

Abstract
Cancers are composed of populations of cells with distinct molecular and phenotypic features, a phenomenon termed intra-tumor heterogeneity (ITH). ITH in lung cancers has not been well studied. We applied multi-region whole exome sequencing (WES) on 11 localized lung adenocarcinomas. All tumors showed clear evidence of ITH. On average, 76% of all mutations and 20/21 known cancer gene mutations were identified in all regions of individual tumors suggesting single-region sequencing may be adequate to identify the majority of known cancer gene mutations in localized lung adenocarcinomas. With a median follow-up of 21 months post-surgery, 3 patients have relapsed and all 3 patients had significantly larger fractions of subclonal mutations in their primary tumors than patients without relapse. These data indicate larger subclonal mutation fraction may be associated with increased likelihood of postsurgical relapse in patients with localized lung adenocarcinomas.
Intra-tumor heterogeneity may have impacts on tumor biopsy strategy, characterization of actionable targets, treatment planning, and drug resistance (1–6). ITH has recently been elucidated in substantial detail in several cancer types using next-generation sequencing (NGS) approaches (7–14). Recent evidence supports a model of branched evolution leading to variable ITH in different tumors (9, 13, 15, 16). Studies in clear cell renal carcinoma (ccRCC) have demonstrated substantial ITH, with the majority of mutations in known cancer genes confined to spatially separated tumor regions except for VHL loss being a ubiquitous event (16, 17). These data suggest that a single biopsy may be inadequate for identifying all cancer gene mutations from a tumor, thus presenting an incomplete view of potential targets for therapy. Critically, the extent to which these observations in ccRCC apply to other solid tumors is currently not clear.
To characterize ITH in localized lung adenocarcinomas, we applied multi-region WES on 48 tumor regions from 11 resected lung adenocarcinomas (8 stage I, 2 stage II and one stage III tumors, tumor size 2 – 4.6 cm), who had surgery with curative intent (Fig. 1 and Table S1). WES was conducted at mean depth of 277×. In total, 7,269 mutations were identified and 7,026 (97%) somatic mutations were validated by a separate bespoke capture sequencing experiment at mean depth of 863x (Table S2). The numbers of mutations varied substantially between tumors (Fig. S1), but no significant correlations were identified between mutation burden and age, gender, tumor size, lymph node status or smoking status.
Fig. 1.
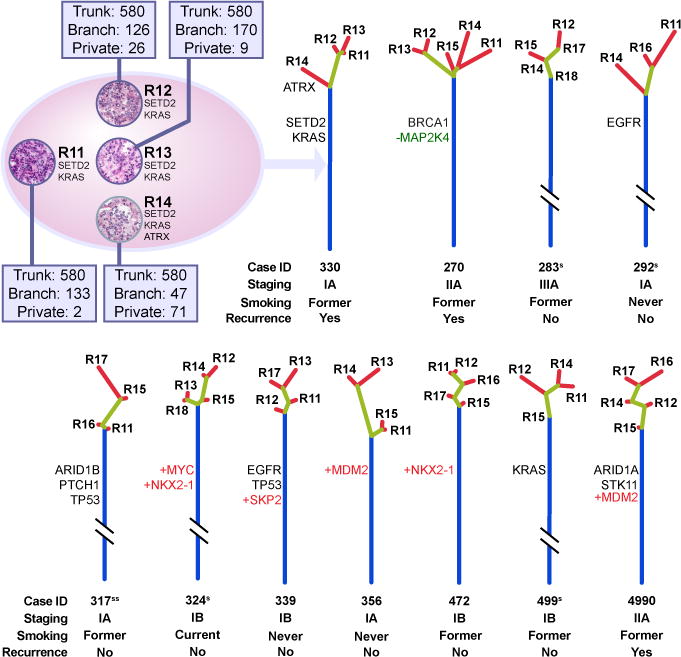
Assessment of ITH of 11 lung adenocarcinomas by multi-region sequencing. An example of regional mutation distribution (case 330) of resected tumors (represented by the ellipsis) is shown at the up left corner. Mutated cancer genes are indicated to the right of representative H&E image of each sequenced tumor region. Numbers of trunk, branch and private branch mutations for each region are indicated in associated windows. A phylogenetic tree was generated from all validated mutations utilizing Wagner parsimony method in PHYLIP. Blue, yellow and red lines represent trunk, branch and private branches respectively. Trees are anchored at germline DNA sequence obtained from peripheral blood of the relevant patients. Known cancer gene mutations are mapped to the trunks and branches as indicated. Point mutations, amplifications and deletions of known cancer genes are presented as black, +red and −green respectively. Trunk and branch lengths are proportional to the numbers of mutations acquired on the corresponding trunk or branch. Note: 5 tumors have their trunk lengths reduced to 10%S or 2%SS of original length for visualization purposes.
A useful approach when considering ITH is to depict a given tumor as a tree structure with the trunk representing ubiquitous mutations present in all regions of the tumor, branches representing heterogeneous mutations present in only some regions of the tumor and private branches representing mutations that are present only in one region of the tumor – analogous to a phylogenetic tree. Placement of mutations on trunks versus branches reflects relative molecular time of acquisition, with branch mutations occurring, by definition, subsequent to trunk mutations. We applied this approach to multi-region sequencing data from these 11 lung adenocarcinomas. Evidence for ITH was found in each tumor studied. On average, 76% of all mutations were detected in all regions of the same tumors. However, the phylogenetic structure varied considerably between tumors (Fig. 1). We then characterized known cancer gene mutations, defined as nonsynonymous mutations identical to those previously reported in known cancer genes (18–23) or truncating mutations in known tumor suppressor genes, in the context of the derived phylogenetic tree structures. Thirteen of 14 known cancer gene point mutations were mapped to the trunks of the phylogenetic trees (Fig. 1, Table S3), indicating these mutations were acquired relatively early during evolution of these 11 tumors. In contrast to ccRCC, these data suggest that single-region sampling may be sufficient to identify the majority of known cancer gene mutations in localized lung adenocarcinomas.
We were also able to evaluate copy number changes relative to ITH. In contrast to ccRCC (16, 17, 24), we did not observe substantial difference in large-scale chromosome aberrations (Fig. S2A) and the log2 ratio profiles were similar between different regions within the same tumors (Fig. S2B and Table S4). Further, amplification or deletion of known cancer genes (22) as well as their relative placement on the phylogenetic trees were delineated for these 11 lung adenocarcinomas. All of these events were mapped to the trunks of the phylogenetic trees (Fig. 1) suggesting that, like known cancer gene point mutations discussed above, amplification/deletion of known cancer genes were also early molecular events for these 11 tumors. Previous work in breast cancer also suggested that known cancer gene mutations were relatively early genetic events shared by all subclones of individual breast cancers (13). Taken together, these results indicate that different cancer types may have different relative timing of acquisition of cancer gene mutations. Further, the data would suggest in this subset of lung adenocarcinomas, there are likely mutations in non-canonical cancer genes that drive tumor development and subclonal divergence.
With a median follow-up of 21 months post surgery at the time of this report, 3 patients have had disease relapse. These 3 patients had significantly larger proportion of subclonal non-trunk mutations (branch plus private branch mutations) in their primary tumors than patients without relapse (average 40% in relapsed patients versus 17% in patients without relapse, p=0.006 by t test, Fig. 1). Although the sample size is small, these findings suggest the possibility that subclonal mutations may be important for cancer progression and that larger subclonal mutation fraction may be associated with an increased likelihood of post-surgical relapse in this subset of lung adenocarcinoma patients.
Analysis of NGS data relies heavily upon adequate sequencing depth to make high accuracy consensus base calls. We compared our WES data (average sequencing depth 277×) to deep sequencing data (average sequencing depth 863×) employed in validation with regards to known cancer gene mutation identification. In the tumor 499, a canonical KRAS p.G12C mutation was detected in only 1 of 4 tumor regions at exome depth but detected in all four tumor regions at increased sequencing depth (Table S2). Extending this analysis, we then compared deep sequencing data to WES data in defining ITH. The result showed many branch and private branch mutations defined by WES were detectable in all regions of individual tumors with increasing sequencing depth (Fig. 2). Taken together, these results indicate that considerable depth of sequencing will be necessary to detect cancer gene mutations and accurately characterize ITH of lung adenocarcinomas.
Fig. 2.
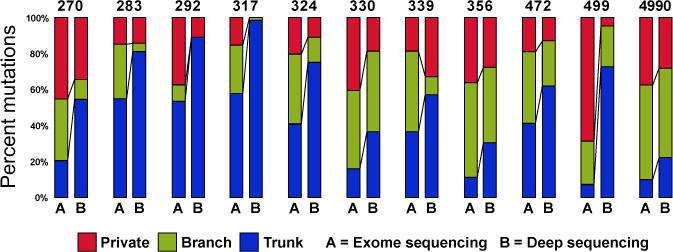
Distribution of trunk, branch and private branch mutations defined by exome sequencing (average sequencing depth of 277×) versus deep sequencing (average sequencing depth of 863×). Only validated mutations meeting the following criteria are included: total counts in tumor DNA≥100; total counts in germ line DNA≥50; variant allele frequency (VAF) ≥5% in tumor DNA and VAF=0 in germ line DNA.
Next, we analyzed the mutational spectrum of these 11 lung adenocarcinomas. Consistent with previous studies (18–20, 25), different mutation spectra were observed in smokers and non-smokers. Three never-smokers (292, 339 and 356) showed C>T-predominant mutation profiles. Three former smokers who quit more than 20 years ago (270, 472 and 4990) and one former smoker who had a 25 pack-year (pack year = number of packs per day x number of years) history of smoking and quit 6 years ago (283) also showed C>T-predominant mutation profiles, as in non-smokers. Two former smokers who had more than a 50 pack-year history of smoking and quit 5 years ago (317 and 499) and one former smoker, who had a 25 pack-year history of smoking and quit only 2.5 years ago (330) showed C>A-predominant mutation profiles consistent with the mutation profile of cigarette smoke exposure. The only current smoker, who had a 20 pack-year history of smoking, but had cut down to 2 cigarettes a day at the time of cancer diagnosis (324) showed an equivalent portion of C>T (26%) versus C>A (21%) substitutions in her tumor (Fig. 3A). These results indicate tumor mutation spectra in former smokers reflect not only quantity of smoking exposure, but also time since smoking cessation.
Fig. 3.
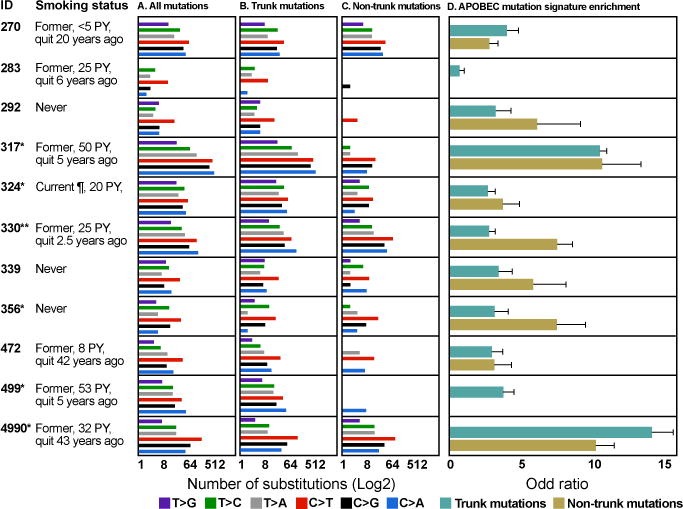
Mutation spectrum of the 11 lung adenocarcinomas. (A) Mutation spectrum of all validated mutations. (B) Mutation spectrum of trunk mutations. (C) Mutation spectrum of non-trunk mutations. The difference of mutation spectrum between trunk and non-trunk mutations in each patient was evaluated with Fisher’s exact test and significant p values are shown as *(P<0.05) and **(P<0.01). (D) APOBEC mutation signature enrichment odds ratio for trunk and non-trunk mutations. 95% confidence intervals for Fisher’s exact test are indicated. PY: pack year. ¶: Patient had cut down to 2 cigarettes a day at the time of cancer diagnosis.
We next compared the mutational spectrum of trunk mutations versus non-trunk to explore the relative contribution of mutational processes over time. Significant differences in mutational spectrum were observed in 6 tumors indicating that specific mutational processes were likely operative at different times during development of these tumors (Fig. 3B and 3C). Of interest, two former smokers (317 and 330) and the current smoker (324) showed significant differences between trunk and non-trunk mutation spectrum with a shift from smoking-associated C>A transversions in trunk mutations to non-smoker-associated C>T transitions in non-trunk mutations.
Recent evidence has suggested APOBEC activity as a major source for C>T and C>G mutations (12, 26). We therefore investigated if there is evidence of an APOBEC mutational process in this subset of lung adenocarcinomas. On average, 28% of all mutations had a specific substitution pattern (C>T/G at TpCpW sites, where W is A or T), consistent with an APOBEC-mediated process (Fig. S3). APOBEC mutation signature enrichment was found to be more pronounced for non-trunk mutations compared to trunk mutations in 7 of the 11 patients, however this difference was statistically significant only for case 330 (Fig. 3D). These data suggest that an APOBEC-like process is contributing substantially to the mutations found in this subset of lung adenocarcinomas and that there is a trend towards this process being more pronounced in later, subclonal mutations – further highlighting the dynamic nature of mutational processes in play.
Substantial variation in the allele frequency of somatic mutations within each individual tumor region from a given tumor was observed in this set of lung adenocarcinomas (Fig. S4A). To more formally characterize subclonal fraction within each tumor region, we employed the ABSOLUTE algorithm (27). These analyses demonstrated that at least 29 out of 48 individual tumor regions showed evidence of intra-regional subclonal populations. The distribution of clonal and subclonal mutations was different among the sampled regions within the same tumors in some patients (Fig. S4B) further suggesting single biopsy analysis would be inadequate to fully represent ITH in these tumors.
To explore the implications of how ITH assessment might be reduced to practice, we repeated the ABSOLUTE analysis on the combined sequencing data from all tumor regions of each patient to assess the global ITH on a per patient level, defined by the relative proportion of subclonal mutations. Similar to the phylogenetic analyses, all 3 patients with relapsed disease had larger subclonal fractions in their primary tumors (average 41% in patients with relapse versus 24% in patients without relapse, p=0.045 by t test, Fig. S5A). Using a complimentary Bayesian Dirichlet process (13) on the per patient combined data showed the same trend (average subclonal mutations 66% in patients with relapse versus 36% in patients without relapse, p=0.035 by t test, Fig. S5B). These results suggest that a measure of overall subclonal fraction may be of interest from a prognostic standpoint in this population of patients.
Resectable localized disease accounts for 30–50% of all non-small cell lung cancers with increasing prevalence as screening is more widely implemented (28–30). Given that this subset of patients has tumors surgically resected as standard of care, there is an opportunity to confirm these preliminary observations by deep sequencing multi-region samples obtained from resected tumors. The question of whether sequencing targeted cancer gene panels versus whole exome will yield sufficient mutation data for meaningful analyses and the most appropriate algorithms for analyses will need to be addressed in order to fully test if the clinical correlation suggested in these data is born out in larger patient cohorts.
Evidence of marked regional ITH in ccRCC suggested substantial challenges to personalized oncology based on single tumor biopsy to portray the mutational landscape. This study, however, provides evidence that ITH patterns may be different between cancer types. With the caveat of limited sample size fully acknowledged, these data suggest that whilst multi-region sampling is needed to fully assess ITH complexity, single biopsy analysis at appropriate depth might be sufficient to identify the majority of known cancer gene mutations in this subset of lung adenocarcinomas. Studies in much larger cohorts, ideally with comprehensive clinical annotation and repeat biopsy at relapse are needed to fully understand the clinical impact of ITH and insights afforded by these types of analyses. Furthermore, extension to epigenetic and phenotypic assessment through regional DNA methylation, chromatin state and RNA/protein expression studies over time and under treatment are needed to fully understand the impact of ITH on the biology of the cancer itself and its impact on the clinical phenotype of cancer patients.
Supplementary Material
Acknowledgments
This study was supported by the Cancer Prevention and Research Institute of Texas (R120501), UT Systems Stars Award (PS100149), the Welch Foundation Robert A. Welch Distinguished University Chair Award (G-0040), Department of Defense PROSPECT grant (W81XWH-07-1-0306), the UT Lung Specialized Programs of Research Excellence grant (P50CA70907), the MD Anderson Cancer Center Support Grant (CA016672), NIH T32 Research Training in Academic Medical Oncology Grant (CA-009666) and the A. Lavoy Moore Endowment Fund. The authors would like to thank Drs. Lynda Chin and Roeland Verhaak for constructive discussions. The DNA sequencing data were submitted to dbGaP (Accession number is pending).
Footnotes
List of supplementary materials
Patients and Methods
References (31–44)
References and Notes
- 1.Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med. 2012 Mar 28;4:127ps10. doi: 10.1126/scitranslmed.3003854. [DOI] [PubMed] [Google Scholar]
- 2.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012 Oct 1;72:4875. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swanton C, et al. Predictive biomarker discovery through the parallel integration of clinical trial and functional genomics datasets. Genome Med. 2010;2:53. doi: 10.1186/gm174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horswell S, Matthews N, Swanton C. Cancer heterogeneity and “The Struggle for Existence”: Diagnostic and analytical challenges. Cancer Lett. 2012 Nov 8; doi: 10.1016/j.canlet.2012.10.031. [DOI] [PubMed] [Google Scholar]
- 5.Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013 Feb 19;108:479. doi: 10.1038/bjc.2012.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders NA, et al. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol Med. 2012 Aug;4:675. doi: 10.1002/emmm.201101131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navin N, et al. Inferring tumor progression from genomic heterogeneity. Genome research. 2010 Jan;20:68. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011 Apr 7;472:90. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson K, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011 Jan 20;469:356. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 10.Sottoriva A, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proceedings of the National Academy of Sciences of the United States of America. 2013 Mar 5;110:4009. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell PJ, et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2008 Sep 2;105:13081. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nik-Zainal S, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012 May 25;149:979. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012 May 25;149:994. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yachida S, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010 Oct 28;467:1114. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009 Oct 8;461:809. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 16.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012 Mar 8;366:883. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerlinger M, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature genetics. 2014 Feb 2; doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imielinski M, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012 Sep 14;150:1107. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008 Oct 23;455:1069. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Govindan R, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012 Sep 14;150:1121. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watson IR, Takahashi K, Futreal PA, Chin L. Emerging patterns of somatic mutations in cancer. Nature reviews Genetics. 2013 Oct;14:703. doi: 10.1038/nrg3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogelstein B, et al. Cancer genome landscapes. Science. 2013 Mar 29;339:1546. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research. 2011 Jan;39:D945. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez P, et al. Parallel evolution of tumour subclones mimics diversity between tumours. The Journal of pathology. 2013 Aug;230:356. doi: 10.1002/path.4214. [DOI] [PubMed] [Google Scholar]
- 25.Hainaut P, Pfeifer GP. Patterns of p53 G–>T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001 Mar;22:367. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 26.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013 Aug 22;500:415. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nature biotechnology. 2012 May;30:413. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulzebruck H, et al. New aspects in the staging of lung cancer. Prospective validation of the International Union Against Cancer TNM classification. Cancer. 1992 Sep 1;70:1102. doi: 10.1002/1097-0142(19920901)70:5<1102::aid-cncr2820700514>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 29.Mahadevia PJ, et al. Lung cancer screening with helical computed tomography in older adult smokers: a decision and cost-effectiveness analysis. JAMA : the journal of the American Medical Association. 2003 Jan 15;289:313. doi: 10.1001/jama.289.3.313. [DOI] [PubMed] [Google Scholar]
- 30.Church TR, et al. Results of initial low-dose computed tomographic screening for lung cancer. The New England journal of medicine. 2013 May 23;368:1980. doi: 10.1056/NEJMoa1209120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010 Mar 1;26:589. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013 Mar;31:213. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009 Nov 1;25:2865. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meacham F, et al. Identification and correction of systematic error in high-throughput sequence data. BMC bioinformatics. 2011;12:451. doi: 10.1186/1471-2105-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loman NJ, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nature biotechnology. 2012 May;30:434. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 36.Kluge AG, Farris JS. Quantitative Phyletics and Evolution of Anurans. Syst Zool. 1969;18:1. [Google Scholar]
- 37.Eck R, Dayhoff M. Atlas of Protein Sequence and Structure 1966. National Biomedical Research Foundation, Silver Spring; Maryland: 1966. [Google Scholar]
- 38.Felsenstein J. PHYLIP – Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164. [Google Scholar]
- 39.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010 Mar 15;26:841. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome research. 2012 Mar;22:568. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts SA, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nature genetics. 2013 Sep;45:970. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolli N, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nature communications. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proceedings of the National Academy of Sciences of the United States of America. 2010 Sep 28;107:16910. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012 Jun 21;486:400. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.