

Abstract
Objective
Non-alcoholic steatohepatitis (NASH) is characterized by hepatic steatosis, inflammation and fibrosis. There are currently no targeted therapies for NASH. We developed a liver-specific LXR inverse agonist, SR9238, which effectively reduces hepatic lipogenesis in models of obesity and hepatic steatosis. We hypothesized that suppression of lipogenesis, which is pathologically elevated in NASH may suppress progression of hepatic steatosis to NASH.
Methods
NASH was induced in B6 V-lep ob/J (ob/ob) mice using a custom complete rodent diet (HTF) containing high amounts of trans-fat, fructose, and cholesterol. Once NASH was induced, mice were treated with SR9238 for one month by i.p. injection. Plasma lipid levels and liver health were analyzed by clinical chemistry. QPCR, western blot, and immunohistochemistry were used to assess disease severity.
Results
Ob/ob mice are obese and diabetic thus they are commonly used as models for the study of metabolic diseases. These mice quickly developed the NASH phenotype when provided the HTF diet. The mice develop hepatic steatosis, severe hepatic inflammation and fibrosis on the HTF diet. Treatment with SR9238 significantly reduced the severity of hepatic steatosis and most importantly reduced hepatic inflammation and ameliorated hepatic fibrosis.
Conclusions
Here, we demonstrate that an LXR inverse agonist, SR9238, is effective in reduction of hepatic steatosis, inflammation and fibrosis in an animal model of NASH. These results have important implications for the development of therapeutics for treatment NASH in humans.
Keywords: NASH, Fibrosis, Lipogenesis, LXR, Inflammation
Abbreviations: NASH, non-alcoholic steatohepatitis; LXR, Liver-X-Receptor; HTF, high trans-fat diet; NR, nuclear receptor
1. Introduction
Non-alcoholic steatohepatitis (NASH) is a severe form of chronic liver disease, characterized by steatosis, inflammation, and pericellular fibrosis [1–4]. It is often associated with obesity, dyslipidemia, insulin resistance, and hyperglycemia, and can progress to cirrhosis or hepatocellular carcinoma. Current therapies often target the associated metabolic disease but not NASH directly [5]. Development of non-alcoholic fatty liver disease (NAFLD) is the first step towards NASH and is characterized by hepatic steatosis that develops due to an imbalance of triglyceride removal and acquisition within the liver and is also associated with elevated hepatic lipogenesis [2,4]. High calorie diets with excessive fats and carbohydrates can lead to this imbalance leading to NAFLD and in some cases, progression to NASH.
The Liver X Receptors (LXRα and LXRβ) are members of the nuclear receptor (NR) superfamily and are widely expressed (LXRα is primarily expressed in the liver, kidneys, intestines, and adipose tissues while LXRβ is ubiquitously expressed) [6,7]. LXRs have been shown to regulate cholesterol efflux and transport, as well as stimulate hepatic lipogenesis [6,8,9]. Synthetic LXR agonists have been shown to display anti-atherogenic properties due to their effects on reverse cholesterol transport mediated by increased cholesterol efflux from peripheral tissues [1,6,8,10,11]. However, the activation of LXR by synthetic ligands results in deleterious effects due to increased hepatic lipogenesis due to increased expression of lipogenic enzymes including fatty acid synthase (Fasn) and sterol regulatory element-binding protein (Srebp1) that are direct target genes of LXR. This has caused significant difficulties in the development of LXR agonists as anti-atherogenic therapeutics [8,10–12].
We believed that regulation of lipogenesis by LXR might be an opportunity to focus on diseases that are characterized by elevated lipogenesis such as NAFLD and NASH. To this end, we developed LXR inverse agonists that have the ability to suppress the expression of LXR target genes such as Fasn and Srepb1 [13]. Previously, we demonstrated that the LXR inverse agonist, SR9238, which displays liver selectivity, is effective in suppressing hepatic steatosis associated with a high fat diet in mice [13]. In this study, we sought to determine if such a therapeutic would have efficacy in reduction of inflammation and fibrosis in a mouse model of NASH.
2. Methods
2.1. Animals
Six-week-old B6 V-Lepob/J (ob/ob) mice were purchased from Jackson Laboratories (Bar Harbor, ME), housed individually in standard cages, and immediately placed on D09100301 (NASH) diet (Research Diets). This is a High Trans-Fat (Primex shortening-based) diet that also contains 20% kcal from fructose and 2% cholesterol. Mice were maintained on the diet for six weeks prior to starting treatment, handled daily for acclimation and weighed every other day during this time. Groups were weight-matched prior to beginning treatment (n = 7). After six weeks, mice continued on the NASH diet and were treated with 30 mg/kg SR9238 q.d. i.p. in 10%DMSO/10%Tween-80/80%water or vehicle for 30 days. Body weight and food intake were monitored daily, blood glucose was determined weekly using a hand-held glucometer, and a final fasting blood glucose was collected at termination. At the termination of dosing, blood was collected by cardiac puncture and analyzed using clinical chemistry (Roche cobas c311) and ELISA. Liver was collected, weighed, and a portion was immediately flash frozen in liquid nitrogen for RNA. The rest was placed in 10% neutral buffered formalin for histology. The Institutional Animal Care and Use Committee at the Scripps Research Institute, Jupiter FL, approved all mouse studies.
2.2. Gene and protein expression studies
RNA was isolated from liver and analyzed by QPCR as previously described in Ref. [9]. Each sample was run in duplicate and measured using the ddCT method, using Gapdh as the reference gene. Liver protein lysate was isolated by standard methods in RIPA buffer containing Roche miniComplete protease inhibitor. Total protein was quantified by a 96-well format BCA assay (ThermoFisher) and diluted in 2× Laemmli Buffer (Amresco) for western blot analysis. Liver lysates (25 μg) were separated on miniTGX anyKd gels (Bio-Rad) and transferred to nitrocellulose membranes. Membranes were blocked in 5% Non-fat Milk in TBST for 1 h, then probed either with FASn (1:1000; Cell Signaling), TGFβ (1:1000; Abcam), SREBP2 (1:750; Abcam) or Actin (1:1000; Cell Signaling). Following incubation with HRP anti-rabbit antibody (1:10,000; Santa Cruz Biotechnology), protein expression was detected with chemiluminescent reagent (Santa Cruz Biotechnology) and analyzed using ImageJ software (NIH). Insulin concentration was determined by an ELISA kit according to manufacturers protocol (EMD Millipore) and used to calculate HOMA-IR.
2.3. Histology and immunofluorescence
Livers were placed in 10% neutral buffered formalin overnight at 4 °C, then transferred to 10% Sucrose in PBS for 2 days. Livers were then transferred to 20% Sucrose in PBS for 2 days, then finally transferred to 30% Sucrose in PBS for 3 days for cryoprotection, then embedded in OCT freezing media in an ethanol-dry ice bath. Livers were sectioned at a thickness of 10 μm and placed on slides (Fisher) or floated in 12-well dishes in 1× PBS. Slides were stained for fibrosis using standard Pico-Sirius Red (Direct Red-80; Sigma) technique, and photographed under polarized light microscopy (Leica). Floated sections were stained with either Bodipy 493/503 (Molecular Probes) or F4/80 (Abd Serotec), and mounted to slides prior to counterstaining with DAPI.
2.4. Expression of data and statistics
All data are expressed as mean ± s.e.m. (n = 3 or more). Statistical analysis was performed using an unpaired Student's t-test in Graphpad Prism Software.
3. Results and discussion
Tresvaskis et al. [14] demonstrated that a high transfat/fructose/cholesterol diet fed to ob/ob mice was sufficient to induce an NASH phenotype that correlated to human disease pathology. We utilized this diet induced NASH model to examine the potential efficacy of SR9238. First, we established this murine model of NASH by feeding young obese diabetic mice (ob/ob) the D09100301 (NASH diet) for 6 weeks prior to treatment [14]. We then initiated treatment with SR9238 (30 mg/kg/day i.p.) for 30 days, but maintained the mice on the NASH diet and monitored body weight and food intake during the treatment period. Although we noted no alterations in food intake during the treatment period, we did observe a significant decrease in body weight beginning the 2nd week of treatment that was maintained until completion of the study (Figure 1A). This is in contrast to our previous study in wt C57Bl6 mice (on an HF diet) where they displayed no change in body weight when subjected to SR9238 treatment [13]. As expected, mice on the SR9238 treatment displayed reduced hepatic steatosis as demonstrated by a significant reduction in hepatic lipids and lipid droplet size as determined by Bodipy 493/503 lipid staining (Figure 1B,C). We also noted significant reduction in expression of genes that regulate lipogenesis and hepatic steatosis, srebf1, scd1, cd36 (Figure 1D). All of these are direct LXR target genes [15–17] and their suppression by SR9238 is consistent with our study in a mouse model of NAFLD [13]. Treatment with SR9238 also resulted in a significant reduction in liver weight (Figure 1E), and reduced hepatocellular damage as demonstrated by reduced plasma liver enzyme levels (Figure 1F). Given that we observed similar reduction in hepatic steatosis in C57Bl6 mice when there was no change in body weight [13], we do not believe that the relatively small reduction in body weight in the ob/ob mice in response to SR9243 treatment is responsible for the reduction in hepatic steatosis. The loss of body weight with no alteration in food intake suggests that there may be an increase in energy expenditure in response to SR9238, but this appears to be specific to the ob/ob mice. LXR has been demonstrated to play a role in regulation of energy expenditure primarily mediated via effects on the brown adipose tissue (BAT) [18]. SR9238 does not display detectable levels outside of the intestine and liver and we previously demonstrated that SR9238 had no effect on LXR target genes in the BAT [13], thus this effect must be mediated via a distinct mechanism.
Figure 1.
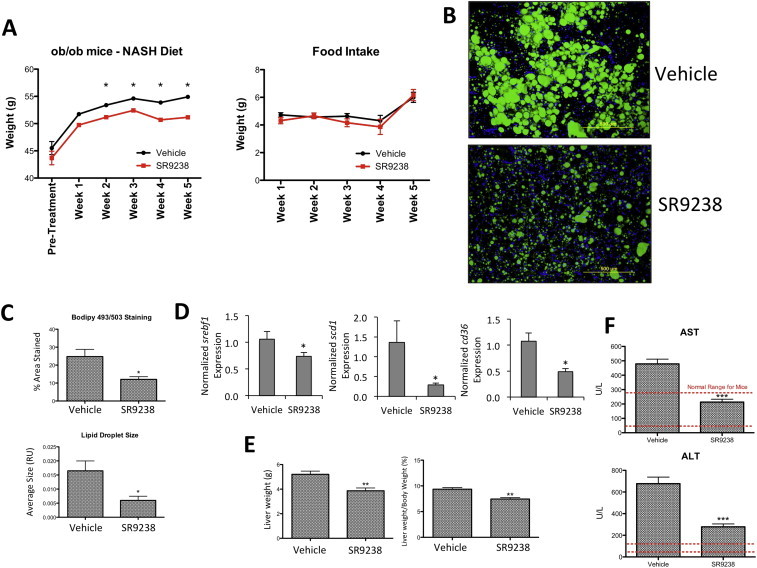
SR9238 significantly reduces hepatic lipids and normalizes liver function in NASH mice. (A) Body weight of mice on NASH diet treated with SR9238 or vehicle (left panel). Averaged daily food intake of mice during treatment period (right panel) (B) Bodipy 493/503 lipid staining of liver sections. (C) Quantitation of bodipy staining by % Area Stained and Lipid Droplet size using ImageJ software (NIH). (D) Hepatic Srepf1, scd1 and cd36 gene expression is suppressed by SR9238 treatment. Gene expression was determined by QPCR and normalized to gapdh expression. (E) SR9238 treatment reduces liver and total body weight. (F) Reduced liver enzyme levels suggest that SR9238 treatment significantly decreases hepatocellular damage. *indicates p < 0.05.
Hematoxylin and eosin (H&E) staining of liver sections revealed very severe disease with inflammatory foci, macrosteatosis, and hepatocellular ballooning in the vehicle treated mice (Figure 2A top). Sections from SR9238 mice showed significant disease but considerably less severe relative to the vehicle treated mice with microsteatosis dominating (relative to macrosteatosis) and limited inflammatory foci (Figure 2A bottom). Hepatitis is key component of NASH [1] and we assessed effects of SR9238 on hepatic inflammation using multiple methods. Kupffer cells play an integral part in the pathogenesis of NASH where they secrete proinflammatory cytokines that contribute to the progression of the disease [19]. We assessed the number of Kupffer cells in the liver by staining liver sections for F4/80 and observed that there is a clear reduction in staining in SR9238 treated livers (Figure 2B). Additionally, hepatic gene expression of several inflammatory markers including Cd68, Tnfa, Il6, Il1b, and Il12, was reduced in SR9238-treated mice (Figure 2C). Hepatic expression of TGFβ expression also displayed a decrease in SR9238-treated mice (Figure 2D).
Figure 2.
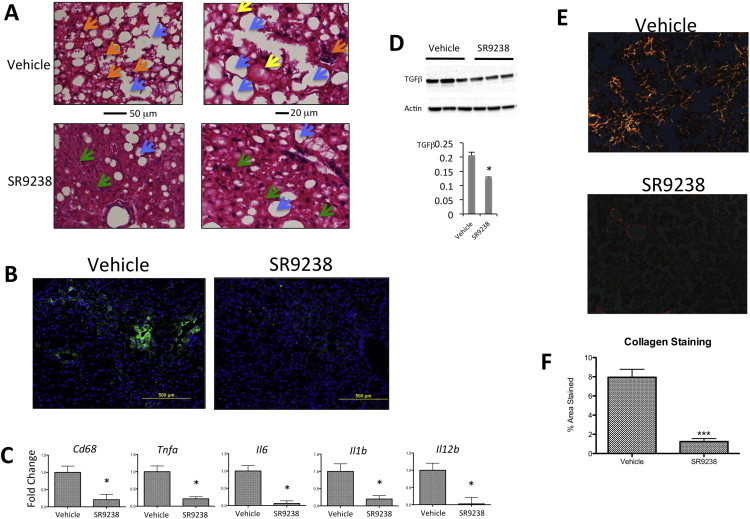
Hepatic inflammation is substantially reduced by SR9238 treatment. (A) Hematoxylin and eosin staining of liver sections from vehicle and SR9238 treated mice revealing inflammatory foci (orange arrows), microsteatosis (green arrows), macrosteatosis (blue arrows) and hepatocellular ballooning (yellow arrows). (B) F4/80 staining of hepatic macrophages (Kupffer cells). (C) SR9238 reduces the hepatic expression of a range of proinflammatory genes (D) Hepatic TGFβ protein expression is reduced by SR9238. (E) SR9238 reduces hepatic fibrosis. Pico-Sirius red staining of collagen was used as a marker of fibrosis in mouse liver sections. (F) Quantification of Pico-Sirius Red staining using ImageJ software (NIH). *indicates p < 0.05.
Progression of NASH is associated with hepatic fibrosis and in order to assess the level of fibrosis in the mice we stained liver sections with pico-sirius red to examine the level of collagen deposition. As illustrated in the top panel of Figure 2E, mice on the NASH diet receiving only vehicle display quite extensive hepatic collagen deposition whereas mice treated with SR9238 (bottom panel) display very little staining. Total hepatic collagen staining was reduced approximately 90% by SR9243 treatment (Figure 2F). In fact, the primary hepatic staining noted SR9238-treated was typically associated with normal structures (e.g. vasculature) and was not the diffuse staining noted in the fibrotic liver vehicle treated mice. These data suggest that SR9243 is quite effective in reducing hepatitis and hepatic fibrosis in this mouse model of NASH.
We also noted a trend towards a decrease in plasma insulin levels (Figure 3A) and fasting glucose levels in SR9243 treated mice (Figure 3A). Determination of HOMA-IR revealed a trend towards increased insulin sensitivity in the SR9238 (Figure 3A). Consistent with our previous study [13], ob/ob mice SR9238-treated display a significant reduction in total cholesterol and LDL cholesterol, however, there is no significant effect on plasma triglycerides (Figure 3B). Previously, we proposed that the decrease in cholesterol may be due to decreased levels of active SREBP2 [13], and we observe that levels of active SREBP2 are reduced in this model as well (Figure 3C). We also considered whether regulation of IDOL might be a component of the mechanism by which an LXR inverse agonist could suppress LDL levels. IDOL is a E3 ubiquitin ligase direct that targets the LDL receptor for ubiquitination and subsequent degradation and is a direct LXR target gene [20], thus it is reasonable that suppression of IDOL expression with an LXR inverse agonist might lead to increased LDL receptor levels and increased LDL clearance. This does not appear to be the case since we observe no alteration in idol gene expression in response to SR9238 treatment (Figure 3D).
Figure 3.
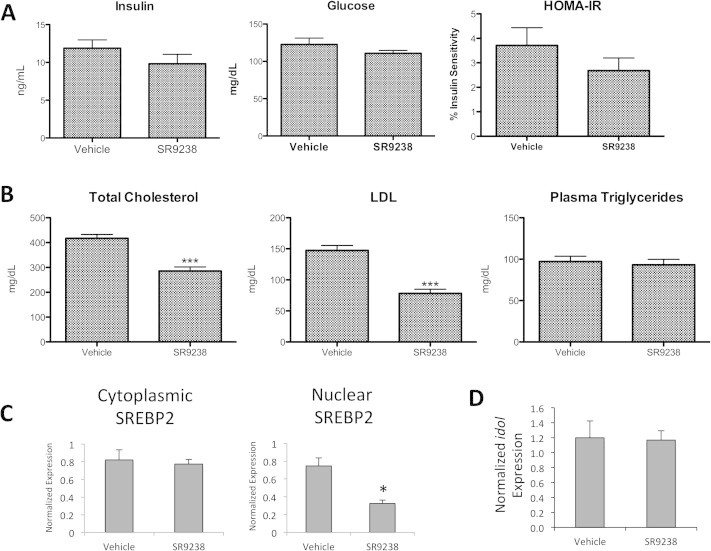
Effect of SR9238 on glucose and lipid levels in a mouse model (A) Fasting insulin levels are shown in the left panel, glucose levels are illustrated in the middle panel. HOMA-IR was measured using fasting blood glucose and derived insulin levels. (B) Total cholesterol, LDL and plasma triglycerides were determined from mouse plasma samples at the termination of the experiment. (C) Since cholesterol was reduced in treated mice, SREBP2 protein expression from liver samples was quantified. SR9238 treatment appears to significantly suppress the expression of active SREBP2, which is consistent with our previous study demonstrating that SREBP2 does not translocate to the nucleus following treatment. (D) Expression of idol was not affected by SR9238 treatment. *indicates p < 0.05.
In summary, the LXR inverse agonist, SR9238, displays efficacy in reduction of hepatic pathology in a mouse model of diet induced NASH. SR9238 is effective in reducing hepatic steatosis and inflammation. Most importantly, SR9238 reduces hepatic fibrosis where we observed an approximate 90% reduction in hepatic collagen deposition. Our data suggests that LXR inverse agonists, such as SR9238 that displays liver specificity, may offer novel therapies to treat NASH.
Author contributions
Conceived and designed experiments: KG, TPB, BAN-T. Performed experiments: KG, CAF, and RDW. Evaluated histology: GRK. Wrote the manuscript: KG. Reviewed the manuscript: KG, TPB, RDW, and BAN-T.
Conflict of interest
None declared.
References
- 1.Ganz M., Szabo G. Immune and inflammatory pathways in NASH. Hepatology International. 2013;7:771–781. doi: 10.1007/s12072-013-9468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hashimoto E., Taniai M., Tokushige K. Characteristics and diagnosis of NAFLD/NASH. Journal of Gastroenterology and Hepatology. 2013;28(Suppl. 4):64–70. doi: 10.1111/jgh.12271. [DOI] [PubMed] [Google Scholar]
- 3.Tacke F., Yoneyama H. From NAFLD to NASH to fibrosis to HCC: role of dendritic cell populations in the liver. Hepatology. 2013;58:494–496. doi: 10.1002/hep.26405. [DOI] [PubMed] [Google Scholar]
- 4.Wree A., Broderick L., Canbay A., Hoffman H.M., Feldstein A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nature Reviews Gastroenterology & Hepatology. 2013;10:627–636. doi: 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
- 5.Ratziu V. Pharmacological agents for NASH. Nature Reviews Gastroenterology & Hepatology. 2013;10:676–685. doi: 10.1038/nrgastro.2013.193. [DOI] [PubMed] [Google Scholar]
- 6.Burris T.P., Solt L.A., Wang Y., Crumbley C., Banerjee S., Griffett K. Nuclear receptors and their selective pharmacologic modulators. Pharmacological Reviews. 2013;65:710–778. doi: 10.1124/pr.112.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savkur R.S., Burris T.P. The coactivator LXXLL nuclear receptor recognition motif. Journal of Peptide Research. 2004;63:207–212. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- 8.Bensinger S.J., Bradley M.N., Joseph S.B., Zelcer N., Janssen E.M., Hausner M.A. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ulven S.M., Dalen K.T., Gustafsson J.A., Nebb H.I. LXR is crucial in lipid metabolism. Prostaglandins Leukotrienes and Essential Fatty Acids. 2005;73:59–63. doi: 10.1016/j.plefa.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Bennett D.J., Cooke A.J., Edwards A.S. Non-steroidal LXR agonists; an emerging therapeutic strategy for the treatment of atherosclerosis. Recent Patents on Cardiovascular Drug Discovery. 2006;1:21–46. doi: 10.2174/157489006775244245. [DOI] [PubMed] [Google Scholar]
- 11.Jaye M. LXR agonists for the treatment of atherosclerosis. Current Opinion in Investigational Drugs. 2003;4:1053–1058. [PubMed] [Google Scholar]
- 12.Bradley M.N., Hong C., Chen M., Joseph S.B., Wilpitz D.C., Wang X. Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. Journal of Clinical Investigation. 2007;117:2337–2346. doi: 10.1172/JCI31909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffett K., Solt L.A., El-Gendy Bel D., Kamenecka T.M., Burris T.P. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chemical Biology. 2012;8:559–567. doi: 10.1021/cb300541g. [DOI] [PubMed] [Google Scholar]
- 14.Trevaskis J.L., Griffin P.S., Wittmer C., Neuschwander-Tetri B.A., Brunt E.M., Dolman C.S. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2012;302:G762–G772. doi: 10.1152/ajpgi.00476.2011. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J., Febbraio M., Wada T., Zhai Y., Kuruba R., He J. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134:556–567. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 16.Repa J.J., Liang G., Ou J., Bashmakov Y., Lobaccaro J.M., Shimomura I. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes & Development. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y., Hao M., Luo Y., Liang C.P., Silver D.L., Cheng C. Stearoyl-CoA desaturase inhibits ATP-binding cassette transporter A1-mediated cholesterol efflux and modulates membrane domain structure. Journal of Biological Chemistry. 2003;278:5813–5820. doi: 10.1074/jbc.M208687200. [DOI] [PubMed] [Google Scholar]
- 18.Korach-Andre M., Archer A., Barros R.P., Parini P., Gustafsson J.A. Both liver-X receptor (LXR) isoforms control energy expenditure by regulating brown adipose tissue activity. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:403–408. doi: 10.1073/pnas.1017884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tosello-Trampont A.C., Landes S.G., Nguyen V., Novobrantseva T.I., Hahn Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. Journal of Biological Chemistry. 2012;287:40161–40172. doi: 10.1074/jbc.M112.417014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zelcer N., Hong C., Boyadjian R., Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]