

Abstract
Post-mortem studies of neurological diseases are not ideal for identifying the underlying causes of disease initiation, as many diseases include a long period of disease progression prior to the onset of symptoms. Because fibroblasts from patients and healthy controls can be efficiently reprogrammed into human induced pluripotent stem cells (hiPSCs), and subsequently differentiated into neural progenitor cells (NPCs) and neurons for the study of these diseases, it is now possible to recapitulate the developmental events that occurred prior to symptom onset in patients. We present a method by which to efficiently differentiate hiPSCs into NPCs, which in addition to being capable of further differentiation into functional neurons, can also be robustly passaged, freeze-thawed or transitioned to grow as neurospheres, enabling rapid genetic screening to identify the molecular factors that impact cellular phenotypes including replication, migration, oxidative stress and/or apoptosis. Patient derived hiPSC NPCs are a unique platform, ideally suited for the empirical testing of the cellular or molecular consequences of manipulating gene expression.
Keywords: Medicine, Issue 96, Induced pluripotent stem cells, neural differentiation, neural progenitor cells, psychiatric disease, lentiviral transduction, neurosphere migration assay
Introduction
Gene expression studies of neurons differentiated in vitro from human induced pluripotent stem cells (hiPSCs) by us 1 and others 2,3 indicate that hiPSC neurons resemble fetal rather than adult brain tissue. At present, hiPSC-based models may be more appropriate for the study of predisposition to, rather than late features of, neurological disease. We have previously reported that a significant fraction of the gene signature of schizophrenia hiPSC-derived neurons is conserved in schizophrenia hiPSC-derived neural progenitor cells (NPCs), indicating that NPCs may be a useful cell type for studying the molecular pathways contributing to schizophrenia 1. We and others have reported aberrant migration, increased oxidative stress and reactive oxygen species, sensitivity to sub-threshold environmental stresses and impaired mitochondrial function in schizophrenia hiPSC NPCs 1,4-6, as well as decreased neuronal connectivity and synaptic function in schizophrenia hiPSC neurons 5,7-10. If the molecular factors contributing to aberrant migration and/or oxidative stress in schizophrenia hiPSC NPCs also underlie the reduced neuronal connectivity in schizophrenia hiPSC-derived neurons, NPCs could be a robust and highly replicative neural population with which to study the mechanisms responsible for disease. Furthermore, because one can rapidly generate large numbers of cells and need not wait weeks or months for neuronal maturation, NPC-based assays are suitable for the study of larger patient cohorts and are more amenable to high throughput screening. We believe that hiPSC NPCs can serve as a proxy for the developmental pathways potentially contributing to disease pathogenesis, as has already been demonstrated in disorders as diverse as schizophrenia 1 and Huntington’s disease 11.
To differentiate NPCs from hiPSCs, initial neural induction is accomplished by dual-SMAD inhibition (0.1μM LDN193189 and 10μM SB431542) 12. By antagonizing BMP and TGFβ signaling with these small molecules, endoderm and mesoderm specification is blocked, accelerating neuronal differentiation and leading to the formation of visible neural rosettes within one week of plating. Neural patterning occurs early in this process, presumably during the period of neural rosette formation and immediately thereafter. In the absence of other cues, these primitive neural cells assume an anterior forebrain-like fate 13. Immediately following neural rosette formation, and ongoing throughout NPC expansion, forebrain NPCs are cultured with FGF2 8,14. They have dual lineage potential and can be differentiated to neural populations of 70-80% βIII-TUBULIN-positive neurons and 20-30% glial fibrillary acidic protein (GFAP)-positive astrocytes (Figure 1). The majority of forebrain hiPSC neurons are VGLUT1-positive, and so are presumably glutamatergic, although approximately 30% of neurons are GAD67-positive (GABAergic) 8.
NPCs are routinely passaged more than ten times in vitro, while maintaining consistent differentiation profiles, and without accumulating karyotype abnormalities. Groups have reported passaging NPCs more than 40 times 15, however, we find that beyond ten passages, NPCs show increased propensity for astrocyte differentiation. NPCs well-tolerate multiple freeze-thaws and can be transitioned to grow as neurospheres by simply culturing in non-adherent plates. NPCs are efficiently transduced by viral vectors, enabling rapid evaluation of the molecular and cellular consequences of genetic perturbation, and easily expandable to yield sufficient material for biochemical studies. Furthermore, because viral vectors permit robust over-expression and/or knockdown of disease-relevant genes, in either control or patient derived neural cells, one can use this platform to test the effect of genetic background on these manipulations. Though not suitable for synaptic or activity-based assays requiring mature neurons, NPCs may be a practical alternative for many straightforward molecular or biochemical analyses of patient-derived neural cells.
Protocol
1. hiPSC Differentiation to Neural Progenitor Cells
Grow and expand hiPSCs in human embryonic stem cell (HES) media (Table 1) co-cultured on a mouse embryonic fibroblast (mEF) feeder layer until large (but subconfluent) colonies are ready for neural differentiation via an embryoid body (EB) intermediate (Figure 2). Routine hiPSC culture conditions are well described elsewhere 16,17; briefly, grow hiPSCs in HES media on a mEF feeder layer until confluent, then enzymatically passage with collagenase (1mg/ml in DMEM) and expand approximately 1:3 every 7 days.
To prepare Poly-L-Ornithine/laminin coated plates, coat plates with 10 μg/ml Poly-L-Ornithine in sterile water for plastic surfaces (50 μg/ml for glass surfaces) and incubate at RT for 3-24 hr. Wash at least once with sterile water and then coat with 5μg/ml Laminin in sterile PBS at RT for 3-24 hr. NOTE: If wrapped in plastic, plates can be stored for up to six months at -20 ºC.
On Day 1, enzymatically lift subconfluent hiPSCs (generally grown on mEFs, or hiPSCs grown in defined media such as TeSR 18 on Matrigel) from the plate as large colonies using 1mg/ml Collagenase IV in DMEM/F12. NOTE: Following 1-2 hr of incubation at 37 ºC, colonies will be floating in the dish.
Gently wash hiPSC colonies (by settling, not centrifugation) 1-2x with DMEM/F12, resuspend in 2ml/well of N2/B27 media (Table 1) and transfer to non-adherent 6-well dishes (combine 3-wells into 1-well) 19. NOTE: Overnight, colonies will form floating spherical clusters termed “embryoid bodies” (EBs). Expect substantial cellular death.
On Day 2, tilt plates and allow EBs to settle. Carefully remove media and wash once with DMEM/F12 to remove debris. Feed with N2/B27 media supplemented with dual SMAD inhibitors (0.1 μM LDN193189 and 10 μM SB431542) 12.
On Days 3-7, neuralization will occur in the context of dual SMAD inhibition; check that EBs are round but not cystic. Feed EBs every second day with N2/B27 media supplemented with 0.1 μM LDN193189 and 10 μM SB431542.
On Days 7-14, following plating of EBs to Poly-L-Ornithine/Laminin coated plates, check using a brightfield microscope that neural rosettes begin to appear within a few days (characterized as round clusters of neuroepithelial cells with apico-basal polarity; Figure 1). Continue to feed adherent EBs every second day with N2/B27 media supplemented with 0.1 μM LDN193189, 10 μM SB431542 and 1 μg/ml laminin.
2. Harvest of Neural Rosettes
NOTE: We recommend that neural rosettes be enzymatically harvested using Neural Rosette Selection Reagent 20 or similar selection reagent. Though neural rosettes can be manually picked into 6-well Poly-L-Ornithine/Laminin coated plate, this methodology takes extensive training to master, and, dependent on user skill, may require a second round of picking at day 20 to further enrich for NPCs and deplete non-neural cell types.
For enzymatic selection of neural rosettes at day 14, aspirate media from adhered EBs and add 1 ml of neural rosette selection reagent per well for a 6-well plate. Incubate at 37 ºC for 1 hr.
With a P1000 pipetman, gently remove enzyme from each well. Add 1 ml of DMEM/F12 per well to wash.
With a P1000, collect the 1 ml of DMEM/F12 and quickly expel the DMEM/F12 back into the well, thus detaching the rosettes from the plate. Collect the rosettes into a falcon tube.
Pipet another 1 ml of DMEM/F12 and quickly expel into the same well to detach remaining rosettes. (Do not triturate. Try not to break up rosette aggregates). Collect the neural rosettes into same falcon tube.
Repeat step 2.4 as necessary. If rosettes do not detach readily, add more enzyme and repeat the process (steps 2.1-2.4)
Spin cells at 300 x g for 3 min. Aspirate the wash, and re-suspend rosettes in 2 ml of NPC media (Table 1). Transfer cells (1:1) into a Poly-L-Ornithine/Laminin coated 6 well plate.
From Days 15-21, the neural rosettes will expand; feed rosettes every second day with NPC media.
Evaluate the quality of the neural rosettes and ensure that flat fibroblast-like cells are not expanding within the culture. NOTE: If non-rosettes persist, the NPC culture can sometimes be salvaged by manual picking, selecting only the best of the picked rosettes into Accutase. Dissociate 15 min at 37 ºC. Pellet, wash and plate in NPC media onto 24-well Poly-L-Ornithine/Laminin coated plate.
3. Expansion of Neural Progenitor Cells
NOTE: hiPSC NPCs can be grown on either Matrigel- or Poly-L-Ornithine/Laminin coated plates. We typically use Matrigel-plates as they can be prepared more quickly and at lower cost.
To prepare Matrigel coated plates, quickly thaw and resuspend a 1mg frozen aliquot of Matrigel in 24 ml cold DMEM/F12 and immediately distribute 2 ml to each well of two six-well plates. Incubate for at least 1 hr at 37 ºC. NOTE: Matrigel needs only to be aspirated, not washed, immediately prior to use.
Feed NPCs every second day and maintain at very high density or they will spontaneously differentiate to neurons. NOTE: Though more established NPCs are typically split 1:4 every week, very low passage NPCs may be split 1:2 as infrequently as once every two weeks.
To split, first aspirate media and add 1 ml warm Accutase (1X) per well of 6-well plate. Incubate at 37 ºC for 10-15 min.
Gently transfer detached cells into a 15 ml tube containing DMEM/F12 with as little mechanical stress as possible. Do not titrate cells while in enzyme. Pellet cells by spinning at 1,000 x g for 5 min.
Aspirate the wash, and re-suspend NPCs in ~1 ml NPC media per original well of 6-well. NOTE: Expect that a confluent well of a 6-well dish has 5-10 million cells; this can be confirmed by counting a 10μl aliquot of cells using a hematocytometer prior to re-plating.
Following re-suspension, re-plate NPCs as NPCs, neurons or neurospheres. To maintain NPCs, plate approximately 1-2 million cells per well of a 6-well plate. The efficiency of neurosphere formation can vary between experiments and cell lines, but generally occurs best if between 200,000 and 1,000,000 cells are seeded per well of a non-adherent 6-well plate; if clumping of the neurospheres occurs, reduce the number of cells seeded. To differentiate to neurons, plate approximately 200,000 cells per well of a 6-well plate, replacing NPC media with Neuron media.
- Immunohistochemically validate every established NPC line. Once sufficient cellular material has been expanded, label NPCs with antibodies for NESTIN and SOX2. Validated NPC lines should also be differentiated into neurons for 4-6 weeks, and labeled with antibodies for βIII-TUBULIN and MAP2AB.
- Fix cells in 4% paraformaldehyde in PBS at 4°C for 10 min. Permeabilize NPCs at RT for 15 min in 1.0% Triton in PBS, and then block in 5% donkey serum with 0.1% Triton at RT for 30 min.
- Incubate with primary antibody in 5% donkey serum with 0.1% Triton, overnight at 4°C. NOTE: We recommend the following antibodies: goat anti-Sox2, 1:200; mouse anti-human Nestin, 1:200; rabbit anti-βIII-tubulin, 1:500; mouse anti-βIII-tubulin, 1:500; mouse anti-MAP2AB, 1:200. (Figure 2).
- Following a wash with PBS, incubate secondary antibodies cells with the appropriate conjugated secondary antibody to goat, mouse or rabbit at 1:300, in in 5% donkey serum with 0.1% Triton for 1-2 hr at RT. To visualize nuclei, stain cells with 0.5 μg/ml DAPI (4',6-diamidino-2-phenylindole) and then mount with Vectashield.
4. NPC Transduction
Use spinfection (centrifugation of cell culture plates in the presence of virus at 1,000 x g for 1 hr) to increase the percentage of transfected cells 21. Aspirate media and replace with the relevant overexpression (or control) lentiviruses (or retroviruses), titered to the desired multiplicity of infection (MOI) (typically 1-10), diluted in NPC media. Use 1.5 ml volume per well of a 6-well plate (or an appropriately scaled volume for other types of tissue culture plates). Spin at 1,000 x g and 37 ºC for 1 hr in a plate centrifuge. NOTE: Ideally, transduce NPCs with lentiviral or retroviral vectors within 1-2 days of splitting. Whether using commercial or laboratory-prepared viruses, it can be helpful to appropriately titer batches of virus in advance by serial dilution. (Figure 3).
To reduce cellular death, replace media within 8 hr of spinfection. NOTE: Viral integration and transgene expression should be detectable within 24 hr by reporter gene fluorescence. (If the virus lacks a fluorescent reporter, this is more difficult to assess; successful transfection can best be validated by immunoblot, immunohistochemistry or qPCR for overexpression products.) Full expression may take 3-7 days; repeated spinfections with additional virus may be required to increase the percentage of transfected cells. Recurrent spinfections can occur daily but need not be so frequent. Ideally, if using a fluorescent reporter, >80% of cells should be labeled.
5. Neurosphere Migration Assay
NOTE: Neurospheres form spontaneously, following the enzymatic dissociation of NPCs (by a manner identical to that used in NPC expansion - steps 3.3-3.6), if cells are cultured in suspension in NPC media.
Briefly, grow dissociated NPCs in NPC media for 48 hr in nonadherent plates in order to generate neurospheres. (Figure 4).
For neurosphere migration, prepare fresh Matrigel plates using cold NPC media, rather than DMEM, in a 96-well plate, 1-2 hr prior to neurosphere picking. Do not aspirate Matrigel from the wells.
As originally published for embryonic stem cell-derived neurospheres 22, manually pick NPC-derived neurospheres under a microscope, after washing once in NPC media to remove cellular debris. Transfer one neurosphere to each well of a Matrigel-coated 96-well plate.
Add an additional 0.5 mg Matrigel, diluted in cold NPC media, to the neurospheres in each 96-well plate.
Manually center each individual neurosphere in the middle of the well using a pipet tip. NOTE: It is important to pick neurospheres of similar size, in order to reduce variability in the results.
Allow migration to occur for 48 hr and then fix cells with 4% paraformaldehyde and stain with desired immunohistochemical markers.
If radial migration is to be determined, photograph neurospheres in their entirely using a 4x microscope objective. Exclude any neurospheres contacting the edge of the well or a second neurosphere from the analysis. NOTE: If migration occurs for longer than 48 hr, the resulting radial migration will likely be too large to image using a 4x objective.
- Measure average migration from each neurosphere using NIH ImageJ software (Figure 5). This can be done using either of the analysis methods described below:
- Radial migration:
- Manually trace the edge of the furthest migrating cells using the freehand selection tool then use the Measure (Ctrl+M) function to calculate the area of the resulting shape. Before measuring the area make sure that Area is checked in the Set Measurements window found under the Analyze tab. Use the value of the area measurement and the equation for the area of a circle (A=πr2) to determine the radius (outer).
- Trace the edge of the original neurosphere, measure the area and calculate the radius (inner) in the same way. Calculate the total radial migration as the difference between the outer and inner radii.
- Greatest cellular migration:
- Measure the distance moved of the five furthest cells from the edge of the inner neurosphere using the straight-line selection tool followed by the Measure (Ctrl+M) function.
Representative Results
Neural rosettes can be identified morphologically, using a brightfield microscope, by their characteristic appearance as round clusters of neuroepithelial cells with apico-basal polarity (Figure 1). Though NPCs are typically cultured at very high cell density, immediately following passaging, slightly pyramidal-shaped soma and bipolar neurite structure is visible (Figure 1D). Validated NPCs express NESTIN and SOX2 in the majority of cells, though βIII-TUBULIN staining is also visible in all NPC populations, indicating that some proportion of NPCs are constantly initiating neuronal differentiation within the culture (Figure 1F). Markers of neural stem cells, such as SOX2 and PAX6, and forebrain neuronal progenitors, such as TBR2, are also expressed in NPCs, while midbrain markers such as LMX1A and FOXA2 are not (Figure 1G-I). A significant proportion of large and flat fibroblast-like cells within an NPC population indicates that poor quality NPCs have been generated, which are unlikely to yield large numbers of neurons following neuronal differentiation (Figure 2).
Following successful transduction with a high-titer lentiviral or retroviral vector, greater than 80% of cells should be labeled by the fluorescent reporter included in the vector (Figure 3). Neurosphere generation should yield a population of neurospheres of relatively homogenous size (Figure 4), which, with regular feeding, can remain healthy in culture for approximately one week. Neurosphere migration occurs robustly in healthy neurospheres (Figure 5), and hiPSC NPCs undergo rapid differentiation during the course of a neurosphere migration assay 1.
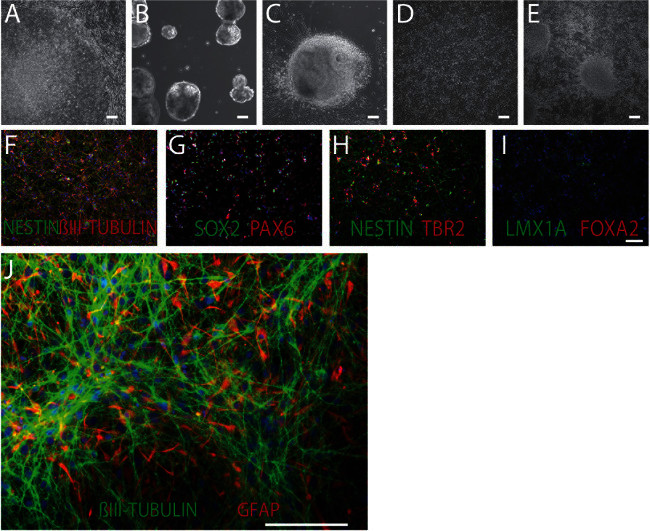
Figure 1. Patient-specific hiPSCs, NPCs and Neurons. Brightfield images of hiPSC neural differentiation, from hiPSCs (A), to embryoid bodies (B), neural rosettes (C), NPCs (D) and neurons (E). 100x, scale bar 100 μm. F-I. hiPSC NPCs are positive for a number of forebrain neural stem cell and neural progenitor cell marker, including the NPC marker NESTIN (green) and the neuronal marker βIII-TUBULIN (red) (F); the neural stem cell transcription factors SOX2 (green) and PAX6 (red) (G); and the forebrain progenitor marker TBR2 (red) (H); but not the midbrain markers LMX1A (green) and FOXA2 (red) (I). DAPI-stained nuclei (blue). 100x, scale bar 100 μm. J. NPCs can differentiate to 70-80% βIII-TUBULIN-positive neurons (green) and 20-30% glial fibrillary acidic protein (GFAP)-positive astrocytes (red). 200x, scale bar 100 μm. Please click here to view a larger version of this figure.
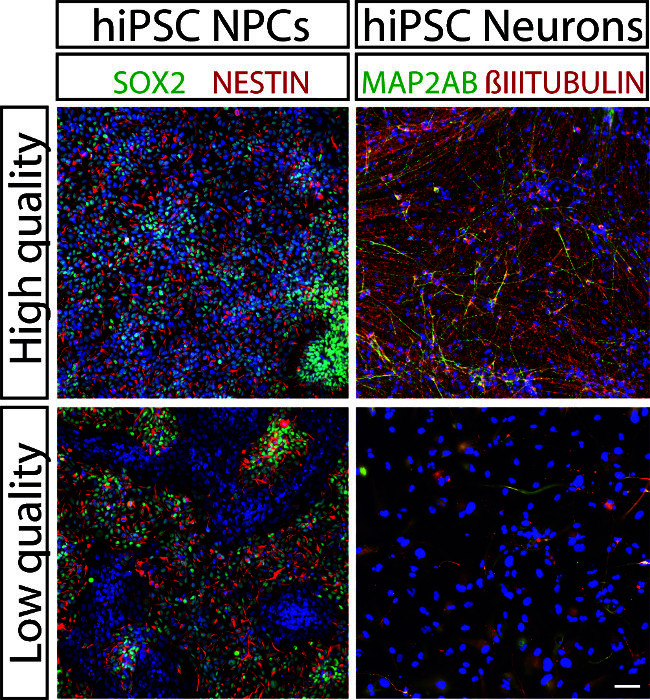
Figure 2. Validating NPC Lines. High quality (top left) hiPSC NPCs express NESTIN (red) and SOX2 (green). in most cells (nuclei stained with DAPI (blue)), whereas low quality (bottom left) NPCs have patches of SOX2-negative and NESTIN-negative cells (right). 4-week-old neurons differentiated from high quality NPCs express MAP2AB (green) and βIII-TUBULIN (red) (top right), but those differentiated from low quality NPCs do not (bottom right). 100x, scale bar 100 μm. Please click here to view a larger version of this figure.
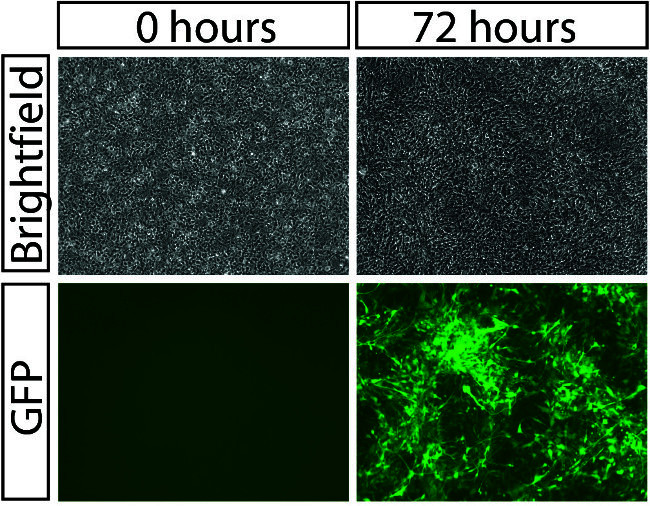
Figure 3. Viral Transduction of NPCs. Brightfield (top) and GFP fluorescent (bottom) images of hiPSC NPCs before (left) and after viral spinfection. 100x, scale bar 100 μm. Please click here to view a larger version of this figure.
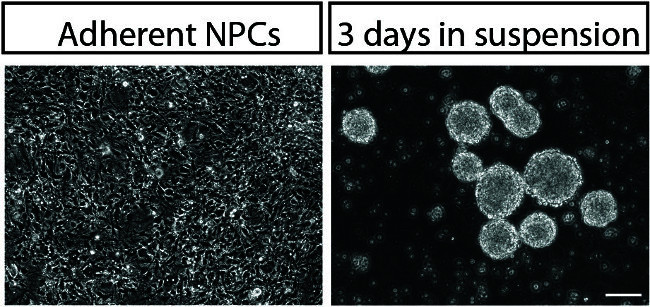
Figure 4. Neurosphere Formation. Brightfield images of adherent NPCs (left) and newly formed neurospheres (right). 100x, scale bar 100 μm. Please click here to view a larger version of this figure.
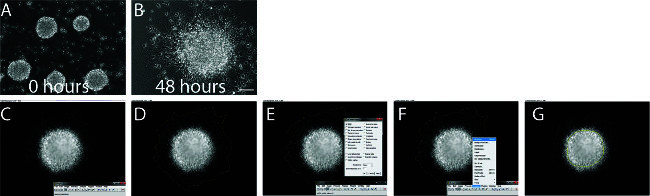
Figure 5. Neurosphere Migration. A-B. Brightfield images of neurospheres before (A) and after (B) 48 hr of migration in Matrigel. 100x, scale bar 100 μm. C-G. Screen capture images demonstrating the methodology for analyzing radial neural migration using NIH ImageJ software. Click on ‘freehand selections’ on ImageJ toolbar (C). Trace the edge of the furthest migrated cells around the neurospheres (D). Make sure that Area is checked in the Set Measurements window (E). Use the Measure function (Ctrl+M) (F). Trace the edge of the original neurospheres (G) and use the Measure function (Ctrl+M). Measurements can then be exported to Excel for analysis. Please click here to view a larger version of this figure.
| Media | Components |
| HES media | DMEM/F12 (Life Technologies #11330) |
| 20% KO-Serum Replacement (Life Technologies #10828) | |
| 1x Glutamax (Life Technologies #35050) | |
| 1x NEAA (Life Technologies #11140) | |
| 1x 2-mercaptoethanol (55mM 1000x) (Life Technologies #21985-023) | |
| 20ng/ml FGF2 (Life Technologies 13256-029) | |
| N2/B27 media | DMEM/F12 (Life Technologies #10565) |
| 1x N2 (Life Technologies #17502-048) | |
| 1x B27-RA (Life Technologies #12587-010) | |
| NPC media | DMEM/F12 (Life Technologies #10565) |
| 1x N2 (Life Technologies #17502-048) | |
| 1x B27-RA (Life Technologies #12587-010) | |
| 20 ng/ml FGF2 (Life Technologies) | |
| 1 mg/ml Natural Mouse Laminin (Life Technologies #23017-015) | |
| Neuron media | DMEM/F12 (Life Technologies #10565) |
| 1x N2 (Life Technologies #17502-048) | |
| 1x B27-RA (Life Technologies #12587-010) | |
| 1 mg/ml Natural Mouse Laminin (Life Technologies) | |
| 20 ng/ml BDNF (Peprotech #450-02) | |
| 20 ng/ml GDNF (Peptrotech #450-10) | |
| 500 μg/ml Dibutyryl cyclic-AMP (Sigma #D0627) | |
| 200 nM L-ascorbic acid (Sigma #A0278) |
Table 1. Media recipes.
Discussion
We have described methods by which to differentiate hiPSCs into NPCs, a neural cell type in which a significant fraction of the gene signature of hiPSC-derived neurons is conserved and that may serve as a proxy for the developmental pathways potentially contributing to disease pathogenesis 8,11. Additionally, as we have detailed, NPCs are a robustly replicative and easily transduced neural population, which we believe may be suitable for molecular and biochemical studies of disease predisposition.
Though we have detailed methods to differentiate and culture forebrain-patterned NPCs, they can be easily adapted to generate NPCs patterned to other cell fates. For example, if floating EBs are first treated with 0.5 mg/ml DKK1, 10 mM SB431542, 0.5 mg/ml Noggin and 1mM cyclopamine, hippocampal NPCs will be generated which can be expanded in NPC media and differentiated in neuronal media supplemented with 20 ng/ml WNT3A 7. Alternately, by replacing FGF2 with 100 ng/ml SHH C25II, 2μM Purmorphamine, 100 μg/ml FGF8 and 3μM CHIR99021, NPCs can be alternately patterned to midbrain dopaminergic fate, capable of generating neuronal populations enriched for FOXA2- and tyrosine hydroxylase-positive dopaminergic neurons 12,23. Given that most subtype specific neuronal differentiation protocols proceed via a NESTIN- and SOX2-positve intermediate, we predict that similar methods may be adaptable for GABAergic- 2,24 and glutamatergic- 3,25,26 specific NPC populations.
There are several critical steps within the protocol worth noting. First, if over-expressing or knocking down endogenous gene expression, it is best to use a control vector expressing a fluorescent protein from the same viral backbone, as we have observed substantial differences in florescence when reporters are expressed from vectors of variable sizes (owing to decreased packaging efficiency with vector size) or with difference promoters (owing to differences in promoter efficiency). Second, we recommend completing viral transduction prior to neurosphere formation, as we have observed poor penetrance of viral vectors into the dense neurosphere structure. Third, as the passage number of the NPCs increases beyond passage ten, we often observe substantially impaired migration in the neurosphere assay. In fact, given the propensity of NPCs to yield a greater percentage of astrocytes with passage, it is crucial to match the passage of NPC lines in each experiment.
Finally, it is important to consider the biological and technical limitations of the techniques described. Though NPCs appear to be morphologically similar by eye, they are a heterogeneous population comprised of cells capable of generating both glutamatergic and GABAergic neurons. While we observe remarkably consistent forebrain patterning of NPC lines between individuals using this method 1, this is only when comparing fully validated high quality NPC lines – there are substantial differences between good and poor quality NPC lines. Additionally, viral transduction yields genetically heterogeneous cells, each with an integration of the viral vector at a unique genomic location; therefore, individual cells will vary both in the location and number of genomic integration sites. More consistent changes in gene expression will always occur when genetic manipulation is targeted to a precise and defined location, whether it occurs by zinc finger, TALEN or CRISPR-based strategies.
Patient hiPSC derived NPCs can be used to study both the global gene expression perturbations occurring in neurological disease and also the molecular and/or cellular effects of manipulating candidate genes or microRNAs in the context of either healthy or diseased patient-derived neural cells. In this way, the consequences of manipulating one or more key risk allele(s) can be characterized in the context of diverse genetic backgrounds.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Kristen Brennand is a New York Stem Cell Foundation - Robertson Investigator. The Brennand Laboratory is supported by a Brain and Behavior Young Investigator Grant, National Institute of Health (NIH) grant R01 MH101454 and the New York Stem Cell Foundation.
References
- Brennand K, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry. 2014. [DOI] [PMC free article] [PubMed]
- Nicholas CR, et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12:573–586. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J, et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:12770–12775. doi: 10.1073/pnas.1202944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Torii K, et al. Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron. 2014;82:560–572. doi: 10.1016/j.neuron.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robicsek O, et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol Psychiatry. 2013;18(10):1067–1076. doi: 10.1038/mp.2013.67. [DOI] [PubMed] [Google Scholar]
- Paulsen BD, et al. Altered oxygen metabolism associated to neurogenesis of induced pluripotent stem cells derived from a schizophrenic patient. Cell Transplant. 2012;21(7):1547–1559. doi: 10.3727/096368911X600957. [DOI] [PubMed] [Google Scholar]
- Yu DX, et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Reports. 2014;2:295–310. doi: 10.1016/j.stemcr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2014. [DOI] [PMC free article] [PubMed]
- Zhang L, Song X, Mohri Y, Qiao L. Role pf Inflammation and Tumor Microenvironment in the Development of Gastrointestinal Cancers: What Induced Pluripotent Stem Cells Can Do. Current stem cell research & therapy. 2014. [DOI] [PubMed]
- An MC, et al. Genetic Correction of Huntington's Disease Phenotypes in Induced Pluripotent Stem Cells. Cell Stem Cell. 2012;11(2):253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz MT, et al. Directed neural differentiation of human embryonic stem cells via an obligated primitive anterior stage. Stem Cells. 2007;25:1511–1520. doi: 10.1634/stemcells.2006-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, et al. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D, et al. Chromosome 7 and 19 trisomy in cultured human neural progenitor cells. PLoS One. 2009;4:e7630. doi: 10.1371/journal.pone.0007630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Cowan CA, et al. Derivation of embryonic stem-cell lines from human blastocysts. N Engl J Med. 2004;350:1353–1356. doi: 10.1056/NEJMsr040330. [DOI] [PubMed] [Google Scholar]
- Ludwig TE, et al. Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol. 2006;24:185–187. doi: 10.1038/nbt1177. [DOI] [PubMed] [Google Scholar]
- Coufal NG, et al. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, et al. Generation of human-induced pluripotent stem cells to model spinocerebellar ataxia type 2 in vitro. Journal of molecular neuroscience : MN. 2013;51:237–248. doi: 10.1007/s12031-012-9930-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aasen T, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Delaloy C, et al. MicroRNA-9 coordinates proliferation and migration of human embryonic stem cell-derived neural progenitors. Cell Stem Cell. 2010;6:323–335. doi: 10.1016/j.stem.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15:477–486. doi: 10.1038/nn.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espuny-Camacho I, et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron. 2013;77:440–456. doi: 10.1016/j.neuron.2012.12.011. [DOI] [PubMed] [Google Scholar]