

Abstract
Aims/Hypothesis
Glucagon release from pancreatic alpha cells is required for normal glucose homoeostasis and is dysregulated in both Type 1 and Type 2 diabetes. The tumour suppressor LKB1 (STK11) and the downstream kinase AMP-activated protein kinase (AMPK), modulate cellular metabolism and growth, and AMPK is an important target of the anti-hyperglycaemic agent metformin. While LKB1 and AMPK have emerged recently as regulators of beta cell mass and insulin secretion, the role of these enzymes in the control of glucagon production in vivo is unclear.
Methods
Here, we ablated LKB1 (αLKB1KO), or the catalytic alpha subunits of AMPK (αAMPKdKO, -α1KO, -α2KO), selectively in ∼45% of alpha cells in mice by deleting the corresponding flox'd alleles with a preproglucagon promoter (PPG) Cre.
Results
Blood glucose levels in male αLKB1KO mice were lower during intraperitoneal glucose, aminoimidazole carboxamide ribonucleotide (AICAR) or arginine tolerance tests, and glucose infusion rates were increased in hypoglycemic clamps (p < 0.01). αLKB1KO mice also displayed impaired hypoglycemia-induced glucagon release. Glucose infusion rates were also elevated (p < 0.001) in αAMPKα1 null mice, and hypoglycemia-induced plasma glucagon increases tended to be lower (p = 0.06). Glucagon secretion from isolated islets was sensitized to the inhibitory action of glucose in αLKB1KO, αAMPKdKO, and -α1KO, but not -α2KO islets.
Conclusions/Interpretation
An LKB1-dependent signalling cassette, involving but not restricted to AMPKα1, is required in pancreatic alpha cells for the control of glucagon release by glucose.
Keywords: LKB1, AMPK, Glucagon secretion, PPG, Knockout, Alpha cell
Abbreviations: AMPK, AMP-activated protein kinase; LKB1, liver kinase B1; T2D, Type 2 diabetes; PPG, preproglucagon promoter; AICAR, aminoimidazole carboxamide ribonucleotide
1. Introduction
Glucagon, secreted by the pancreatic islet alpha cell as blood glucose levels fall, is the key anti-hypoglycemic hormone in mammals, acting as a powerful stimulus for hepatic glucose production [1]. Impaired glucagon release is associated with the episodes of hypoglycemia frequently reported in Type 1 and advanced Type 2 diabetic (T2D) patients treated with insulin or sulphonylureas [2]. On the other hand, elevated circulating glucagon levels contribute to the chronic increase in blood glucose levels characteristic of T2D [1].
Lowered blood glucose levels stimulate glucagon release through multiple mechanisms including changes in parasympathetic [3] and sympathetic tone [4], increases in circulating adrenaline levels [5], as well as through effects of glucose on pancreatic alpha cells [6]. The exact nature of the latter are still debated. Thus, in the mouse, a direct effect of glucose [7,8] and indirect effects of γ-amino butyric acid (GABA) [9,10] and insulin [7,11] released from neighbouring beta cells have all been invoked as regulators of glucagon secretion. Roles for released Zn2+ ions have also been proposed [7,12–14].
AMP-activated protein kinase (AMPK) is a heterotrimeric complex comprising α, β and γ subunits that serves as a master energy sensor [15], highly sensitive to intracellular ATP:AMP [15] and ATP:ADP ratios [16]. Liver kinase B1 (LKB1), also called STK11, is a tumour suppressor whose inactivation leads to Peutz-Jegers syndrome [17], characterized by hamartomatous polyps and an increased risk of all cancers. LKB1 phosphorylates at least 13 different protein kinases including AMPK. LKB1 (and alternative protein kinases including calmodulin kinase kinase β [18,19] and transforming growth factor β-activated kinase-1, TAK1) [20], phosphorylate AMPK catalytic α-subunits at Thr172 in the “T-loop”. A fall in intracellular energy, leading to altered occupancy by AMP and/or ADP of sites in the γ-subunit, leads to conformational changes that render T172 in the α-subunit less susceptible to dephosphorylation [16] by protein phosphatases [21]. Activated AMPK then phosphorylates a range of downstream metabolic targets to promote ATP synthesis and inhibit ATP consumption [15].
Through the use of pharmacological AMPK activators and inhibitors [22], we have previously provided evidence that AMPK is a negative regulator of insulin secretion from pancreatic beta cells [23,24]. Furthermore, activation of AMPK by metformin is associated with enhanced beta cell apoptosis [25]. Correspondingly, mice deleted selectively in the beta cell for LKB1 display beta cell hyperplasia and markedly enhanced insulin release [26–28]. By contrast, and unexpectedly, deletion of both AMPK catalytic subunits (α1 and α2) from beta cells causes impaired insulin secretion and glucose intolerance in vivo [29,30], possibly as a result of rat insulin promoter-2 (RIP2)-Cre-mediated recombination in the brain [31].
We have also recently provided evidence from in vitro studies that AMPK is involved in the regulation of glucagon release. Thus, activation of AMPK stimulated glucagon release from clonal αTC1-9 cells and mouse pancreatic islets, while a dominant-negative form of the kinase blocked the stimulatory effects of low glucose [22]. However, neither the role of LKB1 nor that of individual AMPK isoforms in controlling glucagon secretion has been examined in vivo.
Here, we have generated mice deleted for LKB1 (αLKB1KO), and for one (AMPKα1KO, -α2KO) or both (αAMPKdKO) AMPKα catalytic subunits selectively in pancreatic alpha cells using preproglucagon (PPG)-Cre driven recombination. We show that LKB1 signalling, at least partly mediated via AMPKα1, is essential for the normal stimulation of glucagon secretion at low glucose levels both in vitro and in vivo. Moreover, and in marked contrast to its action in the beta cell, we show that LKB1 plays a limited if any role in the control of alpha cell size or total alpha cell mass [26–28].
2. Methods
2.1. Generation of mutant mice lacking LKB1 selectively in pancreatic alpha cells
Mice heterozygous for flox'd alleles of the Lkb1/Stk11 gene (mixed FVB/129S6 and C57BL/6 background) [32] were obtained from the Mouse Models of Human Cancer Consortium (MMHCC) (www.nih.gov/science/models/mouse/resources/hcc.html) and backcrossed with C57/B6 mice four times. These animals were then bred against PPG-Cre expressing mice [33] and the resulting heterozygous offspring were inter-crossed as siblings to generate αLKB1KO mice (Lkb1fl/fl, Cre positive). αLKB1KO mice were further bred with Lkb1fl/fl mice to generate littermate controls (Lkb1fl/fl). It should be noted that the PPG-Cre transgene is not reported to exert effects on glucagon secretion or glucose homoeostasis [34] or to lead to significant recombination in extra-pancreatic tissues [11]. αLKB1KO mice and littermate controls were born at the expected Mendelian ratios.
2.2. Generation of mutant mice selectively lacking AMPK α1 and α2 in pancreatic alpha cells
Mice homozygous for Ampkα1fl/fl were crossed with mice heterozygous for Ampkα2fl/+. The resulting double heterozygotes (Ampkα1fl/+, α2fl/+) were crossed with PPG-Cre-expressing animals [33] to generate triple heterozygous mice (Ampkα1fl/+, α2fl/+, Cre positive). The latter were then bred with mice homozygous for both flox'd Ampkα1 and α2 alleles (Ampkα1fl/fl, α2fl/fl) to produce αAMPKdKO mice (Ampkα1fl/fl, α2fl/fl, Cre positive). αAMPKdKO mice were further crossed with Ampkα1fl/fl, α2fl/fl mice to generate littermate controls (Ampkα1fl/fl, α2fl/fl). All mice were kept on a C57/B6 background.
2.3. Generation of mice selectively expressing RFP in pancreatic alpha cells
Mice heterozygous for Rosa26tdRFP [35] were crossed with mice heterozygous for PPG-Cre to generate double heterozygous mice.
2.4. Mouse maintenance and diet
Animals were housed two to five per individually ventilated cage in a pathogen-free facility with 12 h light/dark cycle and had free access to standard mouse chow diet. All in vivo procedures described were performed at the Imperial College Central Biomedical Service and approved by the UK Home Office according to the Animals (Scientific Procedures) Act 1986 of the United Kingdom (PPL 70/7349).
2.5. Glucose, insulin, AICAR and arginine tolerance tests
Intraperitoneal glucose and insulin tolerance tests were performed as previously described [28,30]. For AICAR tolerance, mice fasted for 16 h (water allowed) were intraperitoneally injected with 1.75 g AICAR/kg (Toronto Research Chemicals, North York, Canada). Blood (2–4 μl) from the tail vein was obtained at 0, 20, 40, 60, 90 and 120 min after injection [36]. Blood glucose levels were measured with an automatic glucometer (Accuchek; Roche, Burgess Hill, UK). For arginine tolerance, mice fasted for 16 h (water allowed) were intraperitoneally injected with 3 g/kg l-arginine (pH 7.4; Arginine hydrochloride from Sigma) [11]. Blood (50 μl) from the tail vein was collected at 0, 10 and 30 min after injection into EDTA coated tubes containing 1 μl DPP IV inhibitor (final concentration: 100 μmol/l; Millipore, Watford, UK). Plasma insulin and glucagon levels were measured with an ultrasensitive mouse insulin ELISA kit (Mercodia, Uppsala, Sweden) [37] and a glucagon radioimmunoassay kit with a competitive 125I labelled glucagon (Millipore, Watford, UK) [7] respectively. Experiments were performed on mice aged 8–12 weeks.
2.6. Hyperinsulinemic-hypoglycemic clamp
Nine week-old male mice were implanted with catheters in the right jugular vein under general ketamine (100 mg/kg)/xylazine (10 mg/kg) anaesthesia. Catheters were flushed daily with saline containing heparin (0.4U/100 ml). Mice were housed individually in separate cages for post-surgery recovery. On the day of the clamp study, mice were starved for five hours before catheters were connected onto infusion tubing. Mice were infused with bolus insulin (0.033U) at an infusion rate of 30 μl/min for 5 min. To maintain blood glucose levels close to 2.7–3.3 mmol/l, insulin (0.6 U/kg/h) were infused for 120 min, during which 20% glucose was co-infused with adjustable infusion rates. Blood was collected from tail veins for measurement of glucose levels every 10 or 20 min.
2.7. Other methods
Please see Supplementary Materials and Methods.
2.8. Statistical analysis
Significance was tested using unpaired or paired Student's two-tailed t-tests with Bonferroni post-tests for multiple comparisons, or two-way ANOVA with Sidak post-doc tests as required. Analysis was performed using Excel (Microsoft) and Graphpad Prism 4.0 (Graphpad Software). p < 0.05 was considered significant and values represent mean ± SEM.
3. Results
3.1. Generation of mice deleted selectively in the pancreatic alpha cell for LKB1 or AMPKα-subunits
To assess the likely efficiency and specificity of recombination in alpha cells, we first crossed mice bearing a tandem dimer red fluorescent protein transgene at the Rosa26 locus distal to a STOP-LoxP-STOP control cassette (Rosa26tdRFP) [35] against animals bearing Cre recombinase under the control of a 0.6 kb fragment of the rat preproglucagon promoter (PPG) [38]. This led to almost exclusive expression of PPG-Cre in glucagon-positive pancreatic alpha cells, of which ∼45% were positive for tdRFP (Figure 1A(i)). Expression in other cell types was below the limit of reliable detection (<3% of insulin-positive cells showed weak staining, likely attributable to background fluorescence; Figure 1A(ii)).
Figure 1.

Generation of alpha cell specific LKB1 and AMPK α subunit(s) KO mice (A) (i) Representative immunostaining of pancreatic islets isolated from PPG-Cre:RFP mice using antibodies against RFP (1:100) and either glucagon (1:2000; green) or insulin (1:200; green). Nuclei are shown by DAPI (blue) staining. (ii) Images of stained islets (5–8) were used to calculate the percentage of cells positive for glucagon or insulin and co-expressing RFP. (B) RT-PCR analysis of deletion of exon 3–6 on LKB1 transcript levels in pancreatic islets, brain stems, hypothalamus and intestines. The product sizes were 864 and 300bp for the flox'd and null alleles, respectively. (C) Representative immunofluorescence staining of pancreatic sections using mouse anti-glucagon (1:2000; red) and rabbit anti-LKB1 (1:100; green) antibodies. Nuclei are shown with DAPI (blue). Scale bar, 75 μm. (D,E) Total AMPK activity in islets of αLKB1KO (D), αAMPK KOs (E) and their wild type littermate control mice (WT). Islets incubated with RPMI media containing 10 mmol/l glucose for 48–72 h, were further incubated with RPMI media containing 3 mmol/l glucose for 1 h before analysis. Data are expressed as means ± SEM; n = 4, *p < 0.05, **p < 0.01 between groups using unpaired two-tailed Student's t-tests.
We next generated mice deleted for LKB1 in the alpha cell compartment (αLKB1KO) by crossing animals in which exons 3–6 of the Lkb1 gene were flanked with LoxP sites [32] with PPG-Cre mice [33]. The excision of exons 3–6 mediated by Cre was observed in pancreatic islets of αLKB1KO mice, but not evident in other preproglucagon-expressing tissues, such as brain stem, hypothalamus, and intestine or in islets from wild type mice (Figure 1B), consistent with previous findings [38]. Correspondingly, LKB1 immunoreactivity was reduced selectively in glucagon-positive cells in pancreatic sections from αLKB1KO mice (Figure 1C). Moreover, total AMPK activity was reduced by almost 40% in whole islets from these mice (60.5 ± 8.1% of wild type, p < 0.05; Figure 1D) compatible with a higher expression of LKB1 in alpha than beta cells (ratio of Lkb1 to cyclophilin A mRNA: 0.36 ± 0.09 in FACS-purified alpha vs. 0.11 ± 0.03 in beta cells, p < 0.05; Figure S1).
Similarly, we created mice deleted for both (αAMPKdKO) or single (αAMPKα1KO and -α2KO) catalytic α-subunits selectively in pancreatic alpha cells by crossing either double AMPKα1 and α2 or single α-isoform flox'd mice with PPG-Cre deleter mice as above. Cre-mediated recombination led to decreases in AMPK activity of ∼37%, ∼32% and ∼27% in islets from αAMPKdKO, -α1KO and -α2KO mice, respectively (Figure 1E), the more modest change in islets from αAMPKdKO mice, possibly reflecting compensatory changes in the expression or activity of the non-deleted isoform or one or more of the 12 other AMPK-related kinase(s) [39].
Consistent with the above demonstration that no or minimal recombination occurred in enteroendocrine L-cells [38], fed plasma GLP-1 levels were identical between genotypes (19.2 ± 4.8 pg/ml in αLKB1KO mice, n = 3–5 vs. 19.5 ± 1.5 pg/ml in WT, n = 3–5; 11.4 ± 0.23 pg/ml, n = 6 in αAMPKdKO mice vs 13.9 ± 1.97 pg/ml, n = 4 in littermate controls). Likewise, supporting the finding that deletion did not occur in ventromedial hypothalamic feeding centres [11], we observed no significant differences of body weight or daily food intake of αLKB1KO or αAMPK KOs compared to wild-type littermates (Figure S2a–c).
3.2. Pancreatic alpha cell-selective deletion of LKB1, but not AMPKα-subunits moderately lowers blood glucose and glucagon levels
Examined at eight weeks of age, male αLKB1KO mice maintained lower overnight fasting blood glucose levels (Figure 2A), while the levels in αAMPKKOs and their littermate controls were comparable (Figure S2d–f). Consistent with these findings, fasting plasma glucagon levels were significantly decreased after LKB1 deletion from alpha cells (Figure 2B), with tendencies towards decreased plasma glucagon in αAMPKα1KO and αAMPKdKO mice versus controls (Figure S2g-i). Plasma insulin levels were not different between genotypes in the fasted state (0.069 ± 0.022 nmol/l in αLKB1KO, n = 5 vs. 0.078 ± 0.019 nmol/l in WT, n = 5). However, eight week-old male αLKB1KO mice displayed significantly improved tolerance to intraperitoneal (IP) injection of glucose (Figure 2C), with comparable insulin tolerance to wild type littermate controls (15 min post injection: 62.8 ± 3.8% of initial value in αLKB1KO vs. 70.9 ± 4.3% in wild type; Figure S3a). None of the αAMPK KO mice displayed any alterations in insulin (Figure S3b-d) or glucose tolerance (Figure S4a, c, e).
Figure 2.
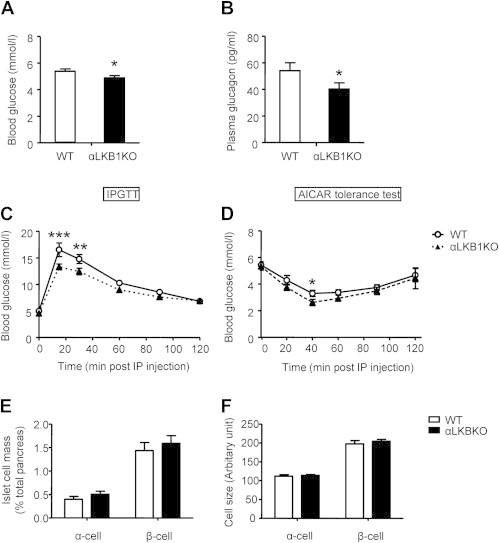
Male αLKB1KO mice display lowered fasting glucose and glucagon levels, and improved glucose and AICAR tolerance. Fasting blood glucose (A) and plasma glucagon levels (B), and intraperitoneal glucose (C) and AICAR (D) tolerance of 8 week-old male mice. Data are expressed as means ± SEM; n = 12–22 (A, B) and 7–12 (C, D) mice per genotype. *p < 0.05, **p < 0.01, ***p < 0.001 between groups using unpaired two-tail t-tests (A, B, E, F) or two-way ANOVA with Sidak post hoc comparison (C, D).
To examine the effects of LKB1 deletion in alpha cells in response to hypoglycemia we first challenged mice with the AMP mimetic aminoimidazole carboxamide ribonucleotide (AICAR) which activates AMPK in extrapancreatic tissues (notably liver and muscle) contributing to the effects of this agent to lower blood glucose levels [40]. As shown in Figure 2D, AICAR sharply reduced blood glucose levels in overnight-fasted eight week-old control male mice from 5.72 ± 0.29 mmol/l at 0 min to 3.37 ± 0.21 mmol/l at 40 min post injection (n = 8). The same manoeuvre lowered blood glucose even more dramatically, from 5.50 ± 0.30 mmol/l to 2.70 ± 0.22 mmol/l in αLKB1KO mice (51 ± 3 vs. 40% ± 3% decrease in KO and wild type respectively, p = 0.04). However, plasma glucagon levels at 40 min post AICAR injection were comparable between genotypes (354.0 ± 62.3 pg/ml in KO vs. 501.7 ± 141.9 pg/ml in WT, n = 5; data not shown). These results suggest that αLKB1KO mice might develop enhanced peripheral sensitivity to AICAR rather than a blunted glucagon response. None of the above differences were apparent in any of the αAMPK KO mice (Figure S4b, d, f).
We next tested whether lower fasting glucagon levels, as observed in αLKB1KO mice, may affect the expression of gluconeogenic genes in the liver [41]. Of three genes (PEPCK, G6Pase and PGC-1α) examined in 24 week-old male αLKB1KO versus control animals, none showed significant differences in expression between genotypes (Figure S4g).
l-arginine stimulates hepatic glucose production, and glucagon and insulin secretion respectively from pancreatic alpha and beta cells [42]. In response to IP injection of l-arginine (3 g/kg) blood glucose levels rose from 5.78 ± 0.27 to 6.95 ± 0.29 mmol/l in wild type mice and from 5.57 ± 0.27 to 5.68 ± 0.41 mmol/l in αLKB1KO mice 10 min after injection (Figure S5a, KO vs. wild type, p = 0.007). Correspondingly, at the same time point, plasma glucagon levels tended to be lower in αLKB1KO (290.5 ± 46.8 pg/ml) vs. wild type mice (389.9 ± 38.9 pg/ml; p = 0.07; Figure S5b). Plasma insulin levels at this time point were not different between genotypes (Figure S5c).
3.3. Enhanced glucose infusion rates in αLKB1KO and αAMPKα1KO mice versus controls under hyperinsulinemic-hypoglycemic clamp and impaired glucagon release during insulin tolerance tests
The above assessments of glucagon release in response to AICAR may conceivably be complicated by impaired AMPK activation in the alpha cell of αLKB1KO mice. We therefore further explored the impact of deleting LKB1 or AMPKα isoforms in the alpha cell using Hyperinsulinemic-hypoglycemic clamps. When αLKB1KO (Figure 3A, B) or αAMPKα1KO (Figure 3C, D) mice were maintained under hypoglycemic conditions by constant intravenous insulin infusion, significantly higher glucose infusion rates were required in each case to maintain ∼3 mmol/l glucose compared to the corresponding littermate controls. Furthermore, and despite showing similar responses in terms of glycaemia during insulin tolerance tests (Figure 3E), αLKB1KO animals showed impaired glucagon secretion under these conditions (Figure 3F). Likewise, αAMPKα1KO mice showed a strong tendency towards lowered glucagon secretion (p = 0.06at 30 min, Figure 3G,H). These data support the view that enhanced glucose disposal observed during clamps in each case was likely due to impaired glucagon release.
Figure 3.

Both αLKB1KO and αAMPKα1KO mice exhibit enhanced glucose sensitivity during hyperinsulinemic-hypoglycemic clamps and impaired glucagon release during insulin tolerance tests. Blood glucose levels (A, C) and glucose infusion rates (B, D) of αLKB1KO (A, B) or αAMPKα1KO mice (C, D) and their wild type littermate controls during insulin (0.6 U/kg/h) and glucose (20%) co-infusion. Arrow heads with dashed lines, bolus insulin infusion; arrow heads with solid lines, constant insulin (0.6 U/kg/h) infusion. (E,F) Changes in plasma glucose (E) and glucagon (F) after injection of 0.75 U/Kg into wild type or αLKB1KO mice. (G,H) as (E,F), using AMPKdKO mice and controls. Data are expressed as means ± SEM; n = 4–7 mice per genotype. *p < 0.05, **p < 0.01, ***p < 0.001 between groups by two-way ANOVA with Sidak post hoc comparison (A–D) or two-tailed Student's t-test with Bonferroni correction (E–H).
No differences were observed in glucose disposal between αAMPKdKO mice vs controls (Figure S6).
3.4. αLKB1KO and αAMPK KO mice display unaltered alpha cell morphology, total mass and single cell size
The above results together suggested that deletion of LKB1 or AMPKα1 from the pancreatic alpha cell impairs glucose deprivation-induced glucagon release in vivo. To determine whether this may be due to a decrease in alpha cell mass, or in the size of individual alpha cells, we measured these parameters by immunohistochemical analysis of pancreatic sections from control, αLKB1KO, αAMPKα1KO or -dKO mice and corresponding controls (Figure 4). This analysis revealed the expected distribution of cell types within islets in each case, with beta cells concentrated in the core and alpha cells located mainly in the mantle (Figure 4A). Quantification of total alpha cell mass (Figure 4B, E, G) and of single alpha cell size after labelling of the plasma membrane with E-cadherin antibodies (Figure 4C) did not reveal significant genotype-dependent differences (Figure 4D, F, H). Correspondingly, growth factor signalling through the phosphatidylinositol 3′ kinase / Akt / mTOR pathway, which is enhanced after deletion of LKB1 from beta cells [28], was unaffected by the same manipulation in alpha cells (Figure S7). Thus, the degree of phosphorylation of ribosomal protein S6 or of its upstream kinase, S6 kinase (both downstream targets of mTOR signalling) [43], was not different in αLKB1KO versus control alpha cells (Figure S7).
Figure 4.

αLKB1KO and αAMPKα1KO, -dKO mouse islets show normal alpha cell morphology, size and mass. (A) Representative immunostaining of pancreatic sections using guinea pig anti-insulin (green, 1:200) and mouse anti-glucagon antibodies (Red, 1:2000). (B, E, G) Quantitation of relative α- and β-cell mass based on immunostaining of pancreatic sections. Relative islet cell mass was calculated by expressing total glucagon (α-cell)- or insulin (β-cell)-positive area as a proportion of total autofluorescent pancreatic area. Three to four sections per pancreas were analyzed. (C) Representative glucagon (red) and E-cadherin (green) staining of pancreatic sections. Nuclei are shown by DAPI (blue) staining. (D, F, H) Quantification of single alpha and beta cell size. Data are expressed as means ± SEM. n = 5 mice per genotype. Scale bar, 40 μm. No statistically significant changes were detected (two-tailed t-test with Bonferroni correction).
3.5. Glucagon secretion is sensitized to inhibition by glucose in islets isolated from αLKB1KO or αAMPKα1KO mice
The above studies suggested that glucagon secretion from alpha cells is likely to be impaired in vivo after the deletion of LKB1 or AMPKα1 by mechanisms which do not affect total alpha cell mass. To explore this possibility, we investigated the effects of genomic deletion of these kinases on the regulation of glucagon secretion from pancreatic alpha cells using islets isolated from αLKB1 KO or αAMPK KO (-α1, -α2, dKO) mice.
Whereas glucagon release at a low, stimulatory concentration (0.1 mmol/l) of glucose was comparable in islets from mice of all genotypes, αLKB1KO or αAMPKα1KO mice released ∼2.5-fold less glucagon in 30 min at 3 mmol/l glucose than the corresponding wild-type islets (Figure 5A,B). Comparable rates of secretion were observed from islets of all genotypes after incubation at 17 mmol/l glucose (Figure 5). Indeed, glucagon secretion from islets of αLKB1KO or αAMPKα1KO mice was maximally inhibited at 3 mmol/l glucose, indicating enhanced sensitivity to the inhibitory effects of the sugar. By contrast, the dose response to glucose of insulin secretion was not different between wild-type or KO islets in either case (data not shown), arguing against the possibility that the enhanced inhibitory effect of 3 mmol/l glucose on glucagon secretion was due to altered insulin or other beta cell secretion [1]. Furthermore, normal stimulatory effects on glucagon secretion in response to 5 μmol/l epinephrine (at 17 mmol/l glucose) were preserved in αLKB1KO (Figure 5A) and in αAMPKαKO (not shown) mouse islets.
Figure 5.
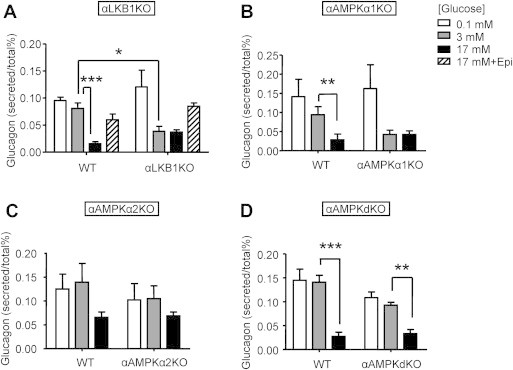
αLKB1 KO and αAMPKα1, -dKO mouse islets secrete less glucagon in response to low (3 mmol/l) glucose. Glucagon secretion from 12 size-matched islets isolated from αLKB1KO (A), α AMPKα1KO (B), α2KO and (C), dKO mice (D) and their respective wild type littermate controls, statically incubated for 1 h at 0.1 mmol/l (white bar), 3 mmol/l (grey bars), 17 mmol/l (black bars) glucose, or 17 mmol/l glucose in the presence of 5 μmol/l epinephrine. Eight week-old male mice were used. Data are expressed as means ± SEM; n = 3–5. *p < 0.05, **p < 0.01, ***p < 0.001 using two-way ANOVA followed by Sidak post-hoc comparison.
Ablation of AMPKα2 (Figure 5C) or of both AMPKα isoforms (Figure 5D) had no effect on the response of glucagon release to 3 mmol/l (vs 0.1 mmol/l) glucose though we noted that the response to 17 mmol/l glucose was impaired after the loss of AMPKα2 (Figure 5C).
4. Discussion
The overall aim of the present study was to explore the cell-autonomous role of LKB1 and of two key downstream kinases in the control of glucagon production in the mouse. The concept that modulation of glucagon levels may represent a useful therapeutic approach to some forms of diabetes has existed for many years [44] and was further supported recently by two separate studies in which mice null for glucagon receptors were shown to be resistant to beta cell destruction by streptozotocin induced diabetes [45,46]. On the other hand, substantial (>90%) [38] ablation of alpha cell mass using diphtheria toxin has relatively modest effects on glucose homoeostasis suggesting alpha cell plasticity under some conditions.
4.1. PPG-Cre expression is localized in alpha cells
The present (Figure 1A) and previous [33] findings suggest that PPG-Cre leads to recombination with high selectivity, but with limited efficiency (∼45%) in alpha cells. Despite this, apparent AMPK activity was markedly (40%) reduced, presumably reflecting the enrichment of LKB1 in alpha and other islet cells [22] and possibly differences between cell types in the expression, stability or regulation of AMPK subunits, or other regulatory enzymes (phosphatases, e.g.). We note that the potential loss of RFP fluorescence during the staining procedure, as well as differences in recombination efficiency at the Rosa26 versus the LKB1 loci, means that these measurements may in any case under (or over-) estimate of the degree of recombination at the latter.
Nonetheless, we observed changes in glucagon secretion in mice deleted for LKB1 or AMPKα1 with this strategy, suggesting an indispensable role of these kinases in the pancreatic alpha cell. Furthermore, it might be speculated that recombination targets a subset of alpha cells which display a greater degree of control of the whole alpha cell population than others, i.e. that these cells play a “hub” or “pacemaker” function [47]. Although a comparison with the effects of more complete deletion of LKB1 or AMPKα1 throughout the alpha cell population would test this hypothesis, alternative alpha cell-targeting strategies, e.g. the “iGlu.Cre” mouse [35], can lead to off-target effects [48].
It is relevant to mention that our studies were limited to examination of the phenotype of animals maintained on the low fat (∼7% of calories) diet usually used in mouse studies, a diet with a fat value much lower than that of the typical westernized human diet (20–25% fat) [49]. Future studies of the knockout models generated here, both in the context of “humanized” diets and after breeding to backgrounds susceptible to diabetes (db/db, e.g.), may also provide interesting information on the role of LKB1 (and AMPK) in controlling glucagon secretion from alpha cells in pathological states in man.
Finally, and although the present findings might seem at odds with the fact that deletion of the majority of the alpha cell complement exerts minor effects on glucose tolerance [38], we note that the effects of hypoglycemia were not examined in this earlier study.
4.2. Deletion of LKB1 in alpha cells does not alter cell size or total pancreatic alpha cell mass
Inactivation of LKB1 selectively in pancreatic beta cells dramatically enhances total insulin output and beta cell mass [26,28]. These changes are accompanied by up-regulation of mTOR signalling [26–28]. Strikingly, deletion of LKB1 from alpha cells here did not affect overall pancreatic alpha cell mass or individual alpha cell size (Figure 4A–D). Such changes, therefore, cannot explain the observed decreases of glucagon output (Figure 2B) and counter-regulation (Figures 2D and 3) seen in vivo in αLKB1KO mice. Analysis of phospho-S6 kinase levels by Western blotting, or staining for phospho-S6 ribosomal protein (rpS6), did not reveal changes in these parameters in alpha cells from αLKB1KO mice compared to controls (Figure S7). Thus, attenuation of mTOR signalling by LKB1 appears to be absent from alpha cells for reasons that are presently unclear.
4.3. LKB1 regulates glucagon secretion from pancreatic alpha cells
The mechanisms through which glucose regulates glucagon release from the pancreatic alpha cell are still incompletely understood. As glucose levels fall, the partial opening of ATP-dependent K+ channels and of low voltage-gated Ca2+ and Na+ channels has been proposed to activate high voltage-gated Ca2+ channels to stimulate glucagon secretion [50]. In an alternative model [51], diminished ATP-dependent Ca2+ accumulation by the endoplasmic reticulum may modulate Ca2+ influx.
We show here that mice null for LKB1 in pancreatic alpha cells exhibit modestly reduced plasma glucose and glucagon levels in vivo (Figure 2A,B) and enhanced insulin sensitivity during hypoglycemic clamps (Figure 3A,B), likely due to reduced glucagon secretion from pancreatic alpha cells. Measurement of portal vein glucagon levels during clamps, which better reflect changes in glucagon secretion from the pancreas than systemic concentrations [52], present a significant technical challenge in the mouse. Moreover, measurement of glucagon from tail vein samples requires the collection of relatively large blood volumes, known to cause substantial increases in catecholamine levels [53]. Consequently, such measurements were not performed here. Nonetheless, examined in isolated islets, inactivation of LKB1 unmasked a clear inhibitory effect of 3 mmol/l glucose on glucagon secretion, a concentration of the sugar at which release was unaffected (versus the rate of release at 0.1 mmol/l glucose) in control islets (Figure 5A). This result is in sharp contrast to the effects of inactivating LKB1 in pancreatic beta cells, where large increases (after one week of deletion in adults) [26,27] or mild decreases (inactivation from E9-11) [28] are seen in glucose-stimulated insulin secretion.
As discussed above, the present results indicate that the absence of LKB1 from alpha cells elicits a marked left-shift in sensitivity to glucose. The mechanism(s) involved will require further investigation but may involve either longer term changes in the expression of key transporters and enzymes involved in glucose sensing (e.g. glucokinase, Ca2+ channels) or a more acute role of LKB1 in mediating the metabolic responses to the sugar, as previously proposed [22]. Detailed transcriptomic and proteomic analyses of purified LKB1-null alpha cells will be required to address the former possibility. Of note, since responses to l-arginine were abnormally reduced in αLKB1KO mice compared to controls (Figure S5b) it seems likely that late events in the triggering of glucagon secretion (e.g. secretory granule fusion at the plasma membrane) require LKB1 and/or its downstream targets.
AMPK is the best studied downstream target of LKB1 and plays key roles in the responses to the anti-diabetic drug, metformin [54]. In contrast to the situation in the beta cell [28,30] we reveal here that AMPKα1 is likely, at least in part, to mediate the effects of LKB1 on glucagon secretion in the alpha cell (Figures 3 and 5). Thus, elimination of AMPKα1 partly mimicked the effects of LKB1 deletion of counter-regulatory responses (Figure 3) and on the control of glucagon release (Figure 5), while loss of AMPKα2 exerted more subtle effects. Paradoxically, elimination of both isoforms had no, or minimal, effects on the above, suggesting that compensatory increases in the expression of other genes may normalize glucagon production. Of note, each of the AMPK-related kinases has similar substrate preferences to the canonical AMPK kinases but is regulated by distinct upstream mechanisms. While MARK2 [26] has been implicated in the effects of LKB1 in beta cells, SIK2 elimination impairs insulin release by permitting the accumulation of the CDK5 activator CDK5R1 (p35) and inhibition of Ca2+ entry [55]. Loss of SIK2 activity in LKB1-depleted alpha cells may thus lead to defective glucagon release in a similar manner. Furthermore, up-regulation of this enzyme in αAMPKdKO islets could conceivably contribute to the normalization of responses in the latter model. Future studies will be needed to test these possibilities.
Acknowledgement
This study was funded by grants to GAR from the Wellcome Trust (Programme 081958/Z/07/Z; Senior Investigator Award WT098424AIA), the MRC (UK; Project GO401641; Programme MR/J0003042/1), and Diabetes UK (BDA 11/0004210). DJH thanks Diabetes UK for an RD Lawrence Fellowship (BDA 12/0004431). The work leading to this publication also received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement n° 155005 (IMIDIA), resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies' in kind contribution. We thank Prof. Nabeel Bardeesy for the kind gift of rabbit anti-LKB1 antibody. GLP-1 assays were performed with the support of the MRC Centre for Obesity and Related Metabolic Diseases (MRC CORD), Cambridge.
Conflict of interest
The authors declare that there is no duality of interest associated with this manuscript.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Gromada J., Franklin I., Wollheim C.B. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocrine Reviews. 2007;28:84–116. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- 2.Cryer P.E. The pathophysiology of hypoglycaemia in diabetes. Diabetes, Nutrition & Metabolism. 2002;15:330–333. [PubMed] [Google Scholar]
- 3.Kaneto A., Miki E., Kosaka K. Effects of vagal stimulation on glucagon and insulin secretion. Endocrinology. 1974;95:1005–1010. doi: 10.1210/endo-95-4-1005. [DOI] [PubMed] [Google Scholar]
- 4.Ahren B., Veith R.C., Taborsky G.J., Jr. Sympathetic nerve stimulation versus pancreatic norepinephrine infusion in the dog: 1). Effects on basal release of insulin and glucagon. Endocrinology. 1987;121:323–331. doi: 10.1210/endo-121-1-323. [DOI] [PubMed] [Google Scholar]
- 5.Taborsky G.J., Jr., Ahren B., Havel P.J. Autonomic mediation of glucagon secretion during hypoglycemia: implications for impaired alpha-cell responses in type 1 diabetes. Diabetes. 1998;47:995–1005. doi: 10.2337/diabetes.47.7.995. [DOI] [PubMed] [Google Scholar]
- 6.Rutter G.A. Regulating glucagon secretion: somatostatin in the spotlight. Diabetes. 2009;58:299–301. doi: 10.2337/db08-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravier M.A., Rutter G.A. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes. 2005;54:1789–1797. doi: 10.2337/diabetes.54.6.1789. [DOI] [PubMed] [Google Scholar]
- 8.Vieira E., Salehi A., Gylfe E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia. 2007;50:370–379. doi: 10.1007/s00125-006-0511-1. [DOI] [PubMed] [Google Scholar]
- 9.Gilon P., Bertrand G., Loubatieres-Mariani M.M., Remacle C., Henquin J.C. The influence of gamma-aminobutyric acid on hormone release by the mouse and rat endocrine pancreas. Endocrinology. 1991;129:2521–2529. doi: 10.1210/endo-129-5-2521. [DOI] [PubMed] [Google Scholar]
- 10.Bailey S.J., Ravier M.A., Rutter G.A. Glucose-dependent regulation of gamma-aminobutyric acid (GABA A) receptor expression in mouse pancreatic islet alpha-cells. Diabetes. 2007;56:320–327. doi: 10.2337/db06-0712. [DOI] [PubMed] [Google Scholar]
- 11.Kawamori D., Kurpad A.J., Hu J., Liew C.W., Shih J.L., Ford E.L. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metabolism. 2009;9:350–361. doi: 10.1016/j.cmet.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishihara H., Maechler P., Gjinovci A., Herrera P.L., Wollheim C.B. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nature Cell Biology. 2003;5:330–335. doi: 10.1038/ncb951. [DOI] [PubMed] [Google Scholar]
- 13.Franklin I., Gromada J., Gjinovci A., Theander S., Wollheim C.B. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes. 2005;54:1808–1815. doi: 10.2337/diabetes.54.6.1808. [DOI] [PubMed] [Google Scholar]
- 14.Ramracheya R., Ward C., Shigeto M., Walker J.N., Amisten S., Zhang Q. Membrane potential-dependent inactivation of voltage-gated ion channels in alpha-cells inhibits glucagon secretion from human islets. Diabetes. 2010;59:2198–2208. doi: 10.2337/db09-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardie D.G. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nature Reviews Molecular Cell Biology. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 16.Xiao B., Sanders M.J., Underwood E., Heath R., Mayer F.V., Carmena D. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 18.Hawley S.A., Pan D.A., Mustard K.J., Ross L., Bain J., Edelman A.M. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metabolism. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Woods A., Dickerson K., Heath R., Hong S.P., Momcilovic M., Johnstone S.R. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metabolism. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Momcilovic M., Hong S.P., Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. The Journal of Biological Chemistry. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Haro L., Garcia-Gimeno M.A., Neumann D., Beullens M., Bollen M., Sanz P. The PP1-R6 protein phosphatase holoenzyme is involved in the glucose-induced dephosphorylation and inactivation of AMP-activated protein kinase, a key regulator of insulin secretion, in MIN6 beta cells. The FASEB Journal. 2010;24:5080–5091. doi: 10.1096/fj.10-166306. [DOI] [PubMed] [Google Scholar]
- 22.Leclerc I., Sun G., Morris C., Fernandez-Millan E., Nyirenda M., Rutter G.A. AMP-activated protein kinase regulates glucagon secretion from mouse pancreatic alpha cells. Diabetologia. 2011;54:125–134. doi: 10.1007/s00125-010-1929-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Silva Xavier G., Leclerc I., Salt I.P., Doiron B., Hardie D.G., Kahn A. Role of AMP-activated protein kinase in the regulation by glucose of islet beta cell gene expression. The Proceedings of the National Academy of Sciences. 2000;97:4023–4028. doi: 10.1073/pnas.97.8.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.da Silva Xavier G., Leclerc I., Varadi A., Tsuboi T., Moule S.K., Rutter G.A. Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochemical Journal. 2003;371:761–774. doi: 10.1042/BJ20021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kefas B.A., Cai Y., Kerckhofs K., Ling Z., Martens G., Heimberg H. Metformin-induced stimulation of AMP-activated protein kinase in beta-cells impairs their glucose responsiveness and can lead to apoptosis. Biochemical Pharmacology. 2004;68:409–416. doi: 10.1016/j.bcp.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Fu A., Ng A.C., Depatie C., Wijesekara N., He Y., Wang G.S. Loss of Lkb1 in adult beta cells increases beta cell mass and enhances glucose tolerance in mice. Cell Metabolism. 2009;10:285–295. doi: 10.1016/j.cmet.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Granot Z., Swisa A., Magenheim J., Stolovich-Rain M., Fujimoto W., Manduchi E. LKB1 regulates pancreatic beta cell size, polarity, and function. Cell Metabolism. 2009;10:296–308. doi: 10.1016/j.cmet.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun G., Tarasov A.I., McGinty J.A., French P.M., McDonald A., Leclerc I. LKB1 deletion with the RIP2.Cre transgene modifies pancreatic beta-cell morphology and enhances insulin secretion in vivo. The American Journal of Physiology – Endocrinology and Metabolism. 2010;298:E1261–E1273. doi: 10.1152/ajpendo.00100.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beall C., Piipari K., Al-Qassab H., Smith M.A., Parker N., Carling D. Loss of AMP-activated protein kinase alpha2 subunit in mouse beta-cells impairs glucose-stimulated insulin secretion and inhibits their sensitivity to hypoglycaemia. Biochemical Journal. 2010;429:323–333. doi: 10.1042/BJ20100231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun G., Tarasov A.I., McGinty J., McDonald A., da Silva Xavier G., Gorman T. Ablation of AMP-activated protein kinase alpha1 and alpha2 from mouse pancreatic beta cells and RIP2.Cre neurons suppresses insulin release in vivo. Diabetologia. 2010;53:924–936. doi: 10.1007/s00125-010-1692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wicksteed B., Brissova M., Yan W., Opland D.M., Plank J.L., Reinert R.B. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010;59:3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bardeesy N., Sinha M., Hezel A.F., Signoretti S., Hathaway N.A., Sharpless N.E. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 33.Herrera P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127:2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- 34.Quoix N., Cheng-Xue R., Guiot Y., Herrera P.L., Henquin J.C., Gilon P. The GluCre-ROSA26EYFP mouse: a new model for easy identification of living pancreatic alpha-cells. FEBS Letter. 2007;581:4235–4240. doi: 10.1016/j.febslet.2007.07.068. [DOI] [PubMed] [Google Scholar]
- 35.Parker H.E., Adriaenssens A., Rogers G., Richards P., Koepsell H., Reimann F. Predominant role of active versus facilitative glucose transport for glucagon-like peptide-1 secretion. Diabetologia. 2012;55:2445–2455. doi: 10.1007/s00125-012-2585-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viollet B., Andreelli F., Jorgensen S.B., Perrin C., Geloen A., Flamez D. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. The Journal of Clinical Investigation. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varadi A., Tsuboi T., Rutter G.A. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 beta-cells. Molecular Biology of the Cell. 2005;16:2670–2680. doi: 10.1091/mbc.E04-11-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thorel F., Damond N., Chera S., Wiederkehr A., Thorens B., Meda P. Normal glucagon signaling and {beta}-Cell function after Near-total {alpha}-Cell ablation in adult mice. Diabetes. 2011;60:2872–2882. doi: 10.2337/db11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lizcano J.M., Goransson O., Toth R., Deak M., Morrice N.A., Boudeau J. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO Journal. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foretz M., Ancellin N., Andreelli F., Saintillan Y., Grondin P., Kahn A. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes. 2005;54:1331–1339. doi: 10.2337/diabetes.54.5.1331. [DOI] [PubMed] [Google Scholar]
- 41.Jiang G., Zhang B.B. Glucagon and regulation of glucose metabolism. The American Journal of Physiology – Endocrinology and Metabolism. 2003;284:E671–E678. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- 42.Unger R.H., Aguilar-Parada E., Muller W.A., Eisentraut A.M. Studies of pancreatic alpha cell function in normal and diabetic subjects. The Journal of Clinical Investigation. 1970;49:837–848. doi: 10.1172/JCI106297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dann S.G., Selvaraj A., Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends in Molecular Medicine. 2007;13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 44.Shah P., Basu A., Basu R., Rizza R. Impact of lack of suppression of glucagon on glucose tolerance in humans. American Journal of Physiology. 1999;277:E283–E290. doi: 10.1152/ajpendo.1999.277.2.E283. [DOI] [PubMed] [Google Scholar]
- 45.Conarello S.L., Jiang G., Mu J., Li Z., Woods J., Zycband E. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia. 2007;50:142–150. doi: 10.1007/s00125-006-0481-3. [DOI] [PubMed] [Google Scholar]
- 46.Lee Y., Wang M.Y., Du X.Q., Charron M.J., Unger R.H. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. 2011;60:391–397. doi: 10.2337/db10-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutter G.A., Hodson D.J. Beta cell connectivity in pancreatic islets: a type 2 diabetes target? Cellular and Molecular Life Sciences. 2014 doi: 10.1007/s00018-014-1755-4. Oct 17. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zac-Varghese S., Trapp S., Richards P., Sayers S., Sun G., Bloom S.R. The Peutz-Jeghers kinase LKB1 suppresses polyp growth from intestinal cells of a proglucagon-expressing lineage in mice. Disease Models & Mechanisms. 2014;7:1275–1286. doi: 10.1242/dmm.014720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drewnowski A., Popkin B.M. The nutrition transition: new trends in the global diet. Nutrition Reviews. 1997;55:31–43. doi: 10.1111/j.1753-4887.1997.tb01593.x. [DOI] [PubMed] [Google Scholar]
- 50.Gopel S.O., Kanno T., Barg S., Weng X.G., Gromada J., Rorsman P. Regulation of glucagon release in mouse -cells by KATP channels and inactivation of TTX-sensitive Na+ channels. The Journal of Physiology. 2000;528:509–520. doi: 10.1111/j.1469-7793.2000.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y.J., Vieira E., Gylfe E. A store-operated mechanism determines the activity of the electrically excitable glucagon-secreting pancreatic alpha-cell. Cell Calcium. 2004;35:357–365. doi: 10.1016/j.ceca.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 52.Pagliassotti M.J., Cherrington A.D. Regulation of net hepatic glucose uptake in vivo. The Annual Review of Physiology. 1992;54:847–860. doi: 10.1146/annurev.ph.54.030192.004215. [DOI] [PubMed] [Google Scholar]
- 53.Ayala J.E., Bracy D.P., McGuinness O.P., Wasserman D.H. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. 2006;55:390–397. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- 54.Long Y.C., Zierath J.R. AMP-activated protein kinase signaling in metabolic regulation. The Journal of Clinical Investigation. 2006;116:1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sakamaki J.I., Fu A., Reeks C., Baird S., Depatie C., Al A.M. Role of the SIK2-p35-PJA2 complex in pancreatic beta-cell functional compensation. Nature Cell Biology. 2014;16:234–244. doi: 10.1038/ncb2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.