

Abstract
The recruitment of chromatin regulators and the assignment of chromatin states to specific genomic loci are pivotal to cell fate decisions and tissue and organ formation during development. Determining the locations and levels of such chromatin features in vivo will provide valuable information about the spatio-temporal regulation of genomic elements, and will support aspirations to mimic embryonic tissue development in vitro. The most commonly used method for genome-wide and high-resolution profiling is chromatin immunoprecipitation followed by next-generation sequencing (ChIP-Seq). This protocol outlines how yolk-rich embryos such as those of the frog Xenopus can be processed for ChIP-Seq experiments, and it offers simple command lines for post-sequencing analysis. Because of the high efficiency with which the protocol extracts nuclei from formaldehyde-fixed tissue, the method allows easy upscaling to obtain enough ChIP material for genome-wide profiling. Our protocol has been used successfully to map various DNA-binding proteins such as transcription factors, signaling mediators, components of the transcription machinery, chromatin modifiers and post-translational histone modifications, and for this to be done at various stages of embryogenesis. Lastly, this protocol should be widely applicable to other model and non-model organisms as more and more genome assemblies become available.
Keywords: Developmental Biology, Issue 96, Chromatin immunoprecipitation, next-generation sequencing, ChIP-Seq, developmental biology, Xenopus embryos, cross-linking, transcription factor, post-sequencing analysis, DNA occupancy, metagene, binding motif, GO term
Introduction
The first attempts to characterize protein-DNA interactions in vivo were reported about 30 years ago in an effort to understand RNA polymerase-mediated gene transcription in bacteria and in the fruit fly1,2. Since then, the use of immunoprecipitation to enrich distinct chromatin features (ChIP) has been widely adopted to capture binding events and chromatin states with high efficiency3. Subsequently, with the emergence of powerful microarray technologies, this method led to the characterization of genome-wide chromatin landscapes4. More recently, chromatin profiling has become even more comprehensive and high-resolution, because millions of co-immunoprecipitated DNA templates can now be sequenced in parallel and mapped to the genome (ChIP-Seq)5. As increasing numbers of genome assemblies are available, ChIP-Seq is an attractive approach to learn more about the genome regulation that underlies biological processes.
Here we provide a protocol to perform ChIP-Seq on yolk-rich embryos such as those of the frog Xenopus. Drafts of the genomes of both widely used Xenopus species—X. tropicalis and X. laevis—have now been released by the International Xenopus Genome Consortium6. The embryos of Xenopus species share many desirable features that facilitate and allow the interpretation of genome-wide chromatin studies, including the production of large numbers of high-quality embryos, the large size of the embryos themselves, and their external development. In addition, the embryos are amenable to classic and novel manipulations like cell lineage tracing, whole-mount in situ hybridisation, RNA overexpression, and TALEN/CRISPR-mediated knockout technology.
The following protocol builds on the work of Lee et al., Blythe et al. and Gentsch et al.7-9. Briefly, Xenopus embryos are formaldehyde-fixed at the developmental stage of interest to covalently bind (cross-link) proteins to their associated genomic DNA. After nuclear extraction, cross-linked chromatin is fragmented to focus subsequent sequencing on specific genomic binding or modification sites, and to minimize the contributions of flanking DNA sequences. Subsequently, the chromatin fragments are immunoprecipitated with a ChIP-grade antibody to enrich those containing the protein of interest. The co-immunoprecipitated DNA is stripped from the protein and purified before creating an indexed (paired-end) library for next-generation sequencing (NGS). At the end, simple command lines are offered for the post-sequencing analysis of ChIP-Seq data.
Protocol
NOTE: All Xenopus work complies fully with the UK Animals (Scientific Procedures) Act 1986 as implemented by the MRC National Institute for Medical Research.
1. Preparations
Estimate the number of embryos required for the ChIP experiment (see discussion).
Prepare the following solutions which are stored at RT: 500 ml of 10x Marc’s Modified Ringers (MMR) without EDTA, pH adjusted to 7.5 and sterilized by autoclaving (1 M NaCl, 20 mM KCl, 20 mM CaCl2, 10 mM MgSO4, 50 mM HEPES pH 7.5)10, 1 ml of SDS elution buffer (50 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% SDS) and 1 ml of 5x DNA loading buffer (0.2% Orange G, 30% glycerol, 60 mM EDTA pH 8.0).
Prepare the following solutions which are stored at 4 °C: 50 ml of HEG buffer (50 mM HEPES–KOH pH 7.5, 1 mM EDTA pH 8.0, 20% glycerol), 500 ml of extraction buffers E1 (50 mM HEPES-KOH pH 7.5, 150 mM NaCl, 1 mM EDTA. 10% glycerol, 0.5% Igepal CA-630, 0.25% Triton X-100), E2 (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA), and E3 (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Igepal CA-630, 0.25% Na-Deoxycholate, 0.1% SDS), 500 ml of RIPA buffer (50 mM HEPES-KOH pH 7.5, 500 mM LiCl, 1 mM EDTA, 1% Igepal CA-630, 0.7% Na-deoxycholate) and 50 ml of TEN buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 150 mM NaCl).
Score and clip a 15 ml conical polystyrene tube at the 7 ml mark. Use this tube to contain nuclear extracts undergoing sonication.
For post-sequencing analysis, use a multicore Unix-style operating computer with at least 8 GB RAM and 500 GB free disk space. Install the following software locally of which most are used at the command line: FastQC, Illumina CASAVA-1.8 quality filter, Bowtie11, SAMtools12, HOMER13, MACS214, IGV15,16, Cluster317, Java TreeView, BLAST+18, and b2g4pipe19. Check the installation instructions and requirements for compilers and third party software.
- Build a Bowtie index for aligning short NGS reads to the Xenopus genome. An example is shown here for the X. tropicalis genome v7.1 (November 2011), which can be downloaded as a FASTA file (genome.fa) from the Xenbase ftp server (/pub/Genomics/JGI). Move the FASTA file to the index subdirectory of Bowtie.
- Use the following command line (here after the prompt character >) to generate xenTro7 index files: > bowtie-build /path/to/bowtie/index/genome.fa xenTro7 > export BOWTIE_INDEXES=/path/to/bowtie/index/
- Download the gene annotation file (GTF) from the UCSC Genome Browser or the mirror site on the NIMR server for latest genome versions (genomes.nimr.mrc.ac.uk) via Tools/Table Browser. Use the genome FASTA file and the GTF file to customize HOMER for Xenopus (e.g., X. tropicalis genome v7.1, -name xenTro7).
- Alternatively, use pre-built HOMER packages for some older versions of the Xenopus genome. > loadGenome.pl -name xenTro7 -org null -fasta /path/to/genome.fa -gtf path/to/genes.gtf
Create a genome track (.genome file) for the genome browser IGV by uploading an indexed FASTA file (genome.fa with genome.fa.fai file in the same folder) and the annotation file (genes.gtf). Create a genome scaffold index (genome.fa.fai) for the Xenopus genome as follows: > samtools faidx /path/to/genome.fa
- Use BLAST+ to pass Gene Ontology (GO) terms from several model species (human, mouse, zebrafish, fruit fly and yeast) onto Xenopus genes as follows:
- Download all coding sequences (CDS) as a single FASTA file (cds.fa) from the UCSC Genome Browser via Tools/Table Browser and update BLAST+ with the pre-formatted BLAST database of non-redundant proteins (nr): > update_blastdb.pl nr
- Search for human (txid9606), mouse (txid10090), zebrafish (txid7955), fruit fly (txid7227) and yeast (txid4932) proteins from the NCBI site via its advanced search function (http://www.ncbi.nlm.nih.gov/protein/advanced) and send the resultant GI (sequence identifier) list (sequence.gi.txt) to the computer.
- Assign Xenopus genes to the most similar proteins from the GI list by executing BLASTx with a certain expect (E) value cutoff (here 10-20). Make sure the output format is xml (-outfmt 5 -out blastx_results.xml). Make use of time-saving threads (-num_threads) which correlate with the number of available computer cores. > blastx -db /path/to/nr -gilist /path/to/sequence.gi.txt -query /path/to/cds.fa -evalue 1e-20 \ -outfmt 5 -out /path/to/blastx_results.xml -num_threads [# threads]
- Open the b2gPipe.properties file of the b2g4pipe folder with a text editor and update the database properties to Dbacces.dbname=b2go_sep13 and Dbacces.dbhost=publicdb.blast2go.com. Run b2g4pipe from the installation folder. > java Xmx1000m -cp *:ext/*: es.blast2go.prog.B2GAnnotPipe -in /path/to/blastx_results.xml \ -out results/xenTro7 -prop b2gPipe.properties -v -annot NOTE: This program extracts GO terms for each BLAST hit and assigns them to corresponding Xenopus genes (xenTro7.annot). The most updated database settings can be found under Tools/General Settings/DataAccess Settings of the Blast2GO Java Web Start application (see 9.11.1).
2. Chromatin Cross-linking
Fertilize Xenopus eggs, de-jelly and culture embryos according to standard protocols20.
Transfer the dejellied embryos (maximum 2,500 X. laevis or 10,000 X. tropicalis) at the developmental stage of interest to an 8 ml glass sample vial with cap and wash them briefly once with 0.01x MMR.
Fix the embryos with 1% formaldehyde in 0.01x MMR (e.g., add 225 µl of 36.5-38% formaldehyde to 8 ml 0.01x MMR) for 15 to 40 min at RT (see Discussion for the fixation time and the number of embryos required per ChIP experiment). NOTE: Formaldehyde is corrosive and highly toxic. It is hazardous in case of eye and skin contact, indigestion, and inhalation. Use the fume hood when adding formaldehyde to the vial.
Stop the fixation by briefly washing the embryos three times with cold 0.01x MMR. Do not let embryos make contact with the liquid surface because surface tension causes them to rupture.
Aliquot the embryos into 2 ml microcentrifuge tubes on ice with a maximum of 250 embryos per tube, which occupy a volume of approximately 250 µl (X. tropicalis) or 600 µl (X. laevis) before hatching.
Pipette away as much 0.01x MMR as possible. Skip the following step if you continue immediately with section 3.
Equilibrate embryos in 250 µl of cold HEG buffer. Once the embryos have settled to the bottom of the tube remove as much liquid as possible and snap-freeze in liquid nitrogen. Store at -80 °C.
3. Chromatin Extraction
NOTE: The following extraction of cross-linked chromatin from Xenopus embryos works most efficiently with the fixation times indicated in step 2.3 and 50 to 80 X. tropicalis or 25 to 40 X. laevis embryos per ml of extraction buffer E1, E2 and E3. Each extraction step is repeated, so that twice the calculated volume of buffer is required. For upscaling, use multiple 2 ml microcentrifuge tubes or 50 ml centrifuge tubes. Keep samples and buffers on ice during the chromatin extraction.
Supplement adequate volumes of buffers E1, E2 and E3 with 1 mM DTT and protease inhibitor tablets. If performing ChIP with a phospho-specific antibody, further supplement buffers with 5 mM NaF and 2 mM Na3VO4.
Homogenize fixed embryos with E1 by pipetting up and down. Centrifuge homogenates in a refrigerated centrifuge (4 °C) at 1,000 x g for 2 min (or 5 min in case of using 50 ml tubes). Aspirate the supernatant and any lipids attached to the wall.
Resuspend pellets in E1. Keep samples on ice for 10 min. Centrifuge and discard supernatants as in step 3.2.
Resuspend pellets in E2. Centrifuge and discard supernatants as in step 3.2.
Repeat step 3.4, but keep samples on ice for 10 min before centrifugation.
Resuspend pellets in E3. Keep samples on ice for at least 10 min. Centrifuge and discard supernatants as in step 3.2. NOTE: At this stage, the resuspensions should become fairly transparent. The anionic detergents in E3 extract cross-linked nuclei by rendering most of the remaining yolk platelets soluble.
Resuspend and pool pellets of cross-linked nuclei (normally colored brown from the insolubilized pigment granules) in a total volume of 1 ml of E3. Dilute sample with E3 to 2 or 3 ml if it appears very viscous and is difficult to pipette. Keep on ice or at 4 °C to proceed with step 4 on the same or following day. Snap-freeze in liquid nitrogen and store at −80 °C for later use.
4. Chromatin Fragmentation
NOTE: Sonication is used both to solubilize and to shear cross-linked chromatin. Here are parameters to run the Misonix Sonicator 3000 equipped with a 1/16 inch tapered microtip and sound enclosure. If using other sonicators, follow the manufacturers’ recommendations to shear cross-linked chromatin or use 6 to 12 W for 4 to 8 min in total.
Transfer the nuclear sample from step 3.7 into a custom-built tube for sonication (step 1.4). Keep the sample chilled during sonication by having the tube attached to an 800 ml plastic beaker filled with ice water via a short thermometer clamp.
Place the beaker on a laboratory jack. Adjust the jack so that the sonicator microtip is submersed in the sample to about two-thirds of the volume depth and centered without touching the tube wall.
Sonicate the sample for 7 min in total, interrupted every 30 sec with 1 min pauses. Set power to 1.0. Start the sonication and immediately increase the power setting (normally 2 to 4) to reach a reading of 9 to 12 W. Pause immediately if the sample begins to froth. Reposition tube and restart when the froth has completely disappeared.
Transfer the sheared chromatin into pre-chilled 1.5 ml microcentrifuge tubes and spin at full speed (>15,000 x g) for 5 min at 4 °C.
Transfer the supernatant to pre-chilled 1.5 ml microcentrifuge tubes. Collect 50 µl of the supernatant (ideally containing the chromatin of around 400,000 or more nuclei) to visualize the degree of chromatin fragmentation (section 5). Use the rest of the supernatant for ChIP (section 6).
Store samples at 4 °C for up to one day. Snap-freeze samples as aliquots (one per ChIP experiment) in liquid nitrogen for long-term storage at -80 °C.
5. Imaging Chromatin Fragmentation
Add 50 µl of SDS elution buffer, 4 µl of 5 M NaCl and 1 µl of proteinase K (20 μg/μl) to 50 µl of the supernatant from step 4.6.
Incubate for 6 to 15 hr (O/N) in a hybridization oven set to 65 °C.
Purify DNA using a commercial PCR purification kit. If necessary, use 3 M of sodium acetate (pH 5.2) to adjust pH as recommended by the manufacturer. Elute the DNA twice with 11 μl of elution buffer (10 mM Tris-HCl pH 8.5).
Add 0.4 µl of RNase A (20 μg/μl) and 5 µl of 5x DNA loading buffer before running the entire sample alongside a 100 bp and a 1 kb DNA ladder on a 1.4% agarose gel by electrophoresis. For optimal results, stain gel with a safe nucleic acid staining solution after electrophoresis.
6. Chromatin Immunoprecipitation
NOTE: In this section, use low-retention 1.5 ml microcentrifuge tubes and at least 1 ml of indicated buffer per tube to wash magnetic beads for 5 min at 4 °C. Before removing the buffer from the beads, leave the tubes in the magnetic rack for 20 to 30 sec each time or until the solution is clear.
Transfer 10 to 30 µl of the supernatant (sheared chromatin) from step 4.6 to a new tube to be used later as input sample, which corresponds to approximately 1% of total chromatin used for ChIP. Store at 4 °C until ChIP samples are ready for reversing cross-links.
Transfer the remaining chromatin to a new tube. For ChIP-qPCR experiments requiring an antibody control, distribute equal volumes of chromatin to two tubes.
- Add the ChIP-grade antibody (or the corresponding antibody control) to chromatin. As a rough guide, use about 1 µg of antibody per one million cells expressing the epitope of interest.
- To more accurately estimate the amount of antibody required per ChIP experiment, run the same ChIP with various amounts of antibody (e.g., 0.25 µg, 1 µg and 2.5 µg) and compare the yield at negative and positive control loci by ChIP-qPCR (see section 10). As an antibody control, use normal serum of the same isotype and host animal species as the antibody.
Incubate on a rotator (10 rpm) O/N at 4 °C.
Wash an adequate amount of antibody-compatible magnetic beads once with E3 for 5 min at 4 °C. Checkthe manufacturer’s specification for the antibody binding capacity of the beads (usually 5 to 20 µl of beads bind 1 µg of IgG antibody).
Add washed beads to the antibody pre-incubated chromatin. Further incubate on a rotator (10 rpm) for 4 hr.
Wash beads four times (ChIP-qPCR) or ten times (ChIP-Seq) with pre-chilled RIPA buffer, and then once with pre-chilled TEN buffer.
- Only carry out this step if performing a ChIP-Seq experiment.
- Resuspend washed beads in 50 µl of TEN buffer per tube. Pool all beads from a single ChIP experiment by transferring them to one new tube. Use the magnetic rack and refrigerated (4 °C) centrifugation at 1,000 x g to collect beads at the bottom of the tube. Discard as much liquid as possible without disrupting the pellet of beads.
Strip ChIP material off the beads by resuspending the beads in 50 to 100 µl of SDS elution buffer and vortexing them continuously with a thermomixer (1,000 rpm) for 15 min at 65 °C. After that centrifuge at full speed (>15,000 x g) for 30 sec. Transfer the supernatant (ChIP eluate) to a new tube.
Repeat the last step and combine the ChIP eluates.
7. Chromatin Reverse Cross-linking and DNA Purification
Add enough SDS elution buffer to the input sample (step 6.1) to reach the volume of the ChIP sample, which is 100 to 200 µl (step 6.10). Supplement both ChIP and input samples with 1/20 volume of 5 M NaCl. Incubate the samples for 6 to 15 hr (O/N) at 65 °C in a hybridization oven.
Add 1 volume of TE buffer and RNase A at 200 µg/ml. Incubate for 1 hr at 37 °C.
Add proteinase K at 200 µg/ml. Incubate for 2 to 4 hr at 55 °C.
Purify DNA by phenol:chloroform:isoamyl alcohol extraction followed by ethanol precipitation as previously outlined9. For ChIP-Seq, add 32 µl of elution buffer (10 mM Tris-HCl, pH 8.5) to dissolve the DNA pellet. Leave samples on ice for 30 min to ensure that the DNA is completely dissolved. NOTE: Commercial PCR purification kits have lower DNA recovery but are more convenient, and can be used for ChIP-qPCR samples.
For ChIP-Seq, determine the concentration of 1 µl of ChIP and input DNA using fluorometry-based methods. Follow the manufacturer’s instructions and make sure that concentration of DNA falls within the reliable detection range of the fluorometer.
8. ChIP-Seq Library Construction and Validation
NOTE: Current methods for DNA library preparation allow construction of high-complexity libraries for NGS from 1 to 2 ng. At the expense of some complexity, libraries can be made from as little as 50 pg of DNA (see Table of Specific Materials/Equipment). Use the same amount of DNA for both ChIP and input library. Briefly, to make indexed (paired-end) ChIP-Seq libraries, ChIP and input DNA need to be end-repaired, ligated to special adaptors (see Table of Specific Materials/Equipment), size-selected and PCR amplified.
Follow the manufacturer’s guidelines to make ChIP-Seq libraries. See discussion for further recommendations.
Elute each library in 12 µl of elution buffer and determine the concentration of 1 µl of each ChIP and input library using a fluorometer. Expect concentrations of 5 to 25 ng/µl. Consider reducing the number of PCR cycles (fewer than 18 cycles) if concentrations are higher than 25 ng/µl. NOTE: Accurate quantification is key to achieve optimal NGS results. Libraries with concentrations as low as 1 ng/µl after 18 PCR cycles can be sequenced, but frequently are of lower complexity.
Use 1 µl of library to determine the fragment size distribution and to check for any adaptor dimer contamination (band around 120 bp) by chip-based capillary electrophoresis. Repeat the solid phase reversible immobilization purification with a beads-to-sample ratio of 1:1 (instead of 1.6:1) if the library contains adaptor dimers.
Perform qPCR on validated positive and negative control loci (see section 10) to check whether similar DNA enrichment trends are observed before and after library preparation. Submit quality control approved libraries for sequencing.
9. Post-sequencing Analysis and Data Visualization
NOTE: Nowadays, NGS is often carried out by in-house or commercial sequencing facilities (see discussion for some NGS guidelines). The standard output are single or multiple gzip-compressed FASTQ files (*.fastq.gz) storing millions of sequencing reads. Normally, multiplexed reads are already separated according to their index and each read contains a sequence identifier and a quality control score (Phred+33 for Illumina 1.8+) for each base call. This approach here is only one out of many ways how to analyze NGS data. The reader is encouraged to check whether any of the following command lines require changes as this field is rapidly advancing and updates are occurring regularly.
Concatenate gzip-compressed FASTQ files and check the quality of the sequencing data using the FastQC script. Execute this and most of the following commands for both ChIP and input sequencing data(examples shown for ChIP) from the terminal: > cat /path/to/*.fastq.gz > ChIP.fastq.gz > fastqc ChIP.fastq.gz NOTE: Raw data from the successful sequencing of a high-complexity ChIP-Seq library should pass most tests. Failures originate mainly from poor sequencing runs and experimental artefacts such as biased PCR amplification or adaptor contamination. A certain degree of duplication (redundancy) is expected as redundant reads can represent bona fide DNA enrichment21. However, one can later restrict read tags - the 5’ end or reads - to one per base pair to eliminate any redundant reads without affecting the detection sensitivity of peaks (step 9.4)21.
Pre-process sequencing data to remove adaptor contamination (homerTools trim -3 <adaptor sequence>) allowing one mismatch (-mis 1). Use the first 20 bases of the (indexed) adaptor (5’ to 3’) proximal to the DNA fragment of interest upon ligation (shown for adaptor listed in the Table of Specific Materials/Equipment). > gzip -cd ChIP.fastq.gz | fastq_illumina_filter -vN > ChIP.fastq > homerTools trim -3 GATCGGAAGAGCACACGTCT -mis 1 -min 36 ChIP.fastq NOTE: The removal of filtered reads (-N) is only required by default in FASTQ files generated by Illumina 1.8. Omit the fastq_illumina_filter command (i.e., ‘| fastq_illumina_filter –vN’) if an older version than 1.8 generated the sequence identifier.
Align pre-processed reads to the reference genome (xenTro7) using Bowtie. Only keep uniquely mapped reads (-m 1) using default settings, i.e., maximal two mismatches in the first 28 bases and a total Phred+33 quality score of all mismatches per read of maximal 70. Report alignment in SAM format (-S). Increase the number of megabytes per thread (--chunkmbs) if the chunk memory is exhausted: > bowtie -m 1 -S -p [# threads] --chunkmbs [e.g. 200] xenTro7 ChIP.fastq.trimmed > ChIP.sam NOTE: Bowtie expects Phred+33 quality scores by default. Include the option --phred64-quals if the FASTQ file was generated with Phred+64 quality scores by Illumina older than 1.8.
Use two HOMER commands to transform the alignment (SAM) file into a bigWig file (.bw): > makeTagDirectory ChIP/ -single -tbp 1 ChIP.sam > makeUCSCfile ChIP/ -bigWig /path/to/ genome.fa.fai -fsize 1e20 -norm 1e7 -o ChIP.bw NOTE: The transformation requires the scaffold index (genome.fa.fai) of the reference genome (step 1.8). Here the profile is limited to one tag per base pair (-tbp 1) and normalized to 10 million reads (-norm 1e7). bigWig is one of the preferred format to dynamically visualize chromatin profiles with a genome browser such as IGV (step 9.12).
Determine the distribution of tags (-d ChIP/) at genomic landmarks (e.g., +/- 10 kb with 25 bp bins, -size 20000 -hist 25) such as the transcription start (tss, example shown here) and termination (tts) sites. Run the HOMER perl script annotatePeaks.pl with Xenopus annotations xenTro7 (step 1.7): > annotatePeaks.pl tss xenTro7 -size 20000 -hist 25 -d ChIP/ > ChIP_tagDensity.tss
Find significant peaks of DNA enrichment between ChIP (-t ChIP.sam) and Input (-c Input.sam) in the X. tropicalis genome using MACS2 with a 1% FDR cutoff (-q 0.01) and DNA fragments (after sonication) of 200 bp (--bw=200) for model building. Add the flag --broad to this command line if expecting a broad distribution of the chromatin feature of interest such as histone marks or RNA polymerase. > macs2 callpeak -t ChIP.sam -c Input.sam -f SAM -n ChIP -g 1.4376e9 -q 0.01 --bw=200 NOTE: The effective size of the X. tropicalis genome assembly v7.1 is about 1.4376 billion bp (-g 1.4376e9). MACS2 generates a BED file (ChIP_peaks.bed) enlisting peaks with their genomic locations.
- Compare several chromatin profiles in form of a clustered heatmap:
- Create a tag distribution matrix from tag density directories of interest (-d ChIP/ other_ChIP/) at MACS2 peaks (e.g., +/- 1 kb with 25 bp bins, -size 2000 -hist 25 -ghist): > annotatePeaks.pl ChIP_peaks.bed xenTro7 -size 2000 -hist 25 -ghist -d ChIP/ other_ChIP/ > ChIP.matrix
- Use the graphical user interface of Cluster3 to upload ChIP.matrix file and hierarchically cluster these tag densities based on the minimal Euclidean distance to the closest centroid. Open the generated CTD file in Java TreeView to visualize the clustering.
Find novel and previously known binding motifs, which are enriched at peak summits +/- 100 bp (-size 200). Use annotatePeaks.pl to map motif occurrences and to plot motif densities: > findMotifsGenome.pl ChIP_peaks.bed xenTro7 ChIP_motifs/ -size 200 -p [# threads] > annotatePeaks.pl ChIP_peaks.bed xenTro7 -m motif1.motif > ChIP_peaks.motif1 > annotatePeaks.pl ChIP_peaks.bed xenTro7 -m motif1.motif -size 800 -hist 25 > motif1.density NOTE: The findMotifsGenome.pl script infers the enrichment from the comparison to randomly selected gene-centric background sequences. The most enriched novel motif is saved under motif1.motif in the format of a position weight matrix. The reader is encouraged to substantiate these results with other de novo motif discovery methods such as cisFinder22 and MEME23.
Annotate peaks by calculating their distance to the nearest gene and by determining their normalized read count within 400 bp windows (-size 400): > annotatePeaks.pl ChIP_peaks.bed xenTro7 -size 400 -d ChIP/ Input/ > ChIP_peaks.genes
Summarize the output using the following awk command to list the number (N), location and normalized read count of individual (R) and all (LR) peaks per nearest (target) gene. > awk 'BEGIN{ FS='\t' } $7 >= -5000 && $7 <= 1000 \ { N[$8]+=1; R[$8]+=$9; LR[$8]=LR[$8]','$7'('$9')' } END{ for(i in N) \ { print i '\t' N[i] '\t' R[i]' \t' substr(LR[i],2)}}' ChIP_peaks.genes > ChIP_peaks.summary NOTE: The number after $ refers to the column number, which may need modifying to fit the ChIP_peaks.genes file created in the previous step. This script exemplar filters out peaks beyond 5 kb upstream and 1 kb downstream of the TSS. $7, $8 and $9 refer to the distance to the TSS, the gene identifier and the normalized read count per peak, respectively.
- Perform the analysis of enriched GO terms among target genes as follows:
- Start the graphical user interface of Blast2GO from the command line via Java Web Start (javaws)19. > javaws http://blast2go.com/webstart/blast2go1000.jnlp
- Follow the developers’ instructions to upload the annotation file for Xenopus genes (xenTro7.annot) as generated in 1.9.4 and a flat file of identified target genes. Make sure that the same gene identifiers are used in both files.
Visualize chromatin profiles by adding bigWig (ChIP.bw, Input.bw) and BED files (ChIP_peaks.bed) into IGV as tracks. Complement data with RNA-Seq tracks if available for the same stage of development. Save results as a session.
Use programming platforms R (www.r-project.org) or MATLAB to further manipulate and visualize data as generated above. Alternatively, plot small datasets with Excel.
10. ChIP-qPCR for Testing ChIP and Confirming ChIP-Seq
Use the on-line platform Primer3 to design primers surrounding approximately 100 bp DNA at 60 °C (Tm) for both positive (peak-specific) and negative control loci. Confirm primer specificity using the in silico PCR search implemented into the UCSC Genome Browser.
Create an 8-point standard curve of three-fold dilutions starting from approximately 1% input or use the 2−ΔΔC(T) method8,24 for the quantification of DNA enrichment.
Execute real-time PCR in technical triplicates for all samples, i.e., ChIP, control and, if need be, standard curve samples.
Plot DNA enrichment as the percentage of input DNA or as a ratio of ChIP versus control sample at both positive and negative control loci.
Representative Results
Equivalent results to those presented here are expected if the protocol is well executed and the antibody in use is of ChIP-grade quality (see discussion). This protocol allows the extraction of nuclei from formaldehyde-fixed Xenopus embryos and the efficient shearing of chromatin by sonication (Figure 1A-C). Sheared chromatin shows an asymmetric distribution of DNA fragments mainly ranging from 100 to 1,000 bp and peaking between 300 and 500 bp (Figure 1C). A minimal 50 pg of immunoprecipitated DNA is required to successfully make an indexed paired-end ChIP-Seq library with similarly sized DNA inserts (Figure 2A). The library should be largely devoid of adaptor dimers, which can be seen on the electropherogram at approximately 120 bp.
Upon sequencing-by-synthesis, pre-processed reads are mapped to the genome (Figure 2B, C). In a successful experiment with X. tropicalis embryos, normally 50 to 70% of single-end reads of 40 bp can be mapped uniquely to the genome assembly of v7.1 with maximally two mismatches. While input reads align quite uniformly across the genome, the alignment of ChIP reads results in strand-specific enrichments that flank the chromatin feature of interest. This is because all fragments are sequenced from the 5’ end (Figure 2C)25. Extending the alignment in reading direction to an average fragment size produces accurate profiles for single chromatin features such as transcription factor binding events. These DNA occupancies appear as peaks when visualized in IGV or any other compatible genome browser. Peak callers like MACS are used to determine the location of these peaks (Figure 3A). This way tens of thousands of binding sites have been determined in the X. tropicalis genome for T-box transcription factors such as VegT26. ChIP-qPCR experiments should confirm the local enrichment found by ChIP-Seq (Figure 3B).
ChIP-Seq experiments allow exploring genome-wide characteristics of chromatin features. For example, calculating the read distribution over genomic elements such as transcription start and termination sites may highlight any spatial binding preferences around genes (Figure 3C). Similarly, a heatmap of read distributions at peak locations is used to compare different chromatin features at a genome-wide scale (Figure 3D). Certain transcription factors bind DNA sequence-specifically. De novo motif analysis of genomic DNA underlying peaks can retrieve this kind of information including co-enriched motifs of potential co-factors (Figure 3E). The great majority of target genes show DNA occupancy at a lower rather than higher level (Figure 3F). This scale-free feature seems to be quite common among transcription factors and suggests that only a small fraction of target genes are directly regulated with biological relevance27,28. The analysis of enriched GO terms or other attributes such as the differential expression of target genes may further reveal insights into the biological function of the chromatin feature in the Xenopus embryo (Figure 3G).
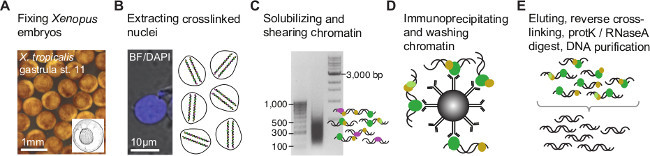 Figure 1. Chromatin immunoprecipitation procedure for Xenopus embryos. (A) Embryos are formaldehyde-fixed at the developmental stage of interest to covalently bind (cross-link) any proteins associated with genomic DNA. Upon nuclear extraction (B), cross-linked chromatin is fragmented to narrow down genomic DNA binding or chromatin modification sites by minimizing the flanking DNA sequence (C). Subsequently, the chromatin fragments are immunoprecipitated with a ChIP-grade antibody to enrich those containing the epitope of interest (D). The co-immunoprecipitated DNA is stripped off the protein and purified (E) before creating the ChIP fragment library for NGS (Figure 2). Please click here to view a larger version of this figure.
Figure 1. Chromatin immunoprecipitation procedure for Xenopus embryos. (A) Embryos are formaldehyde-fixed at the developmental stage of interest to covalently bind (cross-link) any proteins associated with genomic DNA. Upon nuclear extraction (B), cross-linked chromatin is fragmented to narrow down genomic DNA binding or chromatin modification sites by minimizing the flanking DNA sequence (C). Subsequently, the chromatin fragments are immunoprecipitated with a ChIP-grade antibody to enrich those containing the epitope of interest (D). The co-immunoprecipitated DNA is stripped off the protein and purified (E) before creating the ChIP fragment library for NGS (Figure 2). Please click here to view a larger version of this figure.
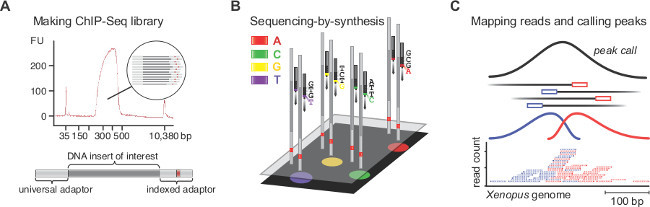 Figure 2. ChIP-Seq library preparation, sequencing-by-synthesis, mapping and peak calling. (A) The electropherogram displays a good ChIP-Seq library with DNA templates of 250 to 450 bp. These templates entail the DNA insert of interest flanked by the universal (58 bp) and the indexed (63 bp) adaptor. (B) Millions of clusters, with each cluster containing identical templates, are sequenced base by base in the presence of all four nucleotides possessing reversible, distinct fluorophore and identical termination properties. Fluorescent images are processed in real time to call corresponding bases, which ultimately are assembled into reads. (C) Only reads that map uniquely to the Xenopus genome are kept. As all fragments are sequenced from the 5’ end, the mapping of ChIP reads results in strand-specific peaks that flank the chromatin feature of interest. Thereby, peak callers detect the enrichment that originates from immunoprecipitation and extend the reads to an average fragment length to accurately localize chromatin features. Please click here to view a larger version of this figure.
Figure 2. ChIP-Seq library preparation, sequencing-by-synthesis, mapping and peak calling. (A) The electropherogram displays a good ChIP-Seq library with DNA templates of 250 to 450 bp. These templates entail the DNA insert of interest flanked by the universal (58 bp) and the indexed (63 bp) adaptor. (B) Millions of clusters, with each cluster containing identical templates, are sequenced base by base in the presence of all four nucleotides possessing reversible, distinct fluorophore and identical termination properties. Fluorescent images are processed in real time to call corresponding bases, which ultimately are assembled into reads. (C) Only reads that map uniquely to the Xenopus genome are kept. As all fragments are sequenced from the 5’ end, the mapping of ChIP reads results in strand-specific peaks that flank the chromatin feature of interest. Thereby, peak callers detect the enrichment that originates from immunoprecipitation and extend the reads to an average fragment length to accurately localize chromatin features. Please click here to view a larger version of this figure.
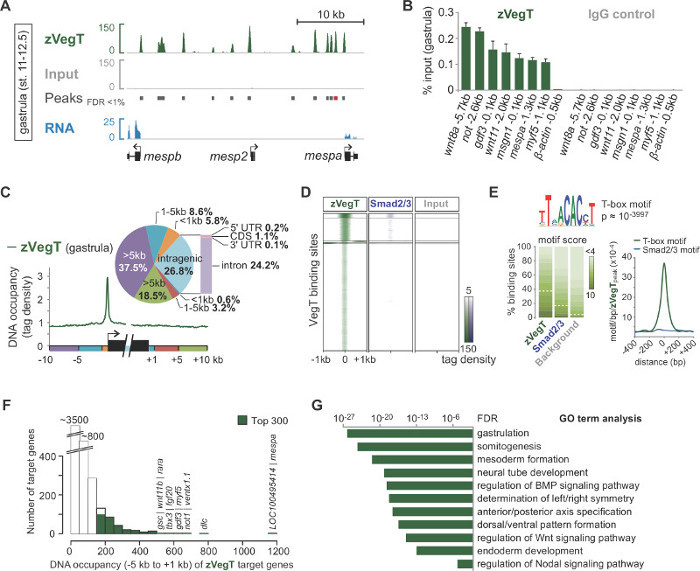 Figure 3. An example of post-sequencing analysis and data visualisation by means of the zygotic T-box transcription factor VegT (zVegT). All read counts shown here are normalised to 10 million uniquely mapped and non-redundant reads. (A) Excerpt of the genome-wide profile of zVegT binding in X. tropicalis gastrula embryos (stage 11 to 12.5 after Nieuwkoop and Faber29). Each peak, a pile-up of extended reads, represents one binding site. These peaks are called by MACS2 with a false discovery rate (FDR) of less than 1%. Each mesp gene shows very proximal and upstream zVegT binding, but only mespa and mespb are expressed by that stage (RNA-Seq data30). (B) DNA occupancy levels of zVegT as determined by ChIP-qPCR at several loci (including a non-bound region 0.5 kb upstream of β-actin) confirm the specific enrichment found by ChIP-Seq. Compare results for mespa with peak called (red bar) in (A). The DNA occupancy level is visualized as a percentage of input for both, the ChIP with the VegT antibody (rabbit polyclonal of IgG isotype) and the ChIP with the antibody control (normal rabbit IgG). Error bars reflect the standard deviation of two biological replicates. (C) Metagene analysis shows preferential zVegT binding (tags binned over 25 bp) to the promoter relative to any other genomic region around and within gene bodies. (D) Heatmap shows k-mean clustered (k=5) DNA occupancy levels (tags binned over 25 bp) of zVegT and Smad2/Smad3 (ChIP-Seq data31) relative to all zVegT-bound regions at gastrula stage. The heatmap is log2 based and centred at 5 tags per bp. (E) De novo motif analysis discovers the canonical T-box transcription factor binding motif in 38% of zVegT-bound regions if the underlying motif score is normalised to a 5% discovery rate in background sequences. The density map shows highest enrichment for the T-box motif in the centre of zVegT binding sites, whereas the canonical Smad2/Smad3 binding motif is hardly enriched. (F) Histogram shows DNA occupancy levels of zVegT, which are calculated for each target gene from all peaks (+/- 200 bp) between 5 kb upstream [-] and 1 kb downstream [+] of corresponding transcription start sites. (G) Top 300 genes with highest DNA occupancy levels within -5 kb and +1 kb are enriched for biological processes of early embryonic development. These GO terms are in line with the putative function of zVegT. The FDR is based on a two-tailed Fisher’s exact test and corrected for multiple testing. Please click here to view a larger version of this figure.
Figure 3. An example of post-sequencing analysis and data visualisation by means of the zygotic T-box transcription factor VegT (zVegT). All read counts shown here are normalised to 10 million uniquely mapped and non-redundant reads. (A) Excerpt of the genome-wide profile of zVegT binding in X. tropicalis gastrula embryos (stage 11 to 12.5 after Nieuwkoop and Faber29). Each peak, a pile-up of extended reads, represents one binding site. These peaks are called by MACS2 with a false discovery rate (FDR) of less than 1%. Each mesp gene shows very proximal and upstream zVegT binding, but only mespa and mespb are expressed by that stage (RNA-Seq data30). (B) DNA occupancy levels of zVegT as determined by ChIP-qPCR at several loci (including a non-bound region 0.5 kb upstream of β-actin) confirm the specific enrichment found by ChIP-Seq. Compare results for mespa with peak called (red bar) in (A). The DNA occupancy level is visualized as a percentage of input for both, the ChIP with the VegT antibody (rabbit polyclonal of IgG isotype) and the ChIP with the antibody control (normal rabbit IgG). Error bars reflect the standard deviation of two biological replicates. (C) Metagene analysis shows preferential zVegT binding (tags binned over 25 bp) to the promoter relative to any other genomic region around and within gene bodies. (D) Heatmap shows k-mean clustered (k=5) DNA occupancy levels (tags binned over 25 bp) of zVegT and Smad2/Smad3 (ChIP-Seq data31) relative to all zVegT-bound regions at gastrula stage. The heatmap is log2 based and centred at 5 tags per bp. (E) De novo motif analysis discovers the canonical T-box transcription factor binding motif in 38% of zVegT-bound regions if the underlying motif score is normalised to a 5% discovery rate in background sequences. The density map shows highest enrichment for the T-box motif in the centre of zVegT binding sites, whereas the canonical Smad2/Smad3 binding motif is hardly enriched. (F) Histogram shows DNA occupancy levels of zVegT, which are calculated for each target gene from all peaks (+/- 200 bp) between 5 kb upstream [-] and 1 kb downstream [+] of corresponding transcription start sites. (G) Top 300 genes with highest DNA occupancy levels within -5 kb and +1 kb are enriched for biological processes of early embryonic development. These GO terms are in line with the putative function of zVegT. The FDR is based on a two-tailed Fisher’s exact test and corrected for multiple testing. Please click here to view a larger version of this figure.
Discussion
Our protocol outlines how to make and analyze genome-wide chromatin profiles from Xenopus embryos. It covers every step from cross-linking proteins to endogenous loci in vivo to processing millions of reads representing enriched genomic sites in silico. Since increasing numbers of genome drafts are available, this protocol should be applicable to other model and non-model organisms. The most important experimental section, which sets this protocol apart from previous work8,31,33,34, is the post-fixation procedure to extract cross-linked nuclei. It facilitates efficient chromatin solubilisation and shearing and easy upscaling. Together with improved efficiencies of library preparation this protocol allows the construction of high-complexity ChIP-Seq libraries from half to two million cells expressing the chromatin-associated epitope of interest. For ChIP-qPCR experiments, a few ten thousand of these cells are normally enough to check for DNA enrichment at perhaps six distinct genomic loci. These numbers are conservative estimates, but may vary depending on protein expression level, antibody quality, cross-linking efficiency, and epitope accessibility. As a guide, a single Xenopus embryo contains about 4,000 cells at the mid-blastula stage (8.5 after Nieuwkoop and Faber29), 40,000 cells at the late gastrula stage (12) and 100,000 cells at the early tailbud stage (20).
The exact fixation time for efficient immunoprecipitation needs to be determined empirically by ChIP-qPCR (section 10). In general, longer fixation times are required if the experiment involves X. laevis embryos, early developmental stages, and weak (or indirect) DNA binding properties. However, it is not recommended fixing Xenopus embryos longer than 40 min, or processing more embryos than indicated (section 3), as chromatin shearing becomes less efficient. It is important not to use any glycine after fixation as this common step for quenching formaldehyde can make nuclear extraction from yolk-rich embryos very difficult. Currently, the reason for this is not known. It is conceivable that the formaldehyde-glycine adduct further reacts with N-terminal amino-groups or arginine residues35.
The antibody is key to any ChIP experiment and sufficient controls need to be carried out to show its specificity for the epitope of interest (see guidelines by Landt et al.36). If no ChIP-grade antibody is available, the introduction of corresponding epitope-tagged fusion proteins may be a legitimate alternative as these proteins can occupy endogeneous binding sites37. In this case, uninjected embryos are best to use as a negative control rather than a ChIP with non-specific serum. This strategy may also be applied if the protein of interest is expressed at low levels resulting in the poor recovery of enriched DNA.
As for making ChIP-Seq libraries, because of the low amount of DNA in use, it is recommended to opt for procedures that reduce the number of cleaning steps and to combine reactions to keep any loss of DNA at a minimum. The adaptors and primers need to be compatible with multiplex sequencing and the NGS platform (see Table of Specific Materials/Equipment). If using Y-adaptors (containing long single-stranded arms), it is critical to pre-amplify the library with three to five rounds of PCR before size-selecting DNA inserts (e.g., 100 to 300 bp) by gel electrophoresis. Single-stranded ends cause DNA fragments to migrate heterogeneously. Trial runs with various amounts of input DNA (e.g., 0.1, 0.5, 1, 2, 5, 10 and 20 ng) are recommended to determine the total number of PCR cycles (less than or equal to 18 cycles) required to make a size-selected library of 100 to 200 ng. Reducing the number of PCR cycles renders the sequencing of redundant reads less likely. Solid phase reversible immobilization beads are good cleaning up reagents to efficiently recover the DNA of interest and reliably remove any free adaptors and dimers from ligation and PCR reactions.
In terms of number, type and length of reads, around 20 to 30 million single-end reads of 36 bp is enough for most ChIP-Seq experiments to cover the whole Xenopus genome with sufficient depth. The most prevalent NGS machines are routinely capable of meeting these criteria. However, it may be beneficial to increase the number of reads if a broad distributions of reads is expected, as observed with histone modifications, rather than sharp peaks. For many ChIP-Seq experiments, 4 to 5 differently indexed libraries can be pooled and sequenced in one flow cell lane using a high-performance NGS machine. Sometimes is also advisable to extend the read length and sequence both ends of the DNA template (paired-end) to increase mappability when analyzing chromatin within repetitive genomic regions.
This protocol has been applied successfully to a wide variety of chromatin features such as transcription factors, signaling mediators and post-translational histone modifications. However, embryos acquire an increasing degree of cellular heterogeneity as they develop and chromatin profiles become harder to interpret. Promising steps have been made in Arabidopsis and Drosophila to tissue-specifically profile chromatin landscapes by extracting cell type-specific nuclei38,39. Our protocol includes a nuclear extraction step, which could pave the way for tissue-specific ChIP-Seq in other embryos.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Chris Benner for implementing the X. tropicalis genome (xenTro2, xenTro2r) into HOMER and the Gilchrist lab for discussions on post-sequencing analysis. I.P. assisted the GO term analysis. G.E.G and J.C.S. were supported by the Wellcome Trust and are now supported by the Medical Research Council (program number U117597140).
References
- Gilmour DS, Lis JT. Detecting protein-DNA interactions in vivo: distribution of RNA polymerase on specific bacterial genes. Proc Natl Acad Sci U S A. 1984;81(14):4275–4279. doi: 10.1073/pnas.81.14.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour DS, Lis JT. In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Mol Cell Biol. 1985;5(8):2009–2018. doi: 10.1128/mcb.5.8.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MJ, Larsen PL, Varshavsky A. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 1988;53(6):937–947. doi: 10.1016/s0092-8674(88)90469-2. [DOI] [PubMed] [Google Scholar]
- Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290(5500):2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Johnson D, Mortazavi A, Myers R, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316(5830):1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Hellsten U, et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328(5978):633–636. doi: 10.1126/science.1183670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nature Protocols. 2006;1(2):729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe SA, Reid CD, Kessler DS, Klein PS. Chromatin immunoprecipitation in early Xenopus laevis embryos. Dev Dyn. 2009;238(6):1422–1432. doi: 10.1002/dvdy.21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentsch GE, Smith JC. Investigating physical chromatin associations across the Xenopus genome by chromatin immunoprecipitation. Cold Spring Harb Protoc. 2014;2014(5) doi: 10.1101/pdb.prot080614. [DOI] [PubMed] [Google Scholar]
- Ubbels GA, Hara K, Koster CH, Kirschner MW. Evidence for a functional role of the cytoskeleton in determination of the dorsoventral axis in Xenopus laevis eggs. J Embryol Exp Morphol. 1983;77:15–37. [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto S, Nolan J, Bioinformatics Miyano S. 2004;20(9):1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Sive H, Grainger R, Harland R. Early development of Xenopus laevis: A laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Chen Y, et al. Systematic evaluation of factors influencing ChIP-seq fidelity. Nat Methods. 2012;9(6):609–614. doi: 10.1038/nmeth.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, Ko MSH. Exhaustive search for over-represented DNA sequence motifs with CisFinder. DNA Res. 2009;16(5):261–273. doi: 10.1093/dnares/dsp014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucl Acids Res. 2009;37(2):W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentsch GE, et al. In vivo T-box transcription factor profiling reveals joint regulation of embryonic neuromesodermal bipotency. Cell Rep. 2013;4(6):1185–1196. doi: 10.1016/j.celrep.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Biggin MD. Animal transcription networks as highly connected, quantitative continua. Dev Cell. 2011;21(4):611–626. doi: 10.1016/j.devcel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin): a systematical and chronological survey of the development from the fertilized egg till the end of metamorphosis. New York & London: Garland; 1994. [Google Scholar]
- Akkers RC, et al. A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus embryos. Dev Cell. 2009;17(3):425–434. doi: 10.1016/j.devcel.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SJ, Wills AE, Chuong E, Gupta R, Baker JC. HEB and E2A function as SMAD/FOXH1 cofactors. Genes Dev. 2011;25(15):1654–1661. doi: 10.1101/gad.16800511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallow Z, Jacobi UG, Weeks DL, Dawid IB, Veenstra GJ. Specialized and redundant roles of TBP and a vertebrate-specific TBP paralog in embryonic gene regulation in Xenopus. Proc Natl Acad Sci U S A. 2004;101(37):13525. doi: 10.1073/pnas.0405536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz DR, Paul BD, Shi Y-B. Gene-specific changes in promoter occupancy by thyroid hormone receptor during frog metamorphosis. Implications for developmental gene regulation. J Biol Chem. 2005;280(50):41222–41228. doi: 10.1074/jbc.M509593200. [DOI] [PubMed] [Google Scholar]
- Wills AE, Guptaa R, Chuonga E, Baker JC. Chromatin immunoprecipitation and deep sequencing in Xenopus tropicalis and Xenopus laevis. Methods. 2014;66(3):410–421. doi: 10.1016/j.ymeth.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz B, et al. Identification of formaldehyde-induced modifications in proteins: reactions with model peptides. J Biol Chem. 2004;279(8):6235–6243. doi: 10.1074/jbc.M310752200. [DOI] [PubMed] [Google Scholar]
- Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, et al. Embryonic stem cell-based mapping of developmental transcriptional programs. Nat Methods. 2011;8(12):1056–1058. doi: 10.1038/nmeth.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell. 2010;18(6):1030–1040. doi: 10.1016/j.devcel.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn S, et al. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet. 2012;44(2):148–156. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]