

Abstract
Hypertension (HTN) is a major worldwide health issue for which only a small proportion of cases have a known mechanistic etiology. Of the defined causes, none have been directly linked to heightened vasoconstrictor responsiveness, despite the fact that vasomotor tone in resistance vessels is a fundamental determinant of blood pressure. Here we reported a previously undescribed role for smooth muscle hypoxia inducible factor 1α (HIF-1α) in controlling blood pressure homeostasis. The lack of HIF-1α in smooth muscle caused hypertension in vivo and hyper-responsiveness of resistance vessels to angiotensin II (AngII) stimulation ex-vivo. These data correlated with an increased expression of angiotensin II receptor type I (ATR1) in the vasculature. Specifically, we show that HIF-1α, through peroxisome proliferator-activated receptor-γ (PPARγ), reciprocally defined ATR1 levels in the vessel wall. Indeed, pharmacological blockade of ATR1 by telmisartan abolished the hypertensive phenotype in SMC-HIF1α-KO mice.
These data revealed a determinant role of a smooth muscle HIF1α/PPARγ/ATR1 axis in controlling vasomotor responsiveness and highlighted an important pathway, the alterations of which may be critical in a variety of hypertensive-based clinical settings.
Keywords: hypertension, vascular smooth muscle, angiotensin, HIF-1α, PPAR-γ
Introduction
HTN is highly prevalent worldwide, affects ~30% of the US population, and is a major risk factor for cardiovascular, renal, and cerebrovascular disease 1, 2. The vast majority of human HTN is of undetermined etiology, with no clearly defined genetic or mechanistic cause.
Many experimental and clinical studies have established that the renin-angiotensin-system (RAS) and its downstream component AngII play a key role in cardiovascular homeostasis 3, regulating vasoconstriction, sodium reabsorption, and renal blood flow. AngII can also control the growth and proliferation of vascular smooth muscle cells (VSMC) 4, 5, and endothelial cells 6, 7. ATR1 activation mediates AngII effects on BP and osmoregulation 8, and constitutes the link between the RAS and the pathogenesis of multiple cardiovascular disorders, including hypertension, atherosclerosis and congestive heart failure 9, 10. Conversely, AngII receptor type II (ATR2) 11, a distinct isoform, is poorly expressed compared to ATR1 and may counteract the effects of ATR1 activation 7, 12, 13.
Focus on the role of RAS in HTN has been directed largely at AngII levels and the determinants thereof. Much less attention has been paid to the determinants of ATR1 levels and activity in the vasculature. Pharmacological and endogenous activators of PPARγ have been shown to decrease ATR1 expression in VSMC in vitro and to attenuate responses to AngII 14. Interestingly, dominant negative mutations in the human PPARγ gene are linked to severe early onset hypertension and insulin resistance 15, 16. Indeed, PPARγ agonists decreased BP and improved vascular function in several animal models of hypertension 16–18, clinically reducing BP in diabetics and in patients with insulin resistance 19.
The HIF pathway coordinates expression of a wide array of genes with oxygen availability and other parameters, and has a key role in multiple pathological processes 20–22. Although HIF-1α induces PPARγ expression in cardiac muscle 23, whether these pathways intersect and play a combined role in the vasculature has remained unestablished. In vitro studies show biological effects of HIF-1α in VSMC 24, 25; however, the role of HIF-1α in VSMC in vivo remains largely unknown, although oxygen content and metabolic substrates are known to significantly affect vascular tone. Here, we show that conditional deletion of HIF-1α from VSMC in mice caused increased systolic, diastolic and mean arterial BP under physiological conditions, and accentuated wall thickness and wall thickness-radius ratio in the mesenteric artery (MA), with no effects on vessel counts or heart function. Ex-vivo experiments demonstrated a specific hyper-contractility of SMC-HIF-1α-KO vs. control MA to AngII. Mechanistically, the loss of HIF-1α in VSMC reduces PPARγ expression, thereby increasing the expression of ATR1 in the vasculature, with no effect on the circulating Ang-II levels. Indeed, a pharmacological blocker of ATR1, telmisartan (telm), reverted the high BP in SMC-HIF-1α-KO to the level of WT mice, suggesting that ATR1 plays a critical role in the onset of hypertension in this mouse model. Thus, our findings underscore the critical role of VSMC HIF1α/PPARγ/ATR1 axis in BP homeostasis and highlight an important pathway the alteration of which may be critical in a variety of hypertensive-based clinical settings.
Methods
Male 8- to 14-week-old congenic SMC-HIF-1α KO mice and their sex and age matched HIF-1α WT littermates were used for all experiments. These lines were created by crossing HIF-1α floxed allele mice26, into SM22α-Cre mice (Tagln-Cre 1Her/J, The Jackson Laboratory, Bar Harbor, Me; cat. 004746; Fig. 1A). SM22α-Cre mice were also crossed with HIF-2α floxed allele mice (Jackson Labs; cat. 009674) to create SMC-HIF2α KO mice. All lines were backcrossed for at least 10 generations into the C57BL6 background. All experiments were approved by the Institutional Animal Care and Use Committee of Yale University.
Figure 1. VSMC-specific deletion of HIF-1α using Sm22α-Cre.

(A) To excise the floxed HIF-1α allele from VSMC, we used the Sm22α-Cre transgenic mouse line. Homozygous knockout mice (HIF-1α loxP/loxP / Sm22α-Cre+) were designated SMC-HIF1α-KO. Littermate mice with the HIF-1αwt/wt / Sm22α-Cre+ genotype, designated HIF1α-WT, were used as controls. PCR analysis shows HIF-1α loxp and Sm22α-Cre genes on DNA extracted from mouse-tails. (B) RT-PCR for HIF-1α mRNA in SMC-HIF- 1α-KO and HIF-1α-WT tissues and blood. (C) To define the spatio-temporal excision of HIF-1α gene, Sm-22α-Cre mice were crossed with a Cre-reporter strain mT/mG mice. As depicted, SM22α-mediated gene excision (green fluorescence, mG cassette) was restricted primarily to VSMC, and to cardiac muscle. Scale bar is 100 μm.
Left ventricular hemodynamic measurements
SMC-HIF-1α-KO and HIF-1α-WT male mice were anesthetized as aforementioned and left-ventricular hemodynamic measurements were performed in a closed-chest preparation. A 1.4 French transducer-tipped catheter (Millar Inc., Houston, TX) was placed through the right carotid artery in the LV. Pressure and volume signals were digitally recorded at 1,000 Hz (Sciences Inc). A solution of dobutamine was infused via left jugular vein at graded doses (0.125, 0.25, 0.5 and 1 ug/kg body weight), each for 3 min. LV peak pressure, including high-fidelity positive and negative dP/dt (dP/dtmax and dP/dtmin), were calculated with analysis software (LabScribe2, iWorx, CB Sciences Inc.).
Detailed methods are provided in the online supplements.
Results
Generation of SMC-HIF-1α-KO mice
Floxed HIF-1α was excised from VSMC by using a sm22α-Cre transgenic mouse line27. Progeny homozygous for HIF-1α deletion (SMC-Cre+ x HIF-1αloxP/loxP) were designated SMC-HIF-1α-KO. The two separate controls used, SMC-Cre+ x HIF-1αwt/wt (control genotype for the data presented) and SMC-Cre- x HIF-1αloxP/loxP showed no phenotypic differences.
SMC-HIF-1α-KO mice were born at expected Mendelian ratios with normal litter sizes, morphology and body weight. HIF-1α mRNA was reduced by approximately 90% in isolated SMC-HIF-1α-KO VSMC. A marked reduction was also observed in the thoracic aorta and heart, while no differences were observed in lung, kidney and blood (Fig. 1B). We also crossed Sm22α-Cre mice into double-fluorescent membrane-Tomato/membrane-Green (mT/mG) mice, a well-described indicator strain (The Jackson Laboratory, Bar Harbor, Me; cat. 007676). In the absence of Cre, cells expressed only the mT cassette (red fluorescence), while Cre+ cells expressed mG cassette (green fluorescence). As shown in Fig. 1C EGFP expression is restricted to VSMC in aorta, carotid, heart, lung, liver, kidney and brain, without evident expression in non-vascular smooth muscle or in the endothelium. These results are consistent with efficient gene deletion in the smooth muscle-rich aorta, and with the previously reported propensity of Sm22α-Cre to also direct gene deletion in cardiac muscle.
SMC-HIF-1α-KO mesenteric arteries were hyper-responsive to AngII ex-vivo
Morphometric analysis revealed significantly increased wall thickness and wall thickness/lumen radius ratios in MA (Fig. 2A–B), but not aorta and carotid (Fig. 1S) in SMC-HIF-1α-KO vs. HIF-1α-WT mice. We next focused on the functional effects of VSMC HIF-1α deletion, analyzing isolated MA using a vessel myograph system (DMT-USA, Inc.). Forces were expressed as a ratio of the maximal response to KCl (60 mM; 0.801±0.098 g and 0.700±0.097 g, SMC-HIF-1α-KO and HIF-1α-WT respectively; n=7 per group). The magnitude of contractile response to AngII in SMC-HIF-1α-KO MA was 2.25-fold higher than MA from HIF-1α-WT mice (0.785±0.150 and 0.349±0.080 respectively; n=7 per group, Fig. 2C). High doses (≥30 nM) of AngII induced the same degree of paradoxical vasodilatation in both groups (due to a previously reported tachyphylaxis effect and ATR2-mediated effects) 28. There were no differences between KO vs. control vessels in PE and 5-HT-induced contraction (10 nM to 30 μM; Fig. 2D–E); in Ach-induced endothelial nitric oxide-dependent relaxation, or in Isop-induced β2 adrenergic receptor-mediated vasorelaxation (Fig. 2F–G). These ex-vivo data strongly suggested a specific role of VSMC HIF-1α in regulating vascular contractility to AngII.
Figure 2. AngII-induced vasoconstriction was increased in SMC-HIF-1α-KO MA.
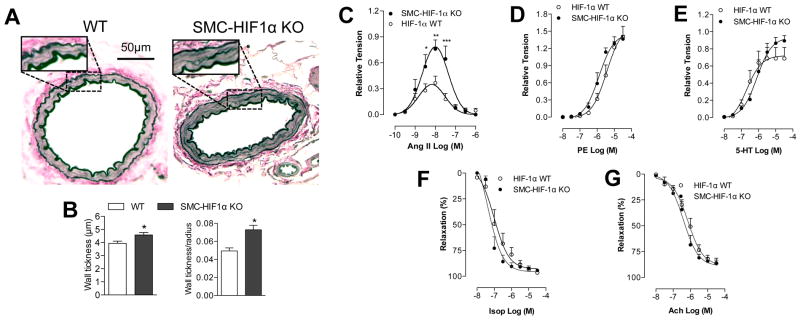
(A) Elastica van Gieson staining of mesesenteric arterery (MA) cross-sections, and (B) quantification of MA wall thickness and wall thickness/radius ratio. (C) Secondary MA function was examined with wire myograph. Cumulative concentration response curves to (C) AngII, (D) PE, (E) 5-HT, (F) Isop and (G) Ach (10−10–3×10−6 M for Ang-II and 10−8–3×10−5 M for the other agents). Data are presented as mean±S.E.M. ***p<0.001 vs. HIF-1α-WT; n=7 per group.
The loss of HIF-1α in VSMC increased systolic, diastolic and mean BP in vivo
Functional studies of the loss of HIF-1α in VSMC on BP in vivo revealed a significant higher systolic BP (SBP), diastolic BP (DBP), and MBP in SMC-HIF-1α-KO versus HIF-1α-WT mice (Fig. 3A–C). Given that our ex vivo data clearly supported a role of VSMC HIF-1α in regulating the responsiveness to AngII, we next evaluated the BP response to increasing doses of AngII, in absence and in presence of telm in vivo. Intravenous administration of AngII induced a dose-dependent increase in MBP in both groups, although MBP in HIF-1α-WT never reached the values of MBP in SMC-HIF-1α-KO mice (Fig. 3D). Telm treatment normalized basal MBP in SMC-HIF-1α-KO mice, resulting in a nearly 3-fold reduction in MBP compared to controls (Fig. 3E), consistent with our ex vivo studies on isolated MA (Fig. 2C), suggesting a clear role for ATR1 in the basal hypertension observed in SMC-HIF-1α-KO mice. Circulating levels of Ang-II determined by ELISA were not different between the two groups of mice (Fig. 3F). Taken together, these results suggest that HIF-1α is directly involved in controlling the vascular tone and thus BP under physiological conditions, by tuning the vascular reactivity to AngII through its ATR1 expression.
Figure 3. VSMC-HIF-1α was critical for BP homeostasis.
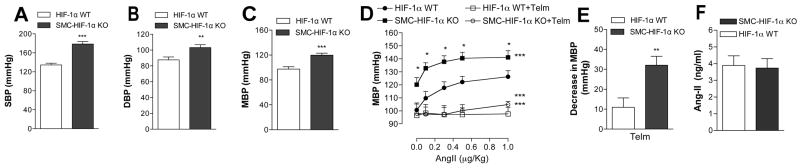
(A–C) Systolic, diastolic, and mean blood pressure (SBP, DBP and MBP) measured by a transducer-tipped catheter in the ascending aorta, were markedly higher in SMC-HIF-1α-KO mice vs. controls (SBP, n≥18 per group; DBP and MBP, n≥10 per group; ** p<-0.005, *** p<0.001, Student’s t-test). (D) Administration of telmisartan (1 mg/Kg, i.v.) resulted in an ~ 3-fold greater reduction in basal MBP in SMC-HIF-1α-KO mice vs. controls (n=8 per group; ** p<0.005). (E) Mice were injected with telmisartan (1 mg/Kg) or vehicle and BP responses to AngII (graduated i.v. dosing) were measured. MBP was significantly greater at baseline and at all concentrations of infused AngII (n=8 per group; ***, p≤0.001 for all data points by Two-way ANOVA; * p<0.05 intergroup comparisons at each AngII concentration by post-hoc t-test).
Increased SBP, DBP and MBP are not attributable to cardiac function in SMC- HIF-1α-KO mice
Developmentally, SM22α is expressed in cardiac muscle as well as VSMC. Thus, SMC-HIF-1α-KO mice exhibit HIF-1α excision also in the heart (Fig. 1B). Heart/body weight ratio, an index of cardiac hypertrophy, was increased in SMC-HIF-1 α-KO vs. control mice (Fig. 4A), with no evidence of fibrosis (Fig. S2). Echocardiographic analysis of lightly sedated mice corroborated hypertrophy, showing increased IVS thickness in the absence of HIF-1α, whereas LVDd and LVSd, ejection fraction (EF) and FS were unchanged between KO and control mice (Fig. 4B–C). Hemodynamic studies at baseline and in response to dobutamine, a β-agonist, showed no differences in the dP/dTmax or dP/dtmin (maximum rates of left ventricular pressure rise or decrease - respectively), or in heart rates between these groups (Fig. 4E–G), establishing that the increase in BP in SMC-HIF-1α-KO mice is not due to altered cardiac function, but is likely due to increased vascular tone. Consistent with this, we have previously reported that cardiac myocyte-specific HIF-1α gene deletion did not increase systemic BP, though did induce mild reductions in cardiac contractile indices 26.
Figure 4. Hypertension was not attributable to changes in cardiac function in SMC-Hif1α-KO mice.

(A) SMC-HIF-1α KO mice had cardiac hypertrophy leading to increased heart-to-body weight ratios vs. HIF-1α-WT. (B–D) Echocardiographic analysis in SMC-HIF-1α-KO and HIF-1α-WT mice showed a significant increase in IVS, without any effect on LVSd, LVDd, or in cardiac function measured as EF and FS. (E) Representative M-mode echocardiogram. Effect of dobutamine on first derivate of peak changes ventricular pressure (F) dP/dtmin, (G) dP/dTmax and (H) heart rate in SMC-HIF-1α-KO and control mice (n=10 per group).
VSMC ATR1 expression is tightly controlled by a HIF-1α-PPARγ axis
ATR1, but not ATR2 (data not shown) mRNA was markedly higher in isolated VSMC, thoracic aorta and hearts from SMC-HIF-1α-KO mice as compared to controls (Fig. 5A). ATR1 expression was significantly increased in SMC-HIF-1α-KO aortas compared to controls as shown by Western blot (Fig. 5B–C) and confirmed by immunostaining for ATR1 on aorta cross-sections (Fig. S3). Immunostaining of MA cross-sections showed a concomitant increase of ATR1 in the absence of HIF-1α (Fig. 5D), suggesting that a reciprocal relationship between HIF-1α and VSMC-ATR1 expression was the fundamental underlying mechanism for the vascular hyper-reactivity to AngII of SMC-HIF-1α-KO MA. Ang-II-induced signaling was unchanged in VSMC isolated from SMC-HIF1α-KO vs. HIF1α-WT thoracic aorta (Fig. S4), strongly suggesting that increased ATR1 expression, and not an intrinsic alteration in Ang-II-induced signaling, was primarily responsible for the hyper-contractility to Ang-II.
Figure 5. The loss of HIF-1α in VSMC induced PPARγ-mediated increase of ATR1 expression.
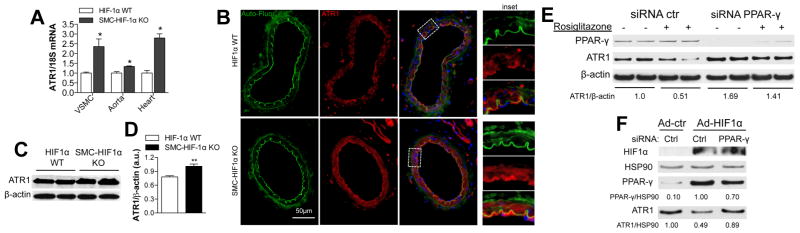
(A) Q-RT-PCR demonstrated a significant increase in ATR1 mRNA in VSMC, aorta, and heart from SMC-HIF-1α KO vs. HIF-1α WT mice (18S normalized). (B–C) Western blot (WB) with quantification of ATR1 and β-actin expression in thoracic aorta from both genotypes. n=5, ** p<0.01. (D) Representative immunofluorescent staining of HIF-1α-WT and SMC-HIF-1α-KO MA cross-sections for ATR1 (red) and DAPI (blue). Auto-fluorescence of the elastic lamina is in green. (E) Q-RT-PCR showing marked reduction of PPARγ mRNA in thoracic aorta from SMC-HIF-1α-KO vs. HIF-1α-WT mice (18S normalized). (F) anti-PPARγ-siRNA and ctr-siRNA treated hVSMC were incubated with vehicle or rosiglitazone (50 μM; 12h), and PPARγ and ATR1 expression evaluated by WB analysis (β-actin loading control). The densitometric ATR1/β-actin ratio for control treated cells (vehicle without rosiglitazone and with control siRNA) is set at 1.00, and all other densitometric ratios are relative to this control baseline ratio. Activation of PPARγ by rosiglitazone decreased ATR1, an effect prevented by siRNA knockdown of PPARγ. (G) WB analysis for HIF-1α, ATR1, PPARγ and HSP90 of control and PPARγ-siRNA treated hVSMC following by infection with Ad-HIF-1α or Ad-ctr. HSP90 was used as loading control (PPARγ/HSP90 and ATR1/HSP90 densitometry ratios are relative to control ratios set at 1.00). WB were representative of four independent experiments. ** P<0.01 vs. HIF-1α WT.
It has been previously reported that PPARγ, but not PPARα activators reduced ATR1 mRNA expression in VSMC in culture 14, and that PPARγ over-expression in pathological cardiac hypertrophy is HIF-1α-dependent 23. Hypothesizing that the reciprocal relationship we observed between HIF-1α and ATR1 levels is mediated via HIF-1α effects on PPARγ, we examined PPARγ expression. PPARγ mRNA was markedly reduced in VSMC lacking HIF-1α versus control (Fig. 5E). Exploring this further, we showed in human VSMC (hVSMC) that the PPARγ agonist rosiglitazone reduced ATR1expression, while siRNA knockdown of PPARγ significantly increased ATR1 levels, preventing the rosiglitazone effects (Fig. 5F). These data confirmed a reciprocal biological link between PPARγ activation and ATR1 expression.
Ad-HIF-1α, expressing a stabilized form of HIF-1α, markedly increased PPARγ protein levels and concomitantly decreased ATR1 levels (up to 50%) in hVSMC. PPARγ knockdown with siRNA abolished the HIF-1α-driven decrease in ATR1 levels, thus demonstrating that HIF-1α effects on ATR1 levels were mediated via PPARγ. Together, these data established the functional importance of a previously unknown HIF-1α/PPARγ/ATR1 axis in VSMC.
Discussion
Despite numerous reports documenting the important role of HIF-1α in transcriptional control of angiogenesis during development and pathological conditions 21, 29–32, the contribution of VSMC-HIF-1α to vascular homeostasis in vivo has not yet been defined. Here we show that HIF-1α is a critical regulator of systemic BP through a HIF-1α/PPAR-γ/ATR1 axis. This conclusion is supported by data showing that the loss of HIF-1α in VSMC increased both the contractility of MA to AngII ex-vivo, and BP in vivo. Telm abolished the latter effect, indicating an important role of ATR1 in the HTN phenotype of SMC-HIF-1α-KO mice. Further, the role of HIF-1α in controlling ATR1 levels was corroborated by in vitro experiments showing that HIF-1α directly controlled PPAR-γ expression, which in turn negatively regulated ATR1 levels. These effects were inhibited by siRNA knockdown of PPAR-γ, and simulated by rosiglitazone, suggesting that these pathways are genetically epistatic 33. Collectively these results support a key role for HIF-1α/PPAR-γ/ATR1 axis in controlling vascular contractility to AngII and BP in vivo.
These findings also underscore the importance of HIF-pathway in vascular cells. Oxygen availability is a critical regulator of vascular tone, with differential effects on pulmonary versus systemic vascular beds 34. Although the link between oxygen and vascular tone is complex, based on our data we propose that oxygen sensing by HIF in VSMC plays a critical role in fine-tuning these direct vascular responses to oxygen levels. As a transcriptional pathway coordinating gene expression with oxygen availability, HIF is not likely to mediate acute/immediate responses to changes in oxygen tension, but is more likely to alter VSMC gene expression in a manner that determines the magnitude of contractile responses of these cells to specific mediators of vascular tone, as we show here for AngII. Interestingly, the HIF-pathway has been shown to be responsive, directly or indirectly, to other factors the vasculature is exposed to in addition to oxygen tension, including reactive oxygen species, nitric oxide and glucose levels. A complication of diabetes is the abnormal response to hypoxia due to an impaired stabilization of HIF-1α under hyperglycemic conditions 35. Considering that diabetes as well as metabolic syndrome are risk factors for the development of HTN, it is reasonable to consider that in this scenario, HIF-1α/PPARγ/ATR1 axis here delineated could be a critical mechanism in the pathogenesis of vascular dysfunction. Consistent with our findings, a recent study reported that mice expressing a smooth muscle-specific dominant-negative PPAR-γ mutant showed increased vascular constriction to Ang-II in mesenteric arteries36.
As with many genes, the biological roles of HIF-1α and HIF2α vary dependent upon cell type. Although pulmonary HTN was reported in endothelial-HIF2α knockout mice, others and we did not observe any systemic HTN in endothelial-HIF-KO mice. Consistent with functionally distinct roles of HIF-1α and HIF2α, we did not observe HTN in SMC-HIF-2α-KO mice (Fig. S5). Despite sm22α-Cre directed excision of HIF-1α also in cardiac muscle, our data clearly demonstrated no cardiac contribution to the HTN phenotype, consistent with our previous study on cardiac-specific-HIF-1α KO mice, which exhibited instead a mildly decreased SBP26.
Finally, experiments on isolated vascular segments from SMC-HIF-1α KO mice showed conclusively that loss of HIF-1α specifically from VSMC accentuated vessel contraction in response to AngII, independent of any cardiac effects, and was linked to increased ATR1 expression. HIF-1α deletion did not alter downstream Ang-II-induced signaling, but this data was limited to aortic VSMC. Future studies of signaling in VSMC isolated from MA and other resistance vessels are thus needed. Interestingly, similar to endothelial-HIF-1α knockout mice 32, the loss of HIF-1α in VSMC did not alter vessel density in multiple tissues studied at baseline (Fig. S6), suggesting hypovascularity did not contribute to the hypertensive phenotype. Although MA thickening was seen, this wasn’t seen in other vessels studied and it remains unclear if this finding was primary to loss of HIF-1α or secondary to the HTN.
Focusing on the parameter of vascular tone and its determinants, it is possible that in addition to increased ATR1 levels and AngII responsiveness, the loss of other HIF-1α associated functions in VSMC contribute to the increased vascular tone in vivo. These might include altered expression of HIF-1α-responsive genes encoding vasoactive autocrine or paracrine factors, or even changes in levels of vasoactive metabolites as a consequence of altered VSMC metabolism caused by the loss of HIF-1α or changes in the expression of genes encoding calcium-handling proteins or other ion-associated proteins. Whereas we cannot exclude contributions by these alternative mechanisms, we did not observe differences in PE or 5-HT-induced contraction in vessel segments from SMC-HIF-1α KO mice.
Perspective
In conclusion, here we defined an important previously unknown role of VSMC HIF-1α in controlling BP homeostasis, through a mechanism involving HIF-1α/PPARγ/ATR1 axis. These findings add to our understanding of how oxygen levels can affect vascular tone in physiological and pathological conditions such as diabetes and metabolic syndrome in which the axis HIF-1α/PPARγ/ATR1 could be a potential link between hyperglycemia and hypertension. This study clearly uncovered an important link between components of HIF-signaling pathway and clinical HTN, thus warranting further investigation in this area.
Supplementary Material
Novelty and Significance.
1. What is new?
This is the first report that a) the major oxygen-sensing transcription factor HIF-1α in smooth muscle is an important regulatory pathway for blood pressure control, and that b) HIF-1α intrinsically controls vascular smooth muscle cell responsiveness to angiotensin II by reciprocally regulating the expression of ATR1 in smooth muscle cells, through a PPARγ mediated effect.
2. What is relevant?
The HIF-pathway is a major cellular environmental sensing pathway that mediates responses to a variety of signals and parameters, including metabolic conditions, reactive oxygen species, glucose levels, and others, in addition to oxygen levels. The involvement of HIF-1α, via PPARγ, in the control of vascular tone provides an important link between these various environmental parameters and control of blood pressure. We propose that this link may play an important role in the alterations in blood pressure noted in clinical metabolic syndromes and obesity, and that alterations in this HIF-1α-PPARγ-ATR1 axis might underlie some cases of clinical hypertension.
Summary.
We demonstrate that the loss of HIF-1α in smooth muscle causes hypertension in vivo and hyper-responsiveness of resistance vessels to angiotensin II (AngII) stimulation ex-vivo. These data correlated with an increased expression of angiotensin II receptor type I (ATR1) in the vasculature through HIF-1α-PPARγ axis.
Acknowledgments
Sources of Funding. This work was funded by NIH grants HL075616-02, HL64001, and the Leducq foundation (FJG), and by an AHA SDG (ADL).
Footnotes
Conflict of Interest/Disclosure Statement.
None.
Disclaimer: The manuscript and its contents are confidential, intended for journal review purposes only, and not to be further disclosed.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2011 update: A report from the american heart association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sliwa K, Stewart S, Gersh BJ. Hypertension: A global perspective. Circulation. 2011;123:2892–2896. doi: 10.1161/CIRCULATIONAHA.110.992362. [DOI] [PubMed] [Google Scholar]
- 3.Savoia C, Burger D, Nishigaki N, Montezano A, Touyz RM. Angiotensin ii and the vascular phenotype in hypertension. Expert Rev Mol Med. 2011;13:e11. doi: 10.1017/S1462399411001815. [DOI] [PubMed] [Google Scholar]
- 4.Geisterfer AA, Peach MJ, Owens GK. Angiotensin ii induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ Res. 1988;62:749–756. doi: 10.1161/01.res.62.4.749. [DOI] [PubMed] [Google Scholar]
- 5.Paquet JL, Baudouin-Legros M, Brunelle G, Meyer P. Angiotensin ii-induced proliferation of aortic myocytes in spontaneously hypertensive rats. J Hypertens. 1990;8:565–572. doi: 10.1097/00004872-199006000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Lucius R, Gallinat S, Busche S, Rosenstiel P, Unger T. Beyond blood pressure: New roles for angiotensin ii. Cell Mol Life Sci. 1999;56:1008–1019. doi: 10.1007/s000180050490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoll M, Steckelings UM, Paul M, Bottari SP, Metzger R, Unger T. The angiotensin at2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J Clin Invest. 1995;95:651–657. doi: 10.1172/JCI117710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasaki K, Yamano Y, Bardhan S, Iwai N, Murray JJ, Hasegawa M, Matsuda Y, Inagami T. Cloning and expression of a complementary DNA encoding a bovine adrenal angiotensin ii type-1 receptor. Nature. 1991;351:230–233. doi: 10.1038/351230a0. [DOI] [PubMed] [Google Scholar]
- 9.Dzau VJ, Antman EM, Black HR, Hayes DL, Manson JE, Plutzky J, Popma JJ, Stevenson W. The cardiovascular disease continuum validated: Clinical evidence of improved patient outcomes: Part i: Pathophysiology and clinical trial evidence (risk factors through stable coronary artery disease) Circulation. 2006;114:2850–2870. doi: 10.1161/CIRCULATIONAHA.106.655688. [DOI] [PubMed] [Google Scholar]
- 10.Stegbauer J, Coffman TM. New insights into angiotensin receptor actions: From blood pressure to aging. Curr Opin Nephrol Hypertens. 2011;20:84–88. doi: 10.1097/MNH.0b013e3283414d40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kambayashi Y, Bardhan S, Takahashi K, Tsuzuki S, Inui H, Hamakubo T, Inagami T. Molecular cloning of a novel angiotensin ii receptor isoform involved in phosphotyrosine phosphatase inhibition. J Biol Chem. 1993;268:24543–24546. [PubMed] [Google Scholar]
- 12.Unger T, Culman J, Gohlke P. Angiotensin ii receptor blockade and end-organ protection: Pharmacological rationale and evidence. J Hypertens Suppl. 1998;16:S3–9. [PubMed] [Google Scholar]
- 13.Nakajima M, Hutchinson HG, Fujinaga M, Hayashida W, Morishita R, Zhang L, Horiuchi M, Pratt RE, Dzau VJ. The angiotensin ii type 2 (at2) receptor antagonizes the growth effects of the at1 receptor: Gain-of-function study using gene transfer. Proc Natl Acad Sci U S A. 1995;92:10663–10667. doi: 10.1073/pnas.92.23.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeda K, Ichiki T, Tokunou T, Funakoshi Y, Iino N, Hirano K, Kanaide H, Takeshita A. Peroxisome proliferator-activated receptor gamma activators downregulate angiotensin ii type 1 receptor in vascular smooth muscle cells. Circulation. 2000;102:1834–1839. doi: 10.1161/01.cir.102.15.1834. [DOI] [PubMed] [Google Scholar]
- 15.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O’Rahilly S. Dominant negative mutations in human ppargamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 16.Ketsawatsomkron P, Pelham CJ, Groh S, Keen HL, Faraci FM, Sigmund CD. Does peroxisome proliferator-activated receptor-gamma (ppar gamma) protect from hypertension directly through effects in the vasculature? J Biol Chem. 2010;285:9311–9316. doi: 10.1074/jbc.R109.025031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan MJ, Didion SP, Mathur S, Faraci FM, Sigmund CD. Ppar(gamma) agonist rosiglitazone improves vascular function and lowers blood pressure in hypertensive transgenic mice. Hypertension. 2004;43:661–666. doi: 10.1161/01.HYP.0000116303.71408.c2. [DOI] [PubMed] [Google Scholar]
- 18.Walker AB, Chattington PD, Buckingham RE, Williams G. The thiazolidinedione rosiglitazone (brl-49653) lowers blood pressure and protects against impairment of endothelial function in zucker fatty rats. Diabetes. 1999;48:1448–1453. doi: 10.2337/diabetes.48.7.1448. [DOI] [PubMed] [Google Scholar]
- 19.Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913. doi: 10.1016/s0140-6736(02)11911-8. [DOI] [PubMed] [Google Scholar]
- 20.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei L, Mason S, Liu D, Huang Y, Marks C, Hickey R, Jovin IS, Pypaert M, Johnson RS, Giordano FJ. Hypoxia inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von hippel-lindau protein. Molecular and cellular biology. 2008;11:3790–3803. doi: 10.1128/MCB.01580-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 23.Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, Perriard JC, Larsen T, Pedrazzini T, Krek W. Activation of a hif1alpha-ppargamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512–524. doi: 10.1016/j.cmet.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Ray JB, Arab S, Deng Y, Liu P, Penn L, Courtman DW, Ward ME. Oxygen regulation of arterial smooth muscle cell proliferation and survival. Am J Physiol Heart Circ Physiol. 2008;294:H839–852. doi: 10.1152/ajpheart.00587.2007. [DOI] [PubMed] [Google Scholar]
- 25.Gao W, Ferguson G, Connell P, Walshe T, Murphy R, Birney YA, O’Brien C, Cahill PA. High glucose concentrations alter hypoxia-induced control of vascular smooth muscle cell growth via a hif-1alpha-dependent pathway. J Mol Cell Cardiol. 2007;42:609–619. doi: 10.1016/j.yjmcc.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Hickey RP, Yeh JL, Liu D, Dadak A, Young LH, Johnson RS, Giordano FJ. Cardiac myocyte-specific hif-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. Faseb J. 2004;18:1138–1140. doi: 10.1096/fj.04-1510fje. [DOI] [PubMed] [Google Scholar]
- 27.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. Lrp: Role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 28.Savoia C, Touyz RM, Volpe M, Schiffrin EL. Angiotensin type 2 receptor in resistance arteries of type 2 diabetic hypertensive patients. Hypertension. 2007;49:341–346. doi: 10.1161/01.HYP.0000253968.95136.b8. [DOI] [PubMed] [Google Scholar]
- 29.Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;342:626–633. doi: 10.1056/NEJM200003023420904. [DOI] [PubMed] [Google Scholar]
- 30.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E. Role of hif-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 31.Semenza GL. Hif-1: Upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS. Loss of hif-1alpha in endothelial cells disrupts a hypoxia-driven vegf autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–495. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 33.Cordell HJ. Epistasis: What it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- 34.Huang Y, Giordano FJ. Chapter 13. Oxygen as a direct and indirect biological determinant in the vasculature. Methods Enzymol. 2008;444:285–304. doi: 10.1016/S0076-6879(08)02813-9. [DOI] [PubMed] [Google Scholar]
- 35.Bento CF, Pereira P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia. 2011;54:1946–1956. doi: 10.1007/s00125-011-2191-8. [DOI] [PubMed] [Google Scholar]
- 36.Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ, Grobe JL, Faraci FM, England SK, Sigmund CD. Ppargamma regulates resistance vessel tone through a mechanism involving rgs5-mediated control of protein kinase c and bkca channel activity. Circ Res. 2012;111:1446–1458. doi: 10.1161/CIRCRESAHA.112.271577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.