

Abstract
Complex neurological phenotypes are inherently difficult to diagnose. Whole-exome sequencing (WES) is a new tool in the neurologist's diagnostic armamentarium. WES can be applied to investigate the “diagnostic odyssey” cases. These cases involve patients with rare diseases of likely genetic etiology who have failed to obtain a diagnosis by clinical evaluation and targeted gene testing.
The 22-year-old adopted proband presented with episodes of jerking ataxic movements that affected his whole body and mild intellectual developmental disability. He underwent numerous multidisciplinary and multicentric evaluations throughout his life that failed to establish a clear diagnosis. Following his visit to the Mayo Clinic, WES was applied for genetic determination of the unknown disorder in the proband, his biological parents and sister. Besides, four other paternal relatives were reported to have similar complaints. Additional clinical evaluation, and magnetic resonance neuroimaging (MRI), electromyography (EMG) and electroencephalography (EEG) of the proband were performed to verify the phenotype after the WES results were available.
Eleven months after the proband's initial visit, WES identified the c.1210G>A (p.V404I) mutation in the potassium voltage-gated channel, shaker-related subfamily, member 1 gene in the proband, his father, and his sister, and thus the diagnosis of episodic ataxia type 1 was established. The proband's MRI demonstrated mild vermian hypoplasia, EMG myokymic discharges, and EEG generalized background slowing. Acetazolamide therapy was beneficial for him at the daily dose of 500 mg.
Keywords: Episodic ataxia type 1, KCNA1 gene mutation, therapy with acetazolamide, whole exome sequencing
Introduction
Complex hereditary neurologic disorders can be difficult to clinically diagnose due to either their atypical phenotype or very rare occurrence. Additionally, these patients have been evaluated by many physicians, so, obtaining a complete medical history is a formidable task. The genetic basis for a number of these inherited disorders has been established; however, selecting the appropriate genetic test can prove to be challenging, time-consuming, and expensive.
Whole-exome sequencing (WES) presents a new clinical diagnostic tool for the neurologist. WES employs high-throughput sequencing to identify disease-causing mutations in an individual's exome.1 The exome comprises 180,000 exons (encoding 22,000 genes), which contain the protein coding information and accounts for approximately 1% of the whole genome.1 It is estimated that WES can evaluate 90-95% of the exome.2 Filtering algorithms based on the results of genomic analyses and clinical data limit the number of possible disease-causing variants and can confirm their relationship with the phenotype. WES has been proven to be an efficient diagnostic tool for identifying mutations and genes that are associated with neurodegenerative diseases with less cost than whole-genome sequencing.3
We describe a scenario where WES was clinically employed to elucidate the underlying genetic cause of a patient's complex movement disorder.
Patients and Methods
When the proband was 22-years-old, he presented to the Mayo Clinic in Florida accompanied by his adoptive mother seeking another neurological opinion for his chronic disorder. At the age of three months, he was placed in foster care and was adopted at the age of four years. He had previously undergone numerous neurological consultations and diagnostic procedures in multiple institutions without a clear diagnosis. Previous medical records were not available at his first visit.
Recently, the Mayo Clinic Center of Individualized Medicine has initiated a program that uses WES to support personalized care through genetic diagnosis. It targets patients with rare complex diseases that are likely genetic, e.g. those with a family history of disease or an earlier age-at-onset. These patients are sometimes referred to as “diagnostic odyssey” cases4 due to numerous prior unsuccessful attempts to establish a definite diagnosis.
As a “diagnostic odyssey” case the patient was offered WES. He agreed to this procedure and signed the authorization form for genetic counseling and collection of his prior medical records. Following first genetic counseling, contact with his biological family was established, and further counseling was provided to all participating biological family members. The assessment was accomplished from August 2013 to July 2014. WES and co-segregation analysis was performed by a Clinical Laboratory Improvement Amendments-certified laboratory utilizing samples collected from the proband, his biological parents, and his sister (Ambry Genetics Corp., Aliso Viejo, United States). Samples were prepared using the SeqCap EZ VCRome 2.0 system (Roche NimbleGen, Madison, WI). Detailed methodology for WES and analysis have been previously described.5 After the WES results were available, additional neurological evaluation, brain magnetic resonance neuroimaging (MRI), electromyography (EMG) and electroencephalography (EEG) were performed on the proband to verify the phenotypic characteristic of the disorder. All exams were performed for diagnostic purposes only; no approval of the institutional review board was sought or required.
Results
Genealogical Investigations
According to the proband's adoptive mother, the proband's sister, father, and four paternal relatives have or had a chronic neurological disorder with symptoms similar to those of the proband, including his already deceased paternal grandfather. No autopsy was performed. The sister's motor complaints worsened after a recent pregnancy, which was her second pregnancy. The proband's mother suffers from a mild intellectual disability and diabetes. Her daughter, who was the proband's half-sister, was reported to have an intellectual disability and possibly seizures. The pedigree structure is presented in Figure 1.
Figure 1.
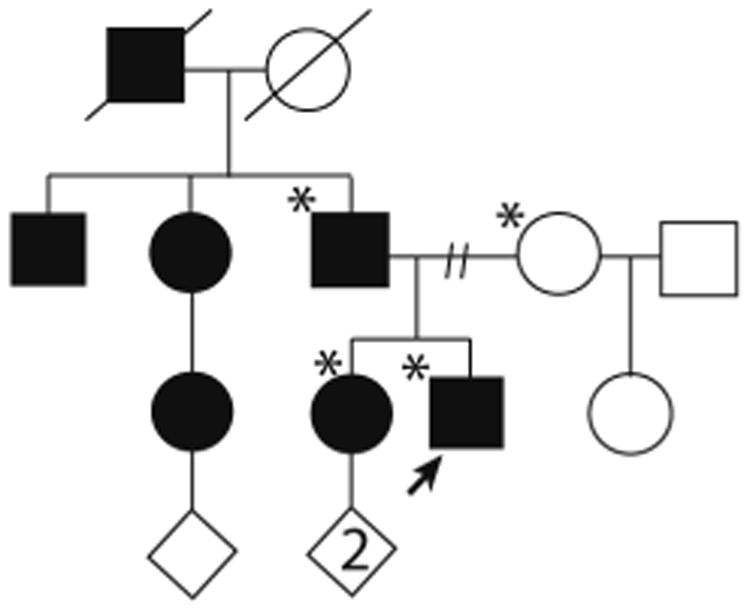
Pedigree structure of the family, Standard symbols were used. Round symbols indicate females, squares indicate males, diagonal lines indicate that the individual is deceased. Diamonds were used to disguise gender, numbers inside symbols indicate number of children. The solid arrowhead indicates the proband. Black full-filled symbols indicate individuals with clinical features of EA1. Asterisks indicate those family members that underwent whole-exome sequencing (proband and parents) or co-segregation analysis (sister).
Molecular Genetic Studies
A review of the proband's medical records showed that prior karyotyping, chromosomal microarray, and screening for mutations in the myotonic dystrophy protein kinase and huntingtin genes did not bring clarification. Exome sequencing of the family trio (proband, mother, and father) resulted in an average of approximately 11 giga base pairs of sequence per sample, with approximately 95% of bases within the captured region covered at least 10-fold. The removal of common single nucleotide polymorphisms, intergenic and 3′/5′-untranslated region variants, non-splice-related intronic variants, and synonymous variants through stepwise filtering resulted in approximately 12,000 variants associated with each trio member. Subsequent filtering based on family history and inheritance modelling (autosomal dominant/recessive, X-linked dominant/recessive, and Y-linked) within the trio narrowed the list of candidates to 165 genes and 171 unique alterations. After manual review of each alteration to rule out sequencing artifacts and polymorphisms, the list of candidates was narrowed to 110 genes and 111 unique alterations. Each gene/variant was then assessed for potential clinical significance and subsequently divided into “characterized” or “novel” classifications depending on previous literature documenting a role in disease.5 Of the 110 genes, only three genes were labeled as “characterized”, and one of these was the potassium voltage-gated channel, shaker-related subfamily, member 1 gene (KCNA1) on chromosome 12p13.32, known to harbor mutations causing episodic ataxia type 1 (EA1). In this trio WES identified the KCNA1 c.1210G>A (p.V404I) heterozygous missense mutation in the proband, inherited from his affected father. Follow-up co-segregation studies showed that the affected sister also carries this mutation.
Clinical and Laboratory Investigations
Only the proband was clinically investigated. Neurological evaluations of other family members for clinical assessment are planned in the near future. The proband was a Caucasian man who developed episodes of “shaking spells” at the age of 18 months. The spells consisted of uncontrollable “jerking” movements of his trunk and limbs such that they would keep him awake at night. Sometimes the movements were seen all over his body. During these episodes the patient had difficulty with swallowing, speaking, and ambulation. The episodes could last for a few minutes and would appear in rapid succession more than 15 times a day, leading to severe exhaustion. They could occur spontaneously or be induced by fatigue, emotional stress, or sudden postural changes. The spells became more severe and frequent. He started to walk at around 14 months of age, but further motor development was delayed. At 12 years of age, he was noted to be short in stature, have a mildly dolichocephalic head shape, and he had facial asymmetry with right-eye downslanting.
Prior evaluations included an ophthalmology exam that did not reveal the Kayser-Fleischer rings. He showed normal 24-hour urine copper excretion. Blood tests were negative for antistreptolysin O and anti-DNAse, and his erythrocyte sedimentation rate was normal. Results from long-term closed-circuit television electroencephalography, brain MRI, and multiple routine EEGs, including studies performed during his spells, were all reported to be negative. He was diagnosed as having attention deficit hyperactivity disorder, anxiety, depression, hereditary spastic paraparesis, chorea, myotonic dystrophy, and “generalized jerking and shaking.” He was treated with unknown doses of multiple medications alone or in combination. These included lisdexamfetamine, risperidone, valproic acid, alprazolam, and clonazepam; he was also given haloperidol for alleged auditory hallucinations. The medications produced no benefit.
At his first visit to Mayo Clinic, multiple spells were observed. During his spells neurological examination revealed high-amplitude and low-velocity, involuntary, uncoordinated movements involving the trunk and upper limbs that increased when he tried to stand up. His gait and pointing tests were ataxic, and the patient needed temporary assistance for mobility. Romberg's test and eye movements were unremarkable (eVideo 1). An increased muscle tone was noted, predominately in his lower extremities, due to mild spasticity with brisk deep tendon reflexes and an increase in tone on passive quick movements of his legs. Babiński's sign, ankle clonus, muscle weakness, or visible myokymias were not observed. The neuropsychological and psychiatric evaluations requested at the time of initial evaluation led to the diagnosis of mild intellectual developmental disorder with a full scale IQ of 59, auditory hallucinations, and lower frustration tolerance.
Brain MRI showed a mild vermian hypoplasia and prominent cisterna magna (Figure 2a and 2b). EEG revealed a generalized background slowing of 7-8 Hz frequency without an epileptiform activity. Nerve conduction studies of the sural, peroneal, and tibial nerves were normal. Concentric needle EMG of the left biceps brachii, triceps brachii, first dorsal interosseous, vastus lateralis, tibialis anterior, and gastrocnemius muscles showed myokymic discharges during and between the ataxic spells (Figure 3). However, they were absent in the left orbicularis oculi muscle.
Figure 2.
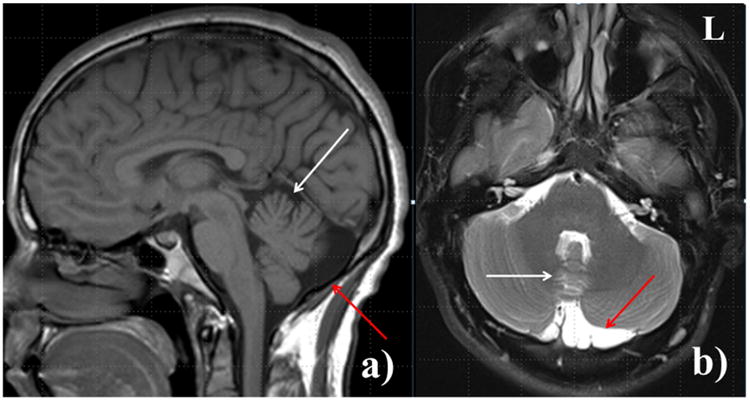
Sagittal T1-weighted magnetic resonance image (a) and axial T2-weighted magnetic resonance image with fat saturation (b) demonstrate mild vermian atrophy (white arrow) and prominent cisterna magna (red arrow).
Figure 3.
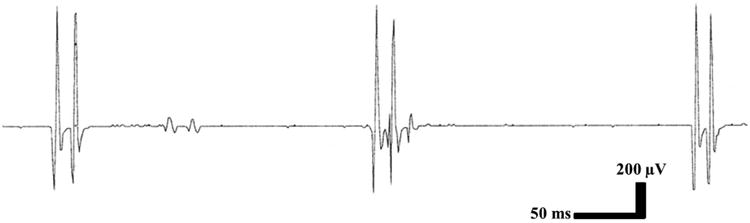
Electromyography showed the myokymic discharges (inner head of left gastrocnemius muscle).
Acetazolamide therapy at the initial dose of 125 mg once daily produced a severe skin rash that appeared after two doses. Following the successful desensitization procedure, the dose was increased to 250 mg twice daily and led to approximately a 50% reduction in the frequency and duration of his spells.
Discussion
Based on successful implementation of the WES studies, the diagnosis of episodic ataxia type 1 (EA1) was accurately established 11 months following the initial visit at the Mayo Clinic. The costs of WES amounted to about $7,000, whereas the previous four genetic tests were estimated to be approximately the same amount. This success with such a low cost underlies the importance of WES to the clinical setting. With this tool, we are able to correctly diagnose complicated neurological disorders that have a potential genetic nature even in difficult circumstances, such as in this case.
Episodic ataxia is a group of seven rare familial disorders characterized by brief attacks of generalized ataxia with normal or almost normal neurological function between attacks.6 EA1 is autosomal dominant, has a childhood or early adolescence onset, and is a potassium channelopathy. Its cardinal symptoms include ataxia with loss of both motor coordination and balance accompanied by constant myokymia of the skeletal muscles of the head and limbs, detectable clinically or on EMG.7 Other symptoms may comprise vertigo, blurred vision, diplopia, nausea, headache, diaphoresis, clumsiness, dysarthric speech, difficulty breathing, epilepsy, delayed motor development, cognitive disability, choreoathetosis, and carpal spasm.6, 7 EA1 was first described in 1975 by van Dyke8, and since then, the EA1 phenotype has been reported in little more than 100 individuals.9 Although most individuals with EA1 present with episodes of ataxia, myokymia on EMG, and normal brain MRI7, establishing a diagnosis may be challenging due to phenotypic variations. The diagnosis will depend on the underlying mutation and a consideration for different presenting phenotypes, which can include only myokymia 10 or distal weakness11 or a lack of episodic ataxia12 or myokymia.13 There is also an intra- and interfamilial phenotypic variability.7
In the described family, analysis of the pedigree suggested an autosomal dominant disorder. However, the family history suggesting mental retardation on both the mother's and father's side and the vermian hypoplasia with prominent cisterna magna on MRI of the proband were difficult to interpret. Additionally, there is only one report of cerebral atrophy in EA1.14 Thus the proband underwent extensive diagnostic tests for Wilson's disease, Huntington's disease, Sydenham's chorea, epilepsy and Curschmann-Steinert myotonic dystrophy, but these did not establish a diagnosis.
The application of WES in a clinical diagnostic setting has accelerated and simplified the diagnosis and management of patients with genetic diseases. Besides, WES is becoming more cost-effective and as a result, more extensively applied.15 Current diagnostic panel testing for complete ataxia evaluation offered by commercial laboratories currently costs almost twice as much as WES and does not include a specific test for EA1. Thus, in the case of clinical atypical ataxia, it may be beneficial to consider WES first, especially if clinical and genealogical data are sparse. However, it should be noted that WES would not detect triplet repeat mutations, which represent an estimated 40% to 50% of genetic ataxias worldwide.15
For the first time in molecular diagnosis, WES can simultaneously interrogate virtually all genes, including those most recently discovered, as well as genes that are both related to and outside of the clinician's differential diagnoses. Therefore, it is particularly useful for diagnosing patients who have genetic disorders with profound and heterogeneous phenotypes and atypical and incomplete presentations. It is also useful for diagnosing patients who are suspected to have a genetic diagnosis but have incomplete histories, and it can help those who have rare or newly-discovered diseases or disorders. However, the limitations of this approach need to be cautioned. WES is not emphasized or optimal for analyzing the following types of mutations: gross deletions/duplications, gross rearrangements, deep intronic variations, long repeat sequences, trinucleotide repeat sequences, mutations involved in tri-allelic inheritance, mitochondrial genome mutations, epigenetic effects, and oligogenic inheritance. Although some of these mutations may be detected by whole-genome sequencing, large expanded-repeat sequences present a considerable diagnostic challenge.
The limitations of WES are not just restricted to technical methodology. In the present case, we had a familial trio available for filtering variants and reached the shortlist of candidate genes. In most cases, only the proband may be available, and each individual carries a substantial number of rare/unique variants that could make genetic diagnosis difficult. In our recent study of 500 unselected families for WES, the diagnostic rate was significantly higher among families undergoing a trio whole-exome testing strategy (37%) as compared with a singleton whole-exome testing strategy (21%).5 In the present study, the likely pathogenic mutation was also identified in a gene known to cause EA1, which fits with the clinical phenotype. However, if the causative mutation is in a “novel” gene that has not been connected with the phenotype, it will fall into a long list of candidates.5 Finally, the KCNA1 c.1210G>A (p.V404I) mutation has previously been reported in a family with EA116, but a scenario could arise where a novel mutation in a “characterized” gene could be identified but with an unclear pathogenicity; functional studies with a pathogenic readout may be helpful in these cases.
To our knowledge, this is the first report of the application of WES to facilitate the diagnosis of EA1. Currently, more than 20 reported KCNA1 mutations have been identified by standard gene analysis in familial cases, and only one de novo mutation has been reported.14 All carriers are heterozygous for KCNA1 mutations that are mostly missense mutations with a very few nonsense and in frame deletion mutations.10, 13, 17
The KCNA1 c.1210G>A (p.V404I) mutation results in a valine-to-isoleucine substitution at a highly conserved position in the sixth transmembrane segment of the potassium voltage gated channel subunit hKv1.1. This leads to a rather subtle disturbance of channel function and a milder phenotype than other mutations, such as the p.R417* mutation.10
Although antiepileptic drugs may significantly reduce the frequency of the attacks in EA1, the response is heterogeneous.10 In a second family with the KCNA1 c.1210G>A (p.V404I) mutation, carbamazepine was reported to have good results.10 In our proband, a trial of two benzodiazepines proved ineffective; acetazolamide therapy first became beneficial at the daily dose of 500 mg. Of note, the first reported family with the KCNA1 c.1210G>A (p.V404I) mutation did not respond well to acetazolamide.16
Conclusions
WES supported by evaluation of clinical data has proven to be a useful, time-saving, and cost-effective diagnostic tool for the diagnosis of a patient with a complex phenotypic presentation of EA1. This diagnostic approach may help determine early therapeutic intervention strategies and directly affect patient care.
Supplementary Material
Supplemental Video 1. “Shaking spells” and ataxic features of the proband. During his spells, there were high-amplitude and low-velocity, involuntary, ataxic movements involving the trunk and upper limbs. His gait and pointing tests were also ataxic, and the patient needed temporary assistance for mobility. Romberg's test was unremarkable. In addition mild spasticity with brisk deep tendon reflexes, more evident in his lower extremities, was noted. Babiński's sign, ankle clonus and visible myokymias were absent on neurological examination.
Acknowledgments
We thank the patients for their assistance and Kelly Viola, ELS, for editorial support. This publication was supported in part by funding from the Mayo Clinic Center for Individualized Medicine.
WES supported by clinical data is a useful and time-saving tool in the evaluation of patients with rare and complex hereditary disorders.
Abbreviations
- WES
whole exome sequencing
- MRI
magnetic resonance Imaging
- EMG
electromyography
- EEG
electroencephalography
- KCNA1
potassium voltage-gated channel, shaker-related subfamily, member 1 gene
- EA1
episodic ataxia type 1
Footnotes
Authors' Diclosure: PT is supported by the Max Kade Foundation.
KJG has nothing to disclose
AJS reports no disclosure.
DB has nothing to disclose
DRJ has nothing to disclose
ST is employed by and receives a salary from Ambry Genetics Corp. Exome sequencing is among its commercially available tests.
DEK is employed by and receives a salary from Ambry Genetics Corp. Exome sequencing is among its commercially available tests.
ASP has nothing to disclose
OAR is partially supported by the NIH/NINDS P50 NS072187 and R01 NS078086.
ZKW is partially supported by the NIH/NINDSP50 NS072187, and the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ng SB, Turner EH, Robertson PD, et al. Nickerson, Jay Shendure Targeted Capture and Massively Parallel Sequencing of Twelve Human Exomes. Nature. 2009;461(7261):272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammer MB, Eleuch-Fayache G, Gibbs JR, et al. Exome sequencing: an efficient diagnostic tool for complex neurodegenerative disorders. Eur J Neurol. 2013;20(3):486–492. doi: 10.1111/j.1468-1331.2012.03883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazaridis KN, McAllister TM, Babovic-Vuksanovic D, et al. Implementing individualized medicine into the medical practice. Am J Med Genet C Semin Med Genet. 2014;166C(1):15–23. doi: 10.1002/ajmg.c.31387. [DOI] [PubMed] [Google Scholar]
- 5.Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. doi: 10.1038/gim.2014.154. [DOI] [PubMed] [Google Scholar]
- 6.Manto M, Marmolino D. Cerebellar ataxias. Curr Opin Neurol. 2009;22(4):419–29. doi: 10.1097/WCO.0b013e32832b9897. [DOI] [PubMed] [Google Scholar]
- 7.Graves TD, Cha YH, Hahn AF, et al. CINCH Investigators. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain. 2014;137(Pt 4):1009–1018. doi: 10.1093/brain/awu012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Dyke DH, Griggs RC, Murphy MJ, Goldstein MN. Hereditary myokymia and periodic ataxia. J Neurol Sci. 1975;25(1):109–118. doi: 10.1016/0022-510x(75)90191-4. [DOI] [PubMed] [Google Scholar]
- 9.Rajakulendran S, Schorge S, Kullmann DM, Hanna MG. Episodic ataxia type 1: a neuronal potassium channelopathy. Neurotherapeutics. 2007;4(2):258–266. doi: 10.1016/j.nurt.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 10.Eunson LH, Rea R, Zuberi SM, et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol. 2000;48(4):647–656. [PubMed] [Google Scholar]
- 11.Klein A, Boltshauser E, Jen J, Baloh RW. Episodic ataxia type 1 with distal weakness: a novel manifestation of a potassium channelopathy. Neuropediatrics. 2004;35(2):147–149. doi: 10.1055/s-2004-817921. [DOI] [PubMed] [Google Scholar]
- 12.Tan SV, Wraige E, Lascelles K, Bostock H. Episodic ataxia type 1 without episodic ataxia: the diagnostic utility of nerve excitability studies in individuals with KCNA1 mutations. Dev Med Child Neurol. 2013;55(10):959–962. doi: 10.1111/dmcn.12236. [DOI] [PubMed] [Google Scholar]
- 13.Zhu J, Alsaber R, Zhao J, Ribeiro-Hurley E, Thornhill WB. Characterization of the Kv1.1 I262T and S342I mutations associated with episodic ataxia 1 with distinct phenotypes. Arch Biochem Biophys. 2012;524(2):99–105. doi: 10.1016/j.abb.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Demos MK, Macri V, Farrell K, et al. A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov Disord. 2009;24(5):778–782. doi: 10.1002/mds.22467. [DOI] [PubMed] [Google Scholar]
- 15.Fogel BL, Lee H, Deignan JL, et al. Exome Sequencing in the Clinical Diagnosis of Sporadic or Familial Cerebellar Ataxia. JAMA Neurol. 2014;71(10):1237–1946. doi: 10.1001/jamaneurol.2014.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheffer H, Brunt ER, Mol GJ, et al. Three novel KCNA1 mutations in episodic ataxia type I families. Hum Genet. 1998;102(4):464–466. doi: 10.1007/s004390050722. [DOI] [PubMed] [Google Scholar]
- 17.Shook SJ, Mamsa H, Jen JC, Baloh RW, Zhou L. Novel mutation in KCNA1 causes episodic ataxia with paroxysmal dyspnea. Muscle Nerve. 2008;37(3):399–402. doi: 10.1002/mus.20904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video 1. “Shaking spells” and ataxic features of the proband. During his spells, there were high-amplitude and low-velocity, involuntary, ataxic movements involving the trunk and upper limbs. His gait and pointing tests were also ataxic, and the patient needed temporary assistance for mobility. Romberg's test was unremarkable. In addition mild spasticity with brisk deep tendon reflexes, more evident in his lower extremities, was noted. Babiński's sign, ankle clonus and visible myokymias were absent on neurological examination.