pull the fur which covers the skull rostrally, towards the nose, and grab it firmly to stabilize the head, dorsal side up.
- Using a sharp scalpel, cut downwards along the edge of the orbit, to transect the orbital muscles and the underlying zygomatic arches.
- Turn head ventral side up and remove attached neck muscles using a rongeur. Crush the first vertebra and remove it from the brainstem.
- Visualize the cochlea, grab the overlying temporal bone with the rongeur and create an opening between the temporal and occipital bone. Peel off the occipital bone from the surface of the cerebellum.
- Crush the bone overlying the nose with the rongeur. Carefully remove the remaining frontal and parietal bones from the surface of the brain being mindful not to damage the surface of the brain. Pay particular attention in the orbital area.
- Turn the head ventral side up. The brain will slide down from the remainder of the skull, connected now solely through the cranial nerves. Using small scissors cut the olfactory, optic and trigeminal nerves. This will release the brain.
Place brain in 4% PFA overnight at 4 °C for post-fixation.
For histological analysis and hematoxylin-eosin staining, transfer the brains to phosphate buffer for 24 hr and then proceed to paraffin embedding.
For immuno-histological analysis, transfer brains into a 30% sucrose solution in phosphate buffered saline for 24-48 h (until the brains sink to the bottom of the tube)
Section brains in half through the middle of the tumor region, place the cut side down into a freezing mold, embed into cryo-embedding media (OCT compound), and flash-freeze in a slurry of dry-ice ethanol, dry-ice isopentane or into liquid nitrogen. Keep frozen blocks at -80 °C until sectioning and staining.
4.2) Hematoxylin-Eosin Staining of Paraffin Embedded Brains for Histo-pathological Analysis of Intrinsic GBMs
Section paraffin embedded brains at 4 µm thickness, drop in a 42 °C water bath and mount onto glass slides.
De-paraffinize slides by placing them for 10 min in a 60 °C oven and then transferring them at 5 min intervals through a series of three slide containers filled with xylene.
Hydrate the slides through a series of ethanol baths: 100% ethanol for 2 min, repeat once, 95% ethanol for 2 min, repeat once, 70% ethanol for 2 min and then transfer slides to water.
Stain the slides by dipping them in a slide container filled with Harris Hematoxylin for 2 min.
Rinse in tap water until water is clear.
Dip slides twice into bluing solution (0.1% Sodium Bicarbonate).
Let slides stand in tap water for 2 min, then transfer to 80% ethanol for 2 min.
Stain the slides by dipping them in a container filled with Eosin for 5 min.
Dehydrate slides in a series of ethanol baths (80% ethanol 3-4 dips, 95% ethanol 2 min, repeat once, 100% ethanol 2 min, repeat once).
Clear with 3 consecutive baths of xylene, 3 min each.
Coverslip with a xylene-based mounting medium and let dry on a flat surface at room temperature until ready for imaging. Store at room temperature.
4.3) Immuno-histochemistry of Cryo-preserved Embedded Brains for Molecular and Cellular Characterization of De Novo Induced GBMs
Retrieve frozen blocks from the -80 °C storage, place into cryostat and allow temperatures to equilibrate for 30 min.
Section 12-14 µm thick sections and mount 2-3 sections onto positively charged glass slides. Cut sections serially, by collecting adjacent sections on different slides. This allows the staining with different antibodies on adjacent tissue sections within the tumor.
Dry slides in a desiccator for at least 1 hr after sectioning to allow for good adherence of the tissue.
Create a hydrophobic barrier (with a hydrophobic marker) at each end of the slide to allow for liquid to cover the sections and to stay on the slide during the washes. Cover the sections with a solution of Phosphate Buffered Saline with 0.1% Triton-X (PBS 0.1% Tx) and gently shake for 5 min on a horizontal shaker to permeabilize the tissue. Repeat twice.
Block nonspecific binding with a solution of 10% normal goat serum (or serum from the animal in which the secondary antibody was raised) for 30 min with gentle shaking
Prepare primary antibody solution in 2% normal goat serum in PBS 0.1% Tx and pipette the solution over the sections. Place slides in a covered humid chamber in the dark and keep at room temperature overnight.
The next day, wash sections three times for 5 min with PBS 0.1% Tx and incubate in fluorescent secondary antibody diluted in 2% normal goat serum PBS 0.1% Tx for one hour.
Wash sections three times for 5 min with PBS 0.1% Tx, counterstain with a nuclear stain (e.g., DAPI) for 2 min, wash once more, clear in water and coverslip with a water based mounting medium that will preserve the fluorescence. Place slides on a flat surface and let them cure for at least 24 hr, in the dark, until ready for imaging. Keep at 4 °C thereafter.
5. Generation of Primary Tumor Cell Lines with Specific Genetic Alterations.
To generate cell lines from Sleeping Beauty tumor-bearing mice select a mouse with confirmed large tumor (bioluminescence 107-108 photons/s/cm2/sr).
Euthanize the mouse with an overdose of isoflurane anesthetic, decapitate the animal and carefully dissect the brain from the skull (as described in section 4). Place the brain in a clean Petri dish.
Using a razor blade, make coronal cuts anterior and posterior to the tumor. The location of the tumor is usually recognizable due to the transparency of the brain.
Dissect the tumor, usually 5-7 mm in diameter with fine forceps and place into a 1.5 ml tube filled with 300 µl Neural Stem Cell Media (NSC Media: DMEM/F12 with B-27 supplement, N2 supplement, and Normocin, supplemented with human recombinant EGF and bFGF at a concentration of 20 ng/ml each).
Gently homogenize tumor with a plastic pestle, which fits to the walls of the tube.
Add 1 ml of enzyme-free tissue dissociation solution and incubate at 37 °C for 5 min.
Pass cell suspension through a 70 µm cell strainer, wash the strainer with 10 ml of NSC media, centrifuge at 300 x g for 4 min, decant supernatant and re-suspend pellet into 7 ml of NSC media.
Plate onto a T25 culture flask and culture at 37 °C in a tissue culture incubator with and atmosphere of 95% air and 5% CO2. After 3 days, some cells have died, some adhered to the bottom of the culture dish, and some are forming neurospheres, freely floating in the media.
Remove these suspended cells and re-plate onto a T75 tissue culture flask.
After another three days of culture, collect neurospheres, dissociate as described in step 5.6, strain through cell strainer and count cells. Freeze aliquots of cells in Fetal Bovine Serum 10% DMSO and passage cells at a density of one million cells in 14 ml of NSC supplemented with EGF and FGF for a T75 flask. The growth rate will depend on the oncogenes used to generate tumors. Adapt cell culture conditions to prevent overgrowth and maintain cells at relatively high density.
Representative Results
To characterize histo-pathological features of SB-induced glioblastomas, C57/ BL6 neonatal mice were injected at P1 with a plasmid encoding luciferase (pT2/SB100x-Luc) in combination with plasmids encoding transposons with oncogenic DNA, i.e., NRAS (pT/CAGGS-NRASV12) and SV40 LgT (pT/CMVSV40-LgT) (Figure 3c) or a plasmid encoding a short hairpin p53 with PDGFβ and a GFP reporter (pT2shp53/GFP4/mPDGFβ) in combination with NRAS (Figure 3d). Animals were monitored for bioluminescence the day after injection (Figure 2a) and periodically until reaching the moribund stage when they were euthanized. Moribund stage is defined as the clinical stage when the animal becomes symptomatic showing impaired mobility, hunched posture, scruffy fur and weight loss. Sometimes, animals develop seizures or abnormal patterns of movement, like walking in circles and sudden jumping. At the end of the experiment animals were anesthetized. The brains were perfused, embedded in paraffin, and processed for hematoxylin and eosin staining. Data show tumors displaying the hallmarks of human GBM (WHO grade IV) with hemorrhages (Figure 3c), pseudo-pallisading necrosis (Figure 3e) and perivascular and diffuse invasion into the brain parenchyma (Figure 3g, f). The formation of pseudopallisades is preceded by the rupture of large glomeruloid vessels with leaky endothelia (Figure 3i) which result in regions of hemorrhage with massive infiltration of mononuclear cells(Figure 3h). Atypical mitoses (Figure 3j) and gigantic multinucleated tumor cells (Figure 3k) are also pathognomonic of human GBM.
Glioblastomas generated using the Sleeping Beauty transposase system can be monitored throughout the progression of tumor growth with bioluminescence, if the plasmids injected encode luciferase. An example experiment is shown in Figure 4a. Animals usually succumb of tumor burden when luminescence reaches an intensity of 107-109 photons/s/cm2/sr. The median survival of animals is predictably dependent on the combinations of oncogenic transposons injected into the neonatal brain, as illustrated in the survival curves presented in Figure 4b. Note that the most aggressive tumors are induced with NRAS and SV40 LgT antigen (median survival of 30 days), whereas the median survival of animals with GBM induced with shp53 NRAS and PDGF is 62.5 days and of animals injected with shp53 and NRAS 83 days.
De novo formed tumors can be characterized immuno-histochemically by their expression of molecules characteristic of glial tumors. Figure 5 shows a nascent tumor (22 dpi, bioluminescence 2x105 photons/s/cm2/sr), induced with shp53 and NRAS. The injected shp53 plasmid encodes for green fluorescent protein (GFP) to allow the identification of transfected cells and their progeny (pT2/shp53/GFP). GFP+ tumor cells express the neural stem cell marker nestin and some GFP+ cells also express glial fibrillary acidic protein (GFAP). Figure 6 illustrates a tumor from a moribund animal at 56 dpi, tumor which was also induced with shp53/GFP and NRAS. Numerous GFAP+ astrocytes are surrounding the tumor. GFP+ tumor cells express nestin, but not GFAP.
A great advantage when using this technique to induce GBMs is the ability to generate novel GBM cell lines with unique genetic alterations by means of the transposons injected. In addition, custom cell lines can be generated using transgenic animals with specific genetic makeup. These cell lines are instrumental in asking many mechanistic questions using biochemical assays. They are ideally suited for cytotoxicity studies with novel chemotherapeutic agents. Figure 7 illustrates a neurosphere from a Sleeping Beauty tumor induced with shp53, PDGF and NRAS, showing expression of GFP, which is encoded on one of the injected plasmids. Note that expression is not equally intense in all the cells, indicating the heterogeneous nature of these primary GBM cells.
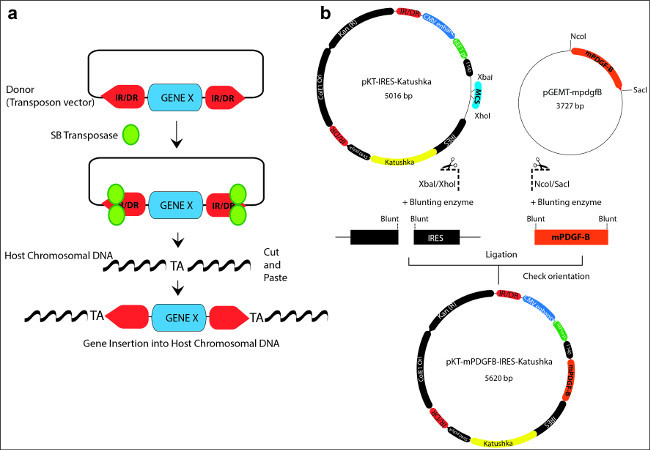 Figure 1: (a) Schematic representation illustrating the “cut and paste” mechanism used by the Sleeping Beauty transposase to integrate transposons into the host chromosomal DNA. A donor transposon plasmid is depicted with the gene of interest flanked by the inverted repeats/direct repeats (IR/DR; red arrow boxes) sequences. The SB transposase (green) binds to the IR/DR, excises the transposon and reintegrates it in between random TA dinucleotide base pairs on the host chromosomal DNA. (b) Example of cloning a gene of interest (mPDGFβ) into the backbone of a SB vector (pKT-IRES-Katushka). The SB vector contains two IR/DR repeat sequences (red) and a gene expression cassette which includes promoter and enhancer sequences, a multiple cloning site (MCS; blue), internal ribosomal entry sites (IRES), a fluorescent reporter marker (Katushka: yellow), and a polyadenilation site (polyA). The oncogene of interest (mPDGFβ; orange) is contained in a cloning vector pGEMT. The oncogene, flanked by specific restriction sites (NcoI/SacI) is released by enzymatic digestion, and blunted by the blunting enzyme. The vector is linearized and blunted as well. Finally, the mPDGFβ oncogene is inserted into the donor vector by means of a blunt-end ligation reaction to generate the new plasmid transposon vector: pKT-mPDGFβ-IRES-Katushka. Please click here to view a larger version of this figure.
Figure 1: (a) Schematic representation illustrating the “cut and paste” mechanism used by the Sleeping Beauty transposase to integrate transposons into the host chromosomal DNA. A donor transposon plasmid is depicted with the gene of interest flanked by the inverted repeats/direct repeats (IR/DR; red arrow boxes) sequences. The SB transposase (green) binds to the IR/DR, excises the transposon and reintegrates it in between random TA dinucleotide base pairs on the host chromosomal DNA. (b) Example of cloning a gene of interest (mPDGFβ) into the backbone of a SB vector (pKT-IRES-Katushka). The SB vector contains two IR/DR repeat sequences (red) and a gene expression cassette which includes promoter and enhancer sequences, a multiple cloning site (MCS; blue), internal ribosomal entry sites (IRES), a fluorescent reporter marker (Katushka: yellow), and a polyadenilation site (polyA). The oncogene of interest (mPDGFβ; orange) is contained in a cloning vector pGEMT. The oncogene, flanked by specific restriction sites (NcoI/SacI) is released by enzymatic digestion, and blunted by the blunting enzyme. The vector is linearized and blunted as well. Finally, the mPDGFβ oncogene is inserted into the donor vector by means of a blunt-end ligation reaction to generate the new plasmid transposon vector: pKT-mPDGFβ-IRES-Katushka. Please click here to view a larger version of this figure.
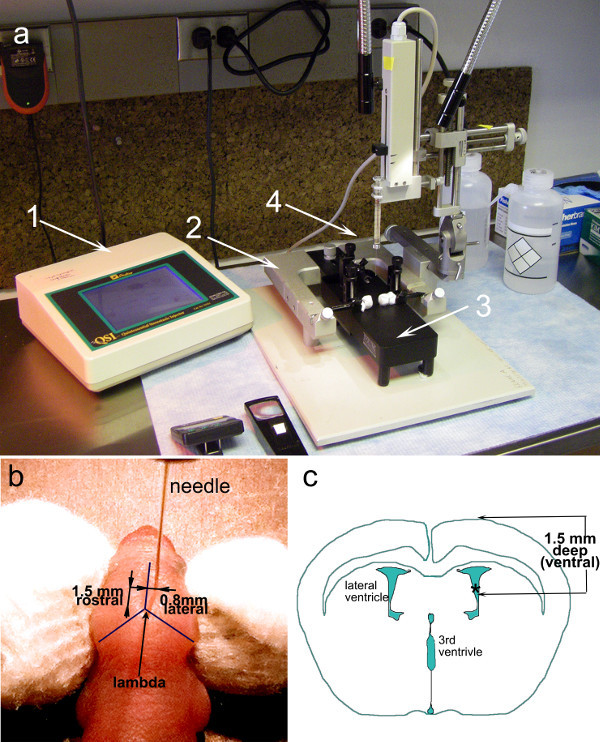 Figure 2: Experimental Setup and guides for intra ventricular injections in neonatal mice (a) A stereotaxic frame (2) with micrometer dials is fitted with an automatic injector holding a 10 µl syringe (4). Inside the U frame, a neonatal adaptor frame is securely fastened (3). The control panel of the automatic injector (1) allows for precise selection of syringe, volume and rate of flow. (b) Photograph of a neonatal mouse (P1) with a needle inserted at the coordinates required for injections into the lateral ventricle: 1.5 mm ventral and 0.8 mm lateral to the lambda. (c) Illustration of a coronal section through the brain of a neonatal mouse (P1) highlighting the relative dimensions and position of the ventricles.
Figure 2: Experimental Setup and guides for intra ventricular injections in neonatal mice (a) A stereotaxic frame (2) with micrometer dials is fitted with an automatic injector holding a 10 µl syringe (4). Inside the U frame, a neonatal adaptor frame is securely fastened (3). The control panel of the automatic injector (1) allows for precise selection of syringe, volume and rate of flow. (b) Photograph of a neonatal mouse (P1) with a needle inserted at the coordinates required for injections into the lateral ventricle: 1.5 mm ventral and 0.8 mm lateral to the lambda. (c) Illustration of a coronal section through the brain of a neonatal mouse (P1) highlighting the relative dimensions and position of the ventricles.
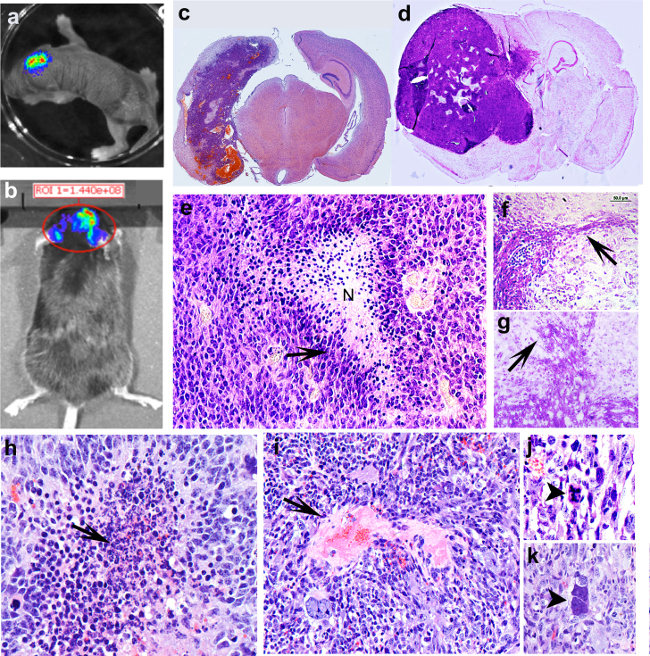 Figure 3: Tumors induced with the Sleeping Beauty transposon system show the histological hallmarks of human GBM (a) Bioluminescence image of a neonatal pup 24 hr after intraventricular injection with plasmids encoding NRAS, SV40 LgT. (b) Bioluminescence image of an adult animal (50 dpi) with a large tumor induced with shp53 NRAS and PDGF. (c) Hematoxilin and eosin stain of a coronal section from a brain of a moribund mouse with a tumor induced with NRAS and LgT (28 dpi). (d) Coronal section from a brain of a moribund mouse with a tumor induces with shp53 NRAS and PDGF. (e) Pseudopalisading necrosis, histological hallmark of human GBM is observed in de novo generated tumors. Arrow points to cells arranged in palisades migrating away from the central area of necrosis (N) (f) and (g) de novo generated tumors are highly invasive, showing invasion along blood vessels (g) and diffuse (f) into the normal brain parenchyma. (h) Region of hemorrhage with massive invasion of mononuclear cells (arrow), the initial stage of an area of pseudopalisading necrosis. (i) Large glomeruloid vessel with leaky endothelium (arrow) at the origin of diffusing hemorrhage inside a tumor induced with NRAS and SV40 –LgT. (j) Atypical mitosis in a tumor cells (arrowhead). (k) Gigantic tumor cell with multiple large nuclei (arrowhead).
Figure 3: Tumors induced with the Sleeping Beauty transposon system show the histological hallmarks of human GBM (a) Bioluminescence image of a neonatal pup 24 hr after intraventricular injection with plasmids encoding NRAS, SV40 LgT. (b) Bioluminescence image of an adult animal (50 dpi) with a large tumor induced with shp53 NRAS and PDGF. (c) Hematoxilin and eosin stain of a coronal section from a brain of a moribund mouse with a tumor induced with NRAS and LgT (28 dpi). (d) Coronal section from a brain of a moribund mouse with a tumor induces with shp53 NRAS and PDGF. (e) Pseudopalisading necrosis, histological hallmark of human GBM is observed in de novo generated tumors. Arrow points to cells arranged in palisades migrating away from the central area of necrosis (N) (f) and (g) de novo generated tumors are highly invasive, showing invasion along blood vessels (g) and diffuse (f) into the normal brain parenchyma. (h) Region of hemorrhage with massive invasion of mononuclear cells (arrow), the initial stage of an area of pseudopalisading necrosis. (i) Large glomeruloid vessel with leaky endothelium (arrow) at the origin of diffusing hemorrhage inside a tumor induced with NRAS and SV40 –LgT. (j) Atypical mitosis in a tumor cells (arrowhead). (k) Gigantic tumor cell with multiple large nuclei (arrowhead).
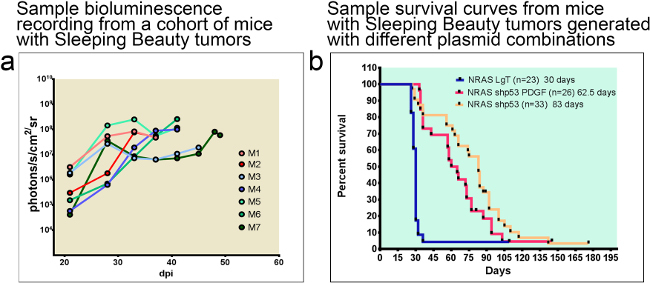 Figure 4: Tumor-bearing animals can be monitored with bioluminescence throughout the duration of disease progression. The median survival of animals is predicted by the combination of oncogenic DNA injected. (a) Example of bioluminescent traces from a cohort of 7 C57/BL6 mice. Note that mice succumb about a week after bioluminescence reaches 108 photons/s/cm2/sr. (b) Sample survival curves comparing median survival of animals with Sleeping Beauty tumors generated with different plasmid combinations. Median survival of tumors induced with NRAS and SV40 LgT is 30 dpi whereas of animals with tumors induced with shp53 NRAS and PDGF or shp53 and NRAS is 62.5 dpi or 83 dpi respectively. dpi: days post injection.
Figure 4: Tumor-bearing animals can be monitored with bioluminescence throughout the duration of disease progression. The median survival of animals is predicted by the combination of oncogenic DNA injected. (a) Example of bioluminescent traces from a cohort of 7 C57/BL6 mice. Note that mice succumb about a week after bioluminescence reaches 108 photons/s/cm2/sr. (b) Sample survival curves comparing median survival of animals with Sleeping Beauty tumors generated with different plasmid combinations. Median survival of tumors induced with NRAS and SV40 LgT is 30 dpi whereas of animals with tumors induced with shp53 NRAS and PDGF or shp53 and NRAS is 62.5 dpi or 83 dpi respectively. dpi: days post injection.
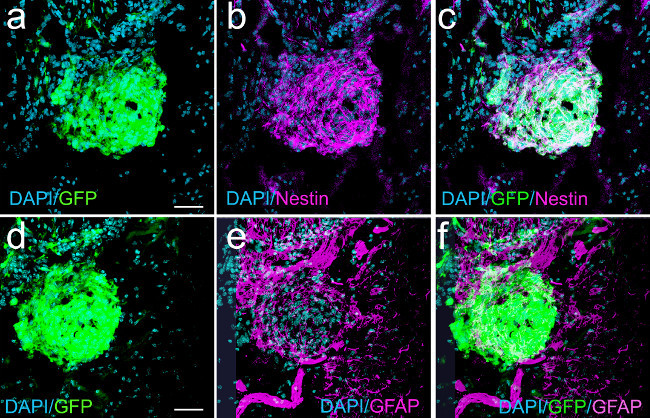 Figure 5: Nascent macroscopic GBM (22 dpi) induced with shp53 and NRAS show expression of nestin and GFAP in GFP+ tumor cells (confocal micrographs). Panel (a) shows a nascent tumor expressing GFP. Panel (b) represents the same field showing nestin expression. Panel (c) is an overlay of (a) and (b) illustrating co-localization of nestin and GFP. (d) GFP expression in tumor cells. (e) GFAP expression in some tumor cells and in cells surrounding the nascent tumor. (f) Overlay projection of (d) and (e) illustrating some tumor cells (white) co-expressing GFAP and GFP. Scale bars in (a) and (d) represent 75 µm.
Figure 5: Nascent macroscopic GBM (22 dpi) induced with shp53 and NRAS show expression of nestin and GFAP in GFP+ tumor cells (confocal micrographs). Panel (a) shows a nascent tumor expressing GFP. Panel (b) represents the same field showing nestin expression. Panel (c) is an overlay of (a) and (b) illustrating co-localization of nestin and GFP. (d) GFP expression in tumor cells. (e) GFAP expression in some tumor cells and in cells surrounding the nascent tumor. (f) Overlay projection of (d) and (e) illustrating some tumor cells (white) co-expressing GFAP and GFP. Scale bars in (a) and (d) represent 75 µm.
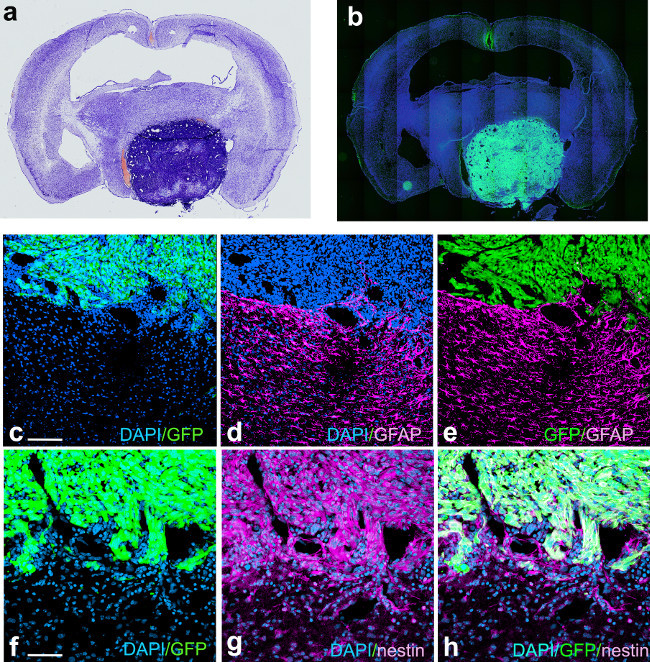 Figure 6: GBM induced with shp53, NRAS and PDGF from a moribund animal (56 dpi) showing GFP and nestin expression in tumor cells and abundant staining for the mature glial marker GFAP surrounding the tumor. Panel (a) represents Nissl stain of acoronal section through the brain of a moribund animal with a tumor (intense blue staining from increased cellularity) induced with the Sleeping Beauty transposon system using shp53, PDGFΒ and NRAS. Panel (b), a coronal adjacent section to the one illustrated in panel (a) stained with the nuclear stain DAPI, showing expression of GFP in tumor cells. Panel (c) represents a confocal micrograph of the tumor border showing GFP expression in the tumor, identified by the high nuclear density with DAPI. Panel (d) illustrates the same field of view as in (a), showing intense GFAP expression in cells adjacent to the tumor. Panel (e) represents the overlay of panels (c) and (d). Panel (f) is a confocal micrograph of the tumor border, showing GFP expression in tumor cells. Panel (g) represents the same field of view as in (f), showing expression of nestin in tumor cells. Panel (h) is the overlay projection of panels (f) and (g). Scale bar in (c) represents 150 µm and in (f) represents 75 µm.
Figure 6: GBM induced with shp53, NRAS and PDGF from a moribund animal (56 dpi) showing GFP and nestin expression in tumor cells and abundant staining for the mature glial marker GFAP surrounding the tumor. Panel (a) represents Nissl stain of acoronal section through the brain of a moribund animal with a tumor (intense blue staining from increased cellularity) induced with the Sleeping Beauty transposon system using shp53, PDGFΒ and NRAS. Panel (b), a coronal adjacent section to the one illustrated in panel (a) stained with the nuclear stain DAPI, showing expression of GFP in tumor cells. Panel (c) represents a confocal micrograph of the tumor border showing GFP expression in the tumor, identified by the high nuclear density with DAPI. Panel (d) illustrates the same field of view as in (a), showing intense GFAP expression in cells adjacent to the tumor. Panel (e) represents the overlay of panels (c) and (d). Panel (f) is a confocal micrograph of the tumor border, showing GFP expression in tumor cells. Panel (g) represents the same field of view as in (f), showing expression of nestin in tumor cells. Panel (h) is the overlay projection of panels (f) and (g). Scale bar in (c) represents 150 µm and in (f) represents 75 µm.
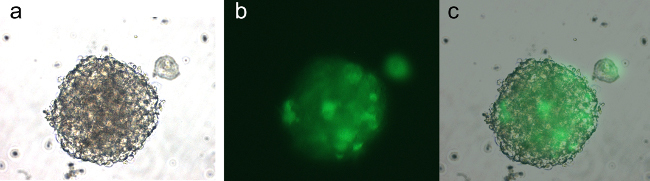 Figure 7: Neurospheres from a Sleeping Beauty tumor induced with shp53, PDGF and NRAS after 5 days in culture, passage 2. The cells are expressing GFP encoded on the shp53 PDGF plasmid (pT2shp53/GFP4/mPDGFβ). Panel (a) is a brightfield micrograph of a neurosphere. Panel (b) represents an epi-fluorescent micrograph of the same view as in (a) showing expression of GFP in the neurosphere cells. Panel (c) represents the overlay of panels (a) and (b).
Figure 7: Neurospheres from a Sleeping Beauty tumor induced with shp53, PDGF and NRAS after 5 days in culture, passage 2. The cells are expressing GFP encoded on the shp53 PDGF plasmid (pT2shp53/GFP4/mPDGFβ). Panel (a) is a brightfield micrograph of a neurosphere. Panel (b) represents an epi-fluorescent micrograph of the same view as in (a) showing expression of GFP in the neurosphere cells. Panel (c) represents the overlay of panels (a) and (b).
Discussion
In this article, we detail a versatile and reproducible method for generating new models of GBM using SB transposase- mediated integration of oncogenic plasmid DNA into cells surrounding the subventricular zone of neonatal mice. We present a protocol to generate transposon plasmids with new genes of interest, illustrate how to monitor the progression of the tumors in live animals, and how to characterize histo-pathological and immuno-histochemical features of these tumors.
As our lab(Figure 3) and others13 have shown, this model reliably creates tumors with the salient characteristics of human GBM including (1) pseudo-palisading necrosis, (2) vascular proliferation, (3) nuclear atypia, (4) abnormal mitoses and (5) perivascular and diffuse invasion. The use of neonatal mice is optimal from a logistical standpoint, providing a relatively easy technical procedure with minimal equipment and sedation/recovery time, allowing for rapid generation of large sample sizes of tumors with specific genetic alterations. These tumors cause animals to succumb to tumor burden with a predictable median survival dependent on the genetic alterations induced. Finally, the SB model has been used as a therapeutic intervention screen for treatment response24.
There are several important factors to consider when preparing the solutions used for the neonatal injections. DNA solutions need to be sterile, endotoxin free and concentrated (2-7 µg/µl) to allow for minimal volume of injections. The optimal ratio of nitrogen residues in the polyethyleneimine (PEI) to the phosphate residues in the DNA (N/P ratio) is 7. This ensures the formation of positively charged particle complexes which will bind anionic moieties on cell surfaces and will be endocytosed. Once in the cytoplasm, osmotic influx will cause the particles to burst and release the encapsulated plasmid DNA. The in vivo jet-PEI solution has a concentration of 150 mM expressed as nitrogen residues. Given that one µg DNA has 3 nmol of anionic phosphate, the amount of transfection reagent necessary can be calculated with the formula:
µl of in vivo jet-PEI = [(µg DNA x 3) x N/P ratio]/150.
For example, to prepare 40 µl of 0.5 µg/µl DNA solution with an N/P ratio of 7, 20 µg of DNA are needed. The volume of PEI required will be 20*3*7/150=2.8 µl.
Up to five different plasmids can be injected simultaneously. Experiments presented herein deliver the Sleeping Beauty transposase in trans on a plasmid that also encodes luciferase (SBLuc). As mentioned in the introduction, the ratio of transposase molecules to transposons is important to ensure optimal transposition. The optimal ratio (empirically established) of the SB100x plasmid to the transposon DNA plasmid is 1:4. Hence, if one uses two different transposon plasmids, T1 and T2 for example, the ratio of SBLuc:T1:T2 will be 1:2:2; or for a total of 20 µg DNA in a 40 µl solution 4 µg of SBLuc will be mixed with 8 µg of T1 and 8 µg of T2. For three different transposon plasmids the ratio will be SBLuc:T1:T2:T3 = 1:1.33:1.33:1.33 and for four transposons: 1:1:1:1:1.
It is useful to verify the uptake of the plasmid DNA by bioluminescence 24-72 hr after injection. As the animals grow, the increased optical density of the fur, skin, skull and brain, prevents monitoring of the tumor progression until the tumor has reached a threshold size, roughly 1-2 mm3 for darkly pigmented C57BL6 mice. Better transmission of the light is possible in these mice if their fur is shaved, or when using mice with white or no fur.
Several limitations need to be considered when using this model. First, the genetic material introduced with the injected plasmids can be integrated randomly in the host genome in different locations and number of copies. This may lead to variability between tumor cells and between tumors. Second, the DNA introduced is inserted primarily into intronic TA repeat sequences DNA25, however, the possibility of interference with coding host genomic DNA remains. Third, intraventricular injections may cause a small but persistent rate of hydrocephalus in young mice, which becomes clinically visible and symptomatic in the 3rd-4th week of life. Some strains of mice are more prone to hydrocephalus than others (i.e., C57/BL6 more than FVB). To minimize the risk of inducing hydrocephalus, it is recommended to inject as small a volume as possible (less than 1 µl in C57/BL6 mice), to ensure that the needles are sharp, the solutions have low salinity and to provide growing pups with enriched food, in order to allow for timely ossification of the skull bones. Finally, late-stage tumors often develop necrosis, which has to be considered when monitoring mice by bioluminescence. A lower reading may indicate an advanced tumor with large areas of necrosis and not necessarily a resolution of the tumor as a consequence of treatment. Ultimately, histological analysis remains the gold standard to assess tumor progression.
The advent of tumor whole genome sequencing in recent years has opened the door to exploration of the role of recurrently mutated genes in GBM. The Sleeping Beauty model is optimal for validating potential oncogenes or tumor suppressors. As in the example given in this paper with PDGFβ ligand, cloning the mouse cDNA sequence of a candidate oncogene into an established SB plasmid backbone will lead to constitutive expression within transfected cells. Evaluation of the role of tumor suppressors can be effectively performed by cloning into the backbone of the SB plasmid with candidate sequences, (available for example in the RNAi codex database26), to create robust expression of second-generation miR short-hairpins.
A current debate in the field of GBM research is related to the identity of the cell-of- origin. It has recently been shown that in mice, tumors induced by down-regulating p53 and Nf1 in neural stem cells, originate from oligodendrocyte precursor cells27. Using the Sleeping Beauty transposon model, similar studies can be designed to test whether other genetic alterations preferentially target other populations of stem cells/precursor cells to generate GBMs. Knowledge from such studies will greatly enhance our understanding of GBM etiology and provide avenues for novel therapeutic approaches.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Dr. John Ohlfest and Dr. Stacey Decker13 for the generous gift of plasmids and for the training provided to master this method. We thank Marta Dzaman for help with standardizing the Hematoxylin-Eosin staining procedure of paraffin-embedded sections. We also thank Molly Dahlgren for perusing this manuscript and providing helpful suggestions. This work is supported by NIH/NINDS grants to MGC and PRL, Leah’s Happy Hearts Foundation grant to MCG and PRL. Alex’s Lemonade Foundation young Investigator Award and the St. Baldrick’s Foundation Fellowship to CK.
References
- Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma--are we there yet. Neuro Oncol. 2013;15(1):4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Holland EC. Glioma models. Biochim Biophys Acta. 2001;1551(1):M19–M27. doi: 10.1016/s0304-419x(01)00027-0. [DOI] [PubMed] [Google Scholar]
- Houchens DP, Ovejera AA, Riblet SM, Slagel DE. Human brain tumor xenografts in nude mice as a chemotherapy model. Eur J Cancer Clin Oncol. 1983;19(6):799–805. doi: 10.1016/0277-5379(83)90012-3. [DOI] [PubMed] [Google Scholar]
- Holland EC. Glioblastoma multiforme: the terminator. Proc Natl Acad Sci U S A. 2000;97(12):6242–6244. doi: 10.1073/pnas.97.12.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candolfi M, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007;85(2):133–148. doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Parada LF, Holland EC, Charest A. Genetic modeling of gliomas in mice: new tools to tackle old problems. Glia. 2011;59(8):1155–1168. doi: 10.1002/glia.21142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojiani AM, Dorovini-Zis K. Glomeruloid vascular structures in glioblastoma multiforme: an immunohistochemical and ultrastructural study. J Neurosurg. 1996;85(6):1078–1084. doi: 10.3171/jns.1996.85.6.1078. [DOI] [PubMed] [Google Scholar]
- Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Translational Oncology. 2009;2(2):89–95. doi: 10.1593/tlo.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann-Morvinski D, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338(6110):1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes J, et al. Lentiviral-Induced High-Grade Gliomas in Rats: The Effects of PDGFB, HRAS-G12V, AKT, and IDH1-R132H. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutic. 2014. [DOI] [PMC free article] [PubMed]
- Uhrbom L, Hesselager G, Nister M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58(23):5275–5279. [PubMed] [Google Scholar]
- Marumoto T, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15(1):110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner SM, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009;69(2):431–439. doi: 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey OT. Genesis of the Percival Bailey-Cushing classification of gliomas. Pediatr Neurosci. 1985;12(4-5):261–265. doi: 10.1159/000120262. [DOI] [PubMed] [Google Scholar]
- Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnihotri S, et al. Glioblastoma, a brief review of history, molecular genetics, animal models and novel therapeutic strategies. Arch Immunol Ther Exp (Warsz) 2013;61(1):25–41. doi: 10.1007/s00005-012-0203-0. [DOI] [PubMed] [Google Scholar]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Izsvak Z, Minter A, Hackett PB. Identification of functional domains and evolution of Tc1-like transposable elements. Proc Natl Acad Sci U S A. 1996;93(10):5008–5013. doi: 10.1073/pnas.93.10.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91(4):501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- Hackett PB, Ekker SC, Largaespada DA, McIvor RS. Sleeping beauty transposon-mediated gene therapy for prolonged expression. Adv Genet. 2005;54:189–232. doi: 10.1016/S0065-2660(05)54009-4. [DOI] [PubMed] [Google Scholar]
- Ammar I, Izsvak Z, Ivics Z. The Sleeping Beauty transposon toolbox. Methods Mol Biol. 2012;859:229–240. doi: 10.1007/978-1-61779-603-6_13. [DOI] [PubMed] [Google Scholar]
- Mates L, et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet. 2009;41(6):753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- Koso H, et al. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc Natl Acad Sci U S A. 2012;109(44):E2998–E3007. doi: 10.1073/pnas.1215899109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Research. 2011;71(7):2664–2674. doi: 10.1158/0008-5472.CAN-10-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts AM, et al. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Molecular Therapy: The Journal Of The American Society Of Gene Therapy. 2003;8(1):108–117. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Dickins RA, et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37(11):1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- Liu C, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146(2):209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]