

Abstract
Objective
Glucagon-like peptide 1 (GLP-1) enhances insulin secretion and protects β-cell mass. Diabetes therapies targeting the GLP-1 receptor (GLP-1R), expressed in numerous tissues, have diminished dose-response in patients with type 2 diabetes compared with healthy human controls. The aim of this study was to determine the mechanistic causes underlying the reduced efficacy of GLP-1R ligands.
Methods
Using primary mouse islets and the β-cell line MIN6, outcomes downstream of the GLP-1R were analyzed: Insulin secretion; phosphorylation of the cAMP-response element binding protein (CREB); cAMP responses. Signaling systems were studied by immunoblotting and qRT-PCR, and PKA activity was assayed. Cell surface localization of the GLP-1R was studied by confocal microscopy using a fluorescein-tagged exendin-4 and GFP-tagged GLP-1R.
Results
Rodent β-cells chronically exposed to high glucose had diminished responses to GLP-1R agonists including: diminished insulin secretory response; reduced phosphorylation of (CREB); impaired cAMP response, attributable to chronically increased cAMP levels. GLP-1R signaling systems were affected by hyperglycemia with increased expression of mRNAs encoding the inducible cAMP early repressor (ICER) and adenylyl cyclase 8, reduced PKA activity due to increased expression of the PKA-RIα subunit, reduced GLP-1R mRNA expression and loss of GLP-1R from the cell surface. To specifically examine the loss of GLP-1R from the plasma membrane a GLP-1R-GFP fusion protein was employed to visualize subcellular localization. Under low glucose conditions or when PKA activity was inhibited, GLP-1R-GFP was found at the plasma membrane. Conversely high glucose, expression of a constitutively active PKA subunit, or exposure to exendin-4 or forskolin led to GLP-1R-GFP internalization. Mutation of serine residue 301 of the GLP-1R abolished the glucose-dependent loss of the receptor from the plasma membrane. This was associated with a loss of an interaction between the receptor and the small ubiquitin-related modifier (SUMO), an interaction that was found to be necessary for internalization of the receptor.
Conclusions
These data show that glucose acting, at least in part, via PKA leads to the loss of the GLP-1R from the cell surface and an impairment of GLP-1R signaling, which may underlie the reduced clinical efficacy of GLP-1R based therapies in individuals with poorly controlled hyperglycemia.
Keywords: GLP-1 receptor, Protein kinase A, Hyperglycemia, Small ubiquitin-related modifier
1. Introduction
Incretins are peptide hormones secreted by intestinal endocrine cells in response to nutrient stimulation [1]. At the β-cells of pancreatic islets of Langerhans, incretins act to enhance insulin synthesis and secretion, reduce apoptosis and, at least in rodents, may stimulate β-cell proliferation [2]. These hormones, principally glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP), bind to their Gαs-coupled receptors, GLP-1R and GIP-R, respectively, and mediate their insulinotropic and β-cell survival effects largely via increased intracellular cAMP [2,3]. This cAMP signal is transduced via the cAMP-dependent protein kinase (PKA) and the exchange proteins activated by cAMP (EPAC) [2]. Novel therapies targeting the β-cell GLP-1R and GIP-R have been introduced over the past decade to treat type 2 diabetes through the derived benefit of enhanced insulin secretion and the possibility of improved preservation of β-cell mass. Therapies targeting the GLP-1R have delivered significant sustained benefits to glucose control and β-cell function out to 4 years of treatment [4–6]. However there is a diminished β-cell response to GLP-1R agonists in pre-diabetic individuals and patients with Type 2 diabetes [7–10]. This reduced efficacy of GLP-1R agonists may involve the β-cells themselves, either through downregulation of the GLP-1R or of signaling systems lying downstream of the receptor [11]. Administration of the GLP-1R agonist, liraglutide, to young db/db mice with only moderate hyperglycemia provides more robust β-cell responses than in older, more hyperglycemic mice [12], indicating that hyperglycemia may be a contributing factor to the diminished efficacy of GLP-1 agonists in type 2 diabetes. Consistent with this, intensive insulin therapy to normalize glucose levels preceding GLP-1R administration improves the insulin secretory response in individuals with type 2 diabetes [13,14], whereas disruption of glucose homeostasis through the induction of insulin resistance diminishes the potentiating effects of GLP-1 upon insulin secretion in human subjects [15]. Understanding the mechanisms by which poorly controlled glucose diminishes GLP-1R signaling at the β-cell raises the potential for developing strategies to improve the effectiveness of GLP-1R targeting therapies. Rodent studies have shown that chronically elevated glucose downregulates both GLP-1R and GIP-R gene expression in vivo [16,17]. Glucose is also considered likely to activate PKC isoforms in the β-cell [18], which may be the stimulus for PKC-mediated phosphorylation of the GLP-1R that leads to its downregulation [19–21]. Homologous GLP-1R activation has been shown to downregulate the receptor rapidly [21,22], consistent with a classical negative feedback system that may be mediated via cAMP signaling. Glucose raises β-cell cAMP levels through the activation of calcium-sensitive adenylyl cyclases [23–25]. Recently we reported that hyperglycemia induces expression of the components of the SUMO (small ubiquitin-related modifier protein) pathway, which is associated with covalent modification of GLP-1R by SUMO-1 [26]. This results in downregulation of GLP1-R expression at the cell surface and impairment of GLP-1R-dependent potentiation of insulin secretion. Here we extend those findings to show that chronically elevated glucose acts via PKA to reduce GLP-1R signaling through a SUMO-1-dependent mechanism.
2. Materials and methods
2.1. Animals
Mice (8–10 weeks old C57BL/6J males) obtained from Harlan Laboratories and housed under conditions approved by the in the University of Chicago IACUC were used for physiological analysis and for islet isolations. Intraperitoneal glucose tolerance tests were performed in mice administered either saline or exendin-4 (at 5 μg/kg) 1 h prior to a bolus of d-glucose (2 g/kg body weight) or in mice which had received exendin-4 at 4–6 h intervals for the preceding 24 h before being administered a final dose of exendin-4 1 h prior to a bolus of d-glucose (2 g/kg body weight).
2.2. Cell culture and islet isolation and transfection
Glucose responsive early passage MIN6 cells (passage <30) used for all the experiments were grown in DMEM supplemented with 15% Fetal Bovine Serum (FBS), 100 IU/ml penicillin, and 100 mg/ml streptomycin and 3–8 (low) or 25 (high) mM glucose. Islets were isolated from 8 to 10 weeks old C57BL/6J wild-type mice (Jackson Laboratory, Bar Harbor, ME) following a protocol approved by The University of Chicago IACUC. Islets, MIN6 and isolated primary cells were transfected with Lipofectamine 2000 (Life Technologies, cat. #116688). Culture media were supplemented with exendin-4 at 10 nM (American Peptide Co. cat. # 46-3-12A), forskolin at 2 μM (Sigma–Aldrich, cat. # F6886), and H89 at 20 μM (Cell Signaling Technologies, cat # 9844). Cells were infected with adenovirus expressing an activated catalytic PKA subunit [27] or a mutated, dominant negative PKA regulatory subunit [28] to manipulate PKA activity according to a previously described protocol [29].
2.3. FRET analysis
FRET measurement of dynamic changes in cAMP was obtained in cells transfected with a plasmid expressing Epac-camps [30]. Islets were trypsinized into small primary cell clusters and cultured in RPMI 1640 medium supplemented with 10% FBS, 2 mM l-glutamine, 100 IU/ml penicillin, and 100 mg/ml streptomycin. Islet cells were co-transfected with the Epac-camps plasmid and a vector expressing monomeric red fluorescent protein driven by rat insulin 2 promoter to identify β-cells. This approach provides inefficient transfection but sufficient co-transfected cells are obtained to perform the analysis. Live cells were imaged 48 h post-transfection in KR2 buffer on a Nikon inverted epifluorescence microscope with a CCD camera. Pancreatic β-cells (Epac-camps and mRFP positive) from multiple transfections had fluorescence recorded before and during stimulation with 50 nM exendin-4. Dynamic changes in cAMP were estimated from the change in FRET ratio (acquired at 5 s intervals) by MetaFluor software (Universal Imaging) following direct addition of stimulatory glucose plus exendin-4 to the culture chamber. GLP-1R/SUMO-1 interactions were determined by live cell imaging using FRET in MIN6 cells transfected with GLP-1R-CFP or GLP-1R(S301A)-CFP and SUMO-1-YFP or SUMO-1(GG)-YFP. Cells were imaged as for analysis of cAMP. Dynamic changes in protein–protein interaction following addition of 2 μM Forskolin was observed as a decrease in FRET ratio.
2.4. Cell surface protein biotinylation
Cell surface proteins of MIN6 cells cultured for 24 h at low or high glucose were biotinylated for 45 min and purified (Thermo FisherScientific EZ-Link Sulfo-NHS-Biotinylation Kit, cat # 21425) from lysates prepared in RIPA buffer (Santa Cruz Biotechnology, cat # sc-24948). Biotinylated GFP-tagged GLP-1R receptor (membrane) and non-biotinylated receptor (cytosolic) were detected using an anti-GFP antibody (Roche Applied Science, cat # 11814460001).
2.5. ELISA and immunoblotting analyses
Insulin was quantified in the media and lysate from cells cultured under the relevant conditions for 2 h (ELISA ALPCO, Salem, NH, cat. # 80-INSMSU-E01). Cyclic AMP was quantified by ELISA (Thermo Scientific, cat # EMSCAMPL) in lysates from MIN6 cells cultured for 20 h at low (3 mM) or high (25 mM) glucose, then exposed to high glucose ± exendin-4 (10 nM) for 10 min. Immunoblotting used the following antibodies: anti-phospho-CREB (Cell Signaling Technologies, cat # 4276, diluted 1:1000); anti-total CREB (Cell Signaling Technologies, cat # 4820, diluted 1:1000); anti-PKA-RIα (Cell Signaling Technologies, cat # 5675, diluted 1:1000); anti-PKA RIIα (BD Transduction Laboratories, cat # 612242, diluted 1:1000); anti-PKA-RIIβ (BD Transduction Laboratories, cat # 610625, diluted 1:1000); anti-PKA-Cα (BD Transduction Laboratories, cat # 610980, diluted 1:1000); anti-FLAG (Cell Signaling Technologies, cat # 8146, diluted 1:500); β-catenin (Cell Signaling Technologies, cat # 9562, diluted 1:1000); anti-tubulin (Cell Signaling Technologies, cat #5346, diluted 1:2000).
2.6. Measurement of RNA levels by qRT-PCR
RNA was isolated from MIN6 cells (Qiagen RNEasy). Equimolar RNA from each sample was reverse transcribed using the First Strand cDNA Synthesis kit (Life Technologies). qPCR was performed using 20 ng of cDNA from replicate samples and the FAST SYBR Master Mix (Life Technologies). Samples were run on an iCycler with MyiQ module (BioRad). Expression was determined by comparative CT, relative to the 18S rRNA internal control (Life Technologies). Primers (Table 1) were designed using Primer3 software.
Table 1.
Oligonucleotide primer pairs used for qRT-PCT analysis of mRNA levels.
| Target RNA | Forward primer | Reverse primer |
|---|---|---|
| ICER | TGAAACTGATGAGGAGACTGACC | AGTTGCTGGGGACTGTGC |
| AC8 | GAGGAATCCCTGGGAGGAT | TCCTCAGGCTGCTTAATCAAA |
| PKA-Cα | CAAGCAGAAGGTGGTGAAGC | CCAGCTACATACTCCATGACCAT |
| PKA-RIα | GTTATTCAGCAAGGTGATGAAGG | TGGCTGCTCTGGGTGTTC |
| PKA-RIIα | ATCCAAGGGTGGTTCATCC | TGCTCGTCAGTTTTGACAATCT |
| PKA-RIIβ | GCGTTCAACGCTCCAGTTA | GCAAGCCTCTTGCAATCTGT |
| GLP-1R A | TTGGCTTCAGACACTTGCAC | CCATCCCACTGGTGTTGC |
| GLP-1R B | CAGCGCATCTTCAAGCTGTA | CCGATAGCAAAGAGAATGGGC |
| GLP-1R C | ACTCTCCTTCACTTCCTTCCA | GACACTTGAGGGGCTTCATG |
2.7. PKA activity assay
PKA activity was measured in lysates prepared from in MIN6 cells cultured for 20 h at low (3 mM) or high (25 mM) glucose using a method previously described [31]. Briefly, 10 μg aliquots of MIN6 lysate were incubated with the kinase substrate, kemptide, cAMP at the indicated concentration, and [32P]γATP for 10 min. Reaction mixes were applied to P10 membranes and washed in phosphoric acid. Membranes were scintillation counted to determine the amount of 32P incorporated into protein. Values were corrected for non-specific 32P incorporation by including control samples incubated in the presence of the PKA inhibiter, PKI. This assay measures PKA activity dependent upon added (exogenous) cAMP that reflects PKA subunit expression and does not reflect PKA activity dependent upon cellular cAMP levels.
2.8. Fluorescein-tagged exendin-4 binding assay
MIN6 cells were fixed with 2% PFA for 5 min, washed 3 times in PBS and incubated with 100 nM fluorescein tagged exendin-4 (Anaspec Inc., cat # 63899) and 0.2 mg/ml BSA for 20 min. Cells were imaged on a Leica STED SP5 laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany). Integrated density was calculated with ImageJ software. Fluorescein-tagged-Exendin-4 binding was quantified in a colorimetric assay using a monoclonal HRP conjugated anti-fluorescein antibody (Jackson Immuno Research, 200-032-037). Upon addition of a colored substrate (Roche, cat # 11484281001) the signal was measured at 450 nm.
2.9. Statistical analyses
Data, expressed as mean ± SD, were analyzed by unpaired Student's t-tests, one-way ANOVA, and two-way ANOVA with Bonferroni post hoc tests using GraphPad Software. P < 0.05 was considered significant.
3. Results
3.1. High glucose downregulates β-cell responses to GLP-1 receptor activation
Pancreatic β-cells secrete insulin in response to elevated glucose concentrations [32–34]. Insulin secretion was stimulated with high glucose, and this was further potentiated with the GLP1-R agonist, exendin-4, in both primary mouse islets and in the immortalized β-cell line, MIN6. However, islets and MIN6 cells chronically maintained at high glucose in vitro showed a significantly reduced potentiation of glucose stimulated insulin secretion in response to GLP-1R activation with exendin-4 (Figure 1A, B).
Figure 1.

Chronic high glucose impairs insulin secretory responses. Primary mouse islets (A) and MIN6 cells (B) were cultured chronically at low glucose (white bars) or high glucose (black bars). Islets and MIN6 cells pre-cultured at low glucose were either left at low glucose (LG) or switched to high glucose in the absence (HG) or presence of exendin-4 (HG + E). Islets and MIN6 cells pre-cultured at high glucose were either left at high glucose (HG) or had exendin-4 added to the medium (HG + E). Insulin values were expressed as a percentage of the cellular insulin content (*, P < 0.05; ns, not significant by 1-way ANOVA).
Elevation of cAMP in β-cells activates PKA, a major target of which is the cAMP-response element binding protein, CREB, which becomes phosphorylated at serine 133. In lysates of cells chronically maintained at low glucose, CREB phosphorylation at serine 133 was shown by immunoblotting to be induced in response to elevated glucose alone and more potently in response to elevated glucose plus exendin-4 (Figure 2A). In contrast, MIN6 cells maintained chronically at high glucose failed to phosphorylate CREB after exendin-4 stimulation. To determine the time course of these events, MIN6 cells were cultured at low glucose then switched to high glucose for 1–20 h and the ability of exendin-4 to induce CREB phosphorylation determined (Figure 2B). Remarkably, CREB phosphorylation was found to be impaired within an hour of culturing at high glucose. Conversely, to determine the time course of the restitution of the CREB phosphorylation response, cells were maintained in high glucose and then transferred to low glucose. CREB phosphorylation in these cells was not responsive to glucose plus exendin-4 prior to 10 h and was not fully restored until 20 h, consistent with a hysteresis effect in this system (Figure 2C).
Figure 2.
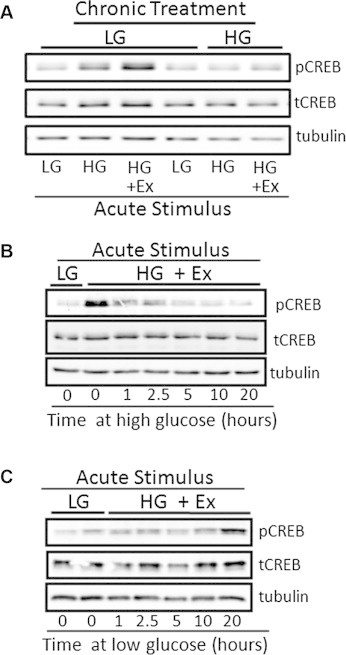
Chronic high glucose impairs CREB phosphorylation activation. The phosphorylation of the cAMP-response element binding protein (pCREB) in lysates of MIN6 cells in response to high glucose + exendin-4 (HG + Ex) for 15 min was determined by immunoblotting. MIN6 cells were pre-cultured chronically (20 h) either at low or high glucose. Immunoblotting was controlled by blotting for total CREB (tCREB) and tubulin. (A). MIN6 cells were chronically maintained at low or high glucose then treated for 15 min with low glucose (LG), high glucose (HG) or high glucose in the presence of the GLP-1R agonist exendin-4 (HG + Ex). (B). MIN6 cells were switched from low glucose to high glucose for the time indicated (0–20 h) to determine the time-course of the loss of the CREB phosphorylation response. (C). MIN6 cells were switched from high to low glucose for the time indicated (0–20 h) to determine the time-course of the restoration of the CREB phosphorylation response.
Since the response to exendin-4 is in large part mediated via cAMP, the ability of β-cells maintained chronically at low or high glucose to respond to exendin-4 by generating cAMP was determined. Using a FRET-based reporter, Epac-camps [30], cAMP levels in islets and MIN6 β-cells maintained at low glucose responded robustly to the addition of exendin-4 (Figure 3A,B). However, in islets and MIN6 cells chronically exposed to high glucose, the cAMP response was impaired. Since the FRET assay measures the relative change in CFP/YFP ratio it cannot be determined whether the reduced response of cAMP levels to GLP-1R activation is due to a high basal level or a reduced response of cAMP generation. To determine actual cAMP concentrations we used a cAMP ELISA (Figure 3C). In MIN6 cells pre-cultured at low glucose, glucose stimulated a rise in cAMP concentrations and this was potentiated by exendin-4. However, in MIN6 cells chronically maintained at high glucose, cAMP levels were elevated above those seen in cells maintained at low glucose and these cells glucose showed a diminished response to the addition of exendin-4. The potentiation of cAMP levels in response to exendin-4 was only 40% in cells maintained chronically at high glucose versus 80% in cells maintained at low glucose (Figure 3C). These data show that chronically elevated glucose raises intracellular cAMP, which is associated with a diminished cAMP response to exendin-4.
Figure 3.

Chronic hyperglycemia reduces the cAMP response to exendin-4. The generation of cAMP was studied using a FRET-based cAMP reporter in isolated islets (A) and MIN6 cells (B) or by cAMP ELISA in MIN6 cells (C). Changes in cAMP were measured using the Epac-cAMP FRET biosensor during a 5 min low glucose period after which high glucose with exendin-4 was added to the culture chamber (A, B). Change in FRET ratio values in the low glucose versus high glucose plus exendin-4 periods were expressed relative to time = 0 values. Changes in Epac-cAMP FRET following high glucose plus exendin-4 addition were analyzed by t-tests (data expressed as mean ± SD; P < 0.0001, n = 7–9). The levels of cAMP were measured using a cAMP ELISA in MIN6 cells pre-cultured for 20 h at low or high glucose. The levels of cAMP were measured 10 min after addition of high glucose (HG) or HG with exendin-4 (HG + E). Data was analyzed by 1-way ANOVA; with *** indicating P < 0.001. n = 5–14).
3.2. Hyperglycemia acts at multiple levels to reduce GLP-1R signaling
The preceding data show that although cAMP levels are elevated under hyperglycemic conditions, the ability of the β-cells to respond with increased insulin secretion and CREB phosphorylation is impaired. This indicates that components of the GLP-1R signaling system are altered by hyperglycemia. Glucose can increase cAMP via a rise in calcium, which activates the calcium sensitive adenylyl cyclase 8 [35,36]. Quantitative RT-PCR analysis of the levels of the mRNA encoding AC8 were elevated 3.3-fold (Figure 4A), consistent with the increased cAMP levels in response to hyperglycemia. Previously, it has been reported that chronic exendin-4 activation of the GLP-1R induces ICER (inducible cAMP early repressor), a negative regulator of CREB binding to CRE DNA sequences [37]. Here ICER mRNA was elevated 5.0-fold by chronic (20 h) exposure of MIN6 cells to hyperglycemia (Figure 4B), consistent with both exendin-4 and hyperglycemia acting to raise cAMP levels.
Figure 4.

Hyperglycemia down-regulates GLP-1R signaling. MIN6 cells cultured chronically at low or high glucose were analyzed for effects of hyperglycemia upon GLP-1R signaling systems. RNA was prepared for qRT-PCR analysis (A, B, D, G) or protein lysates were prepared for PKA activity assays (C) and immunoblotting (E). RNA expression of adenylyl cyclase 8 (AC8; A), the inducible cAMP early repressor (ICER; B), the PKA subunits (PKA-Cα, RIα, RIIα, and RIIβ; D) and the GLP-1R (G) was determined by qRT-PCR. Lysates prepared from MIN6 cells cultured at low glucose (LG; white bars) or high glucose (HG; black bars) were analyzed for: (C) PKA activity in the presence of 100 nm added cAMP and (D) for expression of the PKA subunits PKA-Cα, RIα, RIIα and RIIβ by immunoblotting, with β-tubulin presented as a loading control. (F) MIN6 cells chronically cultured at low or high glucose were fixed in 4% PFA, and the binding of fluorescein-tagged exendin-4 quantified using an anti-fluorescein-HRP antibody. Binding was quantified as the integrated density of HRP activity (Student's t-test, P < 0.0001, n = 4).
The cAMP signal is transduced largely via the cAMP-dependent protein kinase, PKA. PKA activity and PKA subunit expression were analyzed to determine whether hyperglycemia had direct effects upon PKA. Lysates prepared from MIN6 cells chronically cultured at high glucose showed a significant (53%) reduction in PKA activity, in the presence of 100 nM cAMP, compared to lysates prepared from MIN6 cultured at low glucose (Figure 4C). Measurement by qRT-PCR of mRNAs encoding PKA catalytic (PKA-C) and regulatory subunits (PKA-R) showed a significant change only for PKA-RIα (Figure 4D). Consistent with this, protein levels of the PKA-RIα subunit were elevated in response to high glucose, while PKA-C, PKA-RIIα and PKA-RIIβ were unaffected (Figure 4E). To determine whether the GLP-1R itself is downregulated, we used fluorescein-tagged exendin-4 to quantify GLP-1R at the plasma membrane. This assay revealed a dramatic 5-fold reduction in cell surface expression of the GLP-1R after chronic exposure to elevated glucose levels (Figure 4F). However, a previous study has reported that expression of the mRNA encoding the GLP-1R is itself downregulated by hyperglycemia [17]. To determine whether we could observe a similar downregulation, qRT-PCR was performed on cDNA samples prepared from MIN6 cells cultured for 20 h at low (3 mM) or high (25 mM) glucose. Three oligonucleotide primer pairs, covering different regions of the GLP-1R coding region (Table 1), were used to address the possibility of multiple transcripts arising from the GLP-1R gene [38]. Each oligonucleotide pair showed a similar pattern, with the GLP-1R mRNA level reduced by about 50% within an hour of a shift from low to high glucose (Figure 4G). These data show that chronic high glucose, which elevates cAMP, attenuates the ability of β-cells to respond to GLP-1R activation through multiple alterations of the GLP-1R/cAMP/PKA signaling system, including both gene expression and subcellular localization. However, it is unclear whether the loss of GLP-1R from the plasma membrane solely follows the decrease in GLP-1R gene expression, or whether additional regulatory mechanisms are involved.
3.3. The GLP-1R is lost from the plasma membrane with hyperglycemia
To determine whether mechanisms independent of decreased GLP-1R mRNA are responsible for the reduced expression of GLP1-R at the plasma membrane, the receptor was removed from the control of its endogenous promoter. To achieve this, a plasmid construct was prepared in which a GFP-tagged version of the GLP-1R was expressed under the control of the CMV promoter, allowing visualization of the receptor as a GLP-1R-GFP fusion protein. In MIN6 cells maintained under low glucose conditions GLP-1R-GFP was localized to the plasma membrane. In contrast, in cells maintained at high glucose the receptor was predominantly intracellular (Figure 5A), consistent with the data presented above (Figure 4F) that shows loss of fluorescein-tagged exendin-4 binding to the endogenous receptor. Glucose induces generation of cAMP, which activates PKA in pancreatic beta cells [23–25,39]. We hypothesized that glucose, acting via cAMP, induces PKA to down-regulate GLP-1R expression at the cell surface. We tested this hypothesis by evaluating the effect of PKA blockade. Accordingly, treatment of MIN6 cells with high glucose in the presence of the PKA inhibitor, H89, blocked the depletion of GLP-1R-GFP at the plasma membrane (Figure 5A). On the other hand, activating adenylyl cyclases would be expected to act oppositely and enhance the disappearance of GLP-1R from the cell membrane. We indeed found that expression of a constitutively active PKA subunit (PKA-CαRQ) [27] resulted in the severe reduction of plasma membrane localization of the GLP-1R under low glucose conditions, when the receptor is normally present there (Figure 5B). Furthermore, cells cultured in the presence of forskolin, to activate adenylyl cyclases, also exhibited a loss of GLP-1R from the cell surface (Figure 5C). The forskolin effects were more pronounced than the effect of high glucose alone, consistent with the potent effects of forskolin to raise cAMP. The involvement of PKA was further supported by the observation that the PKA inhibitor, H89, prevented the effect of forskolin to deplete GLP-1R-GFP at the plasma membrane (Figure 5C). Likewise, inhibition of PKA activity through the expression of a dominantly negative PKA-RIα subunit (PKA-RIαAB) [31] prevented loss of GLP-1R-GFP from the cell surface (Figure 5C). These data show that the activation of PKA correlates with reduced localization of the GLP-1R to the cell surface, and that blocking PKA activity prevents this change.
Figure 5.

PKA activity correlates with loss of GLP-1R from the cell surface. MIN6 cells were transfected with a GFP-tagged GLP-1R (GLP-1R-GFP) expressed under the control of the constitutive CMV promoter. (A) GLP-1R-GFP transfected MIN6 cells were cultured at low glucose (3 mM; LG) or high glucose (25 mM; HG) for 4 h in the presence or the absence of the PKA inhibitor, H89 (HG + H89). GFP was visualized in green, nuclei stained with dapi (blue) and β-catenin immune-stained to mark the plasma membrane (red). (B) GLP-1R-GFP transfected MIN6 cells were infected with a recombinant adenovirus expressing a constitutively active PKA catalytic subunit (caPKA). Cells were cultured at low glucose (LG), and the localization of the GLP-1R-GFP was determined by fluorescence microscopy. Cells infected with the caPKA adenovirus were identified using an antibody against the FLAG-tag of the caPKA (red). (C) GLP-1R-GFP transfected MIN6 cells were cultured for 4 h at high glucose (HG) with: forskolin (Fsk), to raise cAMP levels through the activation of adenylyl cyclases; H89, to inhibit PKA; or expression of a dominantly negative PKA regulatory subunit (dnPKA). GLP-1-GFP was visualized in green, nuclei by staining with dapi (blue), and the FLAG-tag epitope on the dnPKA in red.
3.4. Loss of GLP-1R from the plasma membrane requires a PKA/SUMO interaction
To determine whether PKA acts directly upon the GLP-1R to cause its loss from the cell surface, three PKA consensus phosphorylation site serine residues (serine 193, serine 301 and serine 433) were individually mutated to alanine residues in the GLP-1R-GFP construct. Cell surface proteins were biotinylated and separated by streptavidin affinity purification from MIN6 cells maintained at low or high glucose for 20 h. Lysates from the biotinylated fraction (plasma membrane) or the non-biotinylated fraction (cytosolic) were analyzed by immunoblotting for GFP (Figure 6A). In MIN6 cells infected with wild type GLP-1R sequence, GFP immunoreactivity was largely in the membrane fraction at low glucose (73 ± 7.2% at the membrane) and largely in the cytosolic fraction at high glucose (16.7 ± 0.3% at the membrane; Figure 6A), consistent with the data presented above. Mutation of serine residues at 193 and 433 of the GLP-1R similarly showed localization of the receptor to the plasma membrane at low glucose and with the cytosolic fraction at high glucose (membrane localization 71.2 ± 1.8% and 65.3 ± 1.6% at low glucose, 28.4 ± 10.4 and 20 ± 6.2% at high glucose, respectively). However, mutation of serine 301 resulted in the loss of this regulation, with the receptor largely found at the cell surface regardless of glucose concentration (membrane localization 63.9 ± 3.1% at low glucose versus 69.1 ± 3.3% at high glucose; Figure 6A,B).
Figure 6.
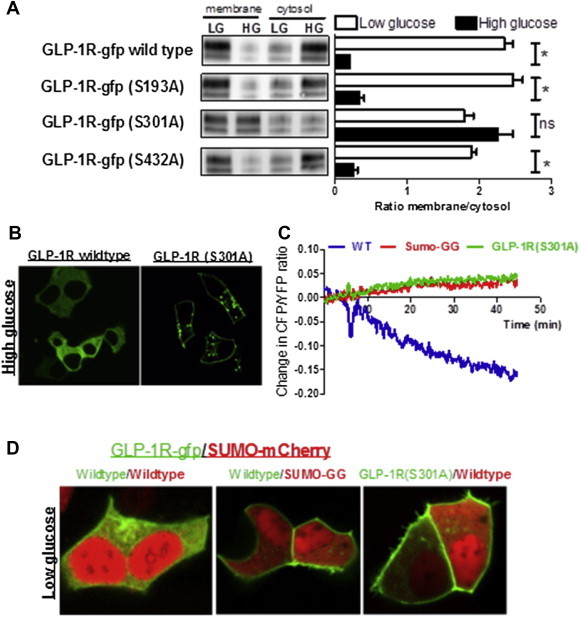
GLP-1R serine 301 mediates the glucose-dependent membrane downregulation. (A) MIN6 cells expressing wild type GLP-1R-GFP or the mutant gfp-tagged receptors S193A, S301A, or S430A, were chronically cultured at low glucose (LG) or high glucose (HG) before cell surface proteins were biotinylated and purified by streptavidin affinity. Biotinylated (membrane) and non-biotinylated (cytosol) proteins were immunoblotted for GFP and quantified by densitometry (A. t-test, P = 0.002, n = 3). (B) MIN6 cells infected with either the wild type GLP-1R-GFP or the S301A mutated GLP-1R-GFP were chronically maintained at high glucose and GLP-1R-GFP localization determined by fluorescence microscopy. (C) Determination of GLP-1R/SUMO-1 interaction using FRET of CFP-tagged GLP-1R (GLP-1R-GFP) and YFP-tagged SUMO-1 (SUMO-YFP); Blue, MIN6 cells expressing wild type GLP-1R-GFP and wild type SUMO-1-mCherry; Red, MIN6 cells expressing wild type GLP-1R-GFP and SUMO-GG mutant SUMO-1-mCherry; Green, MIN6 cells expressing S301A mutant GLP-1R-GFP and wild type SUMO-1-mCherry. Data collected from 8 to 10 cells on multiple plates. (D) Fluorescence microscopy image of MIN6 cells cultured at low glucose showing localization of GLP-1R-GFP (wild type or S301A mutant, green) with the expression of SUMO-1-mCherry (wild type or SUMO-GG mutant, red).
SUMO proteins are covalently conjugated to target proteins in order to modify their function. Previously, we showed that glucose increases expression of SUMO-1 in β-cells, resulting in the intracellular retention of the GLP-1R through covalent SUMO-1modification (SUMOylation) of the receptor [26]. To determine whether SUMOylation and PKA phosphorylation of GLP1-R were linked, a CFP-tagged GLP-1 receptor (GLP1-R-CFP) and a YFP-tagged SUMO-1 (SUMO-YFP) were co-expressed in MIN6 cells. Upon the addition of forskolin, the CFP/YFP fluorescence ratio decreased, indicating increased interaction between GLP-1R-CFP and SUMO-1-YFP (Figure 6C). We then examined the effects of SUMO(GG)-YFP, a conjugation deficient SUMO-1 mutant in which a stop codon introduced at G96 removes the last 4 amino acids, which are essential for covalent modification of target proteins. This truncated construct abolished the change in CFP/YFP ratio in response to forskolin. Similarly, the GLP1-R-CFP with the S301A mutation was unable to interact with SUMO-YFP in response to forskolin, indicating the specificity of cAMP dependent increase in interaction between GLP-1R-CFP and SUMO-1-YFP (Figure 6C). Consistent with these data, over-expression of SUMO using an mCherry-tagged SUMO-1 led to intracellular retention of the GLP1-R-GFP in MIN6 cells maintained at low glucose (Figure 6D). Expression of SUMO(GG), to prevent SUMOylation of target proteins, or the GLP1-R(Ser301A)-GFP, to prevent PKA phosphorylation of the GLP1-R, abolished both forskolin and SUMO-1 overexpression-induced intracellular retention of GLP-1R. These results indicate that the phosphorylation of serine 301 by PKA promotes SUMOylation of the GLP1-R, thereby resulting in intracellular localization of the receptor under chronic high glucose conditions.
3.5. Chronic GLP-1R activation downregulates GLP-1R signaling
The data presented above show that the activation of PKA by glucose downregulates GLP-1R expression at the plasma membrane. This indicates that other stimuli of the cAMP/PKA signaling system are also likely to lead to loss of the GLP-1R from the cell surface. Since GLP-1R activation itself raises cAMP and activates PKA [2,40], chronic exposure to GLP-1R agonists should also reduce cell surface GLP-1R and impair downstream signaling. Consistent with this hypothesis, chronic exposure of MIN6 cells cultured at low glucose in the presence of the GLP-1R agonist exendin-4, led to the dramatic loss of fluorescein-tagged exendin-4 binding to the cell surface (Figure 7A). Consistent with this being due to PKA activity, MIN6 cells expressing the GLP-1R-GFP and exposed to exendin-4 in the present of high glucose, had GFP expression restored to the plasma membrane upon the addition of the PKA inhibitor, H89 (Figure 7B). These data indicate that chronic exendin-4 mediated PKA activation should, like chronic hyperglycemia, reduce the effects of GLP-1R activation upon insulin secretion.
Figure 7.

Chronic exposure of β-cells to the GLP-1R agonist exendin-4. (A) MIN6 cells were chronically cultured (20 h) at low glucose in the absence (LG) or the presence (LG + Ex4) of the GLP-1R agonist exendin-4, fixed and incubated with fluorescein-tagged exendin-4 to bind cell surface (but not internal) GLP-1R. (B) MIN6 cells transfected with GLP-1R-GFP were cultured for 4 h at high glucose with exendin-4 in the absence (HG + Ex) or the presence of H89 (HG + Ex + H89), and GLP-1R-GFP localization was determined by fluorescence microscopy (green). Cells were stained for β-catenin to mark membranes (red) and dapi to identify nuclei (blue). (C) To determine the in vivo effects of chronic exendin-4 exposure, male mice were administered exendin-4 at 4–6 h intervals for 24 h or were administered saline at each time-point (controls). Mice were then given a 5 μg/kg body exendin-4 dose and an hour later a 3 g/kg intraperitoneal glucose bolus. Plasma insulin levels were measured for 15 min following the glucose challenge (C) and blood glucose levels for 120 min (D). Data were analyzed by 2-way ANOVA with Bonferroni post hoc tests. *, P < 0.05; ***, P < 0.001. n = 6–8 mice for both C and D).
To test this hypothesis in vivo, mice were chronically administered exendin-4 (5 μg/kg body weight) at 4–6 h intervals for 24 h, and an acute exendin-4 bolus was administered 1 h prior to a 3 g/kg i.p. glucose challenge (Chronic-Ex4 mice). Control mice were administered saline throughout this period and then were administered either the acute exendin-4 (Acute-Ex4 mice) or another saline bolus (Saline Control mice) 1 h prior to the glucose challenge. Acute-Ex4 mice exhibited potentiated insulin release, compared to Saline Control mice, most notably at 2 min after glucose administration (Figure 7C). This enhanced insulin release led to significantly enhanced glucose tolerance (Figure 7D). In contrast, Chronic-Ex4 mice showed a significant loss of the potentiation of insulin secretion by exendin-4 (P < 0.01, 2-way ANOVA Bonferroni post-tests), and their glucose tolerance was poorer than that observed in mice receiving exendin-4 acutely (P < 0.0001, 2-way ANOVA). These data show that the insulinotropic effect of GLP-1R signaling is diminished after chronic activation of cAMP signaling in vivo. Moreover, the data are consistent with the in vitro observation that chronic exendin-4, acting via PKA, decreases GLP-1R expression at the cell surface.
4. Discussion
The beneficial effects of cyclic AMP signaling upon β-cell function and mass have been the basis for the development of therapies that activate the GLP1-R. Hyperglycemia was shown to downregulate GLP-1R signaling in animal models of diabetes [17]. Consistent with this, targeting the GLP1-R in the setting of poorly controlled diabetes has consistently shown impaired efficacy in β-cell responses [7–9]. The data presented here provide a possible explanation for this phenomenon. Chronic hyperglycemia was shown to downregulate responses to exendin-4 at the β-cell, with reduced insulin secretion, a diminished cAMP response and impaired phosphorylation of CREB. This was a rapid effect, leading to suppression of CREB phosphorylation within one hour, consistent with previous analysis of GLP1-R downregulation [21]. The effects of hyperglycemia upon the β-cells were found to lie at multiple points of the GLP-1R signaling system. The repressor of CREB activity, ICER, was induced; PKA activity was suppressed; the GLP-1R mRNA fell rapidly; and exendin-4 binding to the plasma membrane was reduced. This reduced binding of exendin-4 was found to be due to loss of GLP1-R from the plasma membrane, which required PKA activity.
Expression of an activated PKA catalytic subunit [27] replicated this effect, while a dominant negative PKA subunit [28] blocked the glucose dependent loss of GLP-1R from the plasma membrane. Mutation of serine 301 of the GLP-1R to alanine, which lies within a PKA consensus site, also prevented loss of the receptor from the plasma membrane at high glucose. FRET-based analysis of MIN6 cells showed that SUMO-1 interacted with GLP-1R in response to forskolin. Overexpression of SUMO, which promotes SUMOylation of target proteins [41,42], mimicked the effects of constitutive PKA activity in promoting the loss of the receptor from the plasma membrane at low glucose. Consistent with these data, the S301A mutation of the GLP-1R abolished the ability of the receptor to interact with SUMO-1 in response to rising cAMP. Likewise expression of a mutated SUMO protein (SUMO-GG), which is unable to covalently modify target proteins, abolished this interaction. Previously, we found that the GLP-1R is SUMOylated and that SUMO proteins and the E2 conjugating enzyme, ubc9, are upregulated upon chronic glucose exposure [26]. In addition, several studies have shown an association between PKA phosphorylation and SUMOylation of proteins [43,44], including the similar downregulation of β2-adrenergic signaling by PKA-mediated SUMOylation of PDE4D5 that activates its phosphodiesterase activity [45]. Thus, we propose a model whereby activation of adenylyl cyclase by glucose raises cAMP to activate PKA leading to phosphorylation of GLP1-R on serine 301, which promotes SUMOylation of the receptor and its loss from the plasma membrane.
This model also accommodates the activation of PKA by other mechanisms. Chronic GLP-1 administration, which has been shown to downregulate β-cell responses in vitro and in vivo [21,46–49], is also likely to act via the activation of PKA and fits the classical endocrine paradigm of negative feedback regulation. In support of this, chronic exendin-4 administration to MIN6 cells led to loss of GLP-1R from the plasma membrane and in mice led to a diminished ability to respond with potentiated insulin release to an exendin-4 stimulus. Since negative feedback regulation of homeostatic endocrine systems is a well-established paradigm, it is likely that other β-cell Gαs-coupled receptors are downregulated by a similar mechanism. It is also likely that the activation of other Gαs-coupled receptors will raise cAMP and activate PKA to downregulate the GLP-1R. Although these data reveal both a decrease in PKA activity in response to hyperglycemia and a role for PKA activity in the loss of the GLP-1R from the plasma membrane, these findings are not incompatible. PKA signaling is highly compartmentalized [50], and so whole cell reduction in PKA activity need not be reflective of localized signaling complexes. Moreover, the assay used here to measure PKA activity is an in vitro assay that uses cell lysates and measures PKA activity in response to exogenously added cAMP [31]. This assay provides a reflection of the kinase activity as a product of the concentration of the added cAMP, and the catalytic and regulatory subunits present in the lysate. Thus, these PKA assay data should be interpreted primarily as supporting the observed increase in PKA-RIα expression. The increase in PKA-RIα subunit expression is likely to lead to the redistribution of PKA catalytic subunits to PKA-RIα complexes at the expense of PKA-RII containing signaling complexes. PKA-RI and PKA-RII subunits are differentially distributed to A-kinase anchoring complexes, where they regulate distinct cellular processes (Supplementary Figure 1 and [51,52]). PKA regulatory subunits inhibit the kinase activity of the catalytic subunits and so increased PKA-RIα expression may decrease PKA activity, although PKA-RI subunits have a high affinity for cAMP, thereby lowering the cAMP threshold for PKA kinase activation of PKA-RI holoenzymes compared to PKA-RII holoenzymes. However, the overall finding that increase PKA-RIα expression is associated with decreased PKA activity is consistent with genetic deletion of the PKA-RIα subunit in mice and in individuals with Carney complex, who have PKA-RIα inactivating mutations, leading to increased PKA activity and enhanced insulin release [53].
GLP1-R based therapies were initially developed to treat diabetes mellitus through improved β-cell function and protection of β-cell mass. These therapies have sustained efficacy in reducing circulating glucose and improving β-cell function [4–6]. However, their ability to reduce weight and lower glucagon levels, combined with their limited efficacy in promoting β-cell function in individuals with type 2 diabetes, has led to speculation that their effects lie largely outside the β-cell [54]. This is supported by studies suggesting improved glucose control in individuals with type 1 diabetes who lack residual β-cell function [55,56]. In addition, chronic GLP-1 administration suppresses its potentiation of the insulin secretory response but the inhibitory effect upon glucagon levels is preserved [47]. Thus, it is unclear whether the benefits to glucose control and β-cell function in the setting of type 2 diabetes lie in the effects of GLP-1R therapies upon the β-cell directly or via an improved environment for β-cell function or both. The data presented in this study indicate that therapeutically targeting the GLP-1R to gain the benefits of cAMP signaling upon the β-cell faces the problem of loss of efficacy by downregulation via this same cAMP pathway. Whether this can be mitigated simply by alternate or intermittent treatment strategies should be explored. The study of Larsen and co-workers [47], showing that removing a GLP-1 infusion for 8 h restored the insulin secretory response, indicates a possible route for effective use of these therapies. However, that same study highlights that continuous infusion of GLP-1 delivers more sustained benefits to glucose control. Moreover, since the study was conducted over 7 days it may not exhibit the long-term benefits to β-cell function that appear to accrue over a longer timeframe [4]. In addition, GLP-1R therapies may best be used when glucose is moderately well controlled, either early in the pre-diabetic state or through the initial use of other therapies, for example insulin [13,14]. Although GLP1-R agonists were developed for their effects upon the β-cell they have considerable glucose lowering effects at other tissues, including acting centrally to promote satiety, affecting gastric emptying and suppressing inappropriately elevated glucagon levels. The retention of the glucagon suppressive effects of GLP-1R agonists, even when the β-cell response has been lost by chronic activation, indicate that the inhibitory effects of GLP-1 therapies upon glucagon secretion may not be downregulated by the same mechanism.
In summary, this study shows that alteration of receptor levels is a likely mechanism for the reduced efficacy of incretin therapies in type 2 diabetes. The indication that this may be a broadly applicable mechanism of Gαs-coupled receptor regulation raises an issue that warrants further examination in the development of therapies that target these receptors. Novel therapy administration regimes that involve intermittent or cyclic receptor stimulation may be able to minimize such downregulation. Alternatively, other strategies to raise cAMP may prove more effective at the β-cells, such as inhibition of Gαi-coupled receptors or finding specific targets downstream of cAMP/PKA. The benefits of activating cAMP signaling are one of the most attractive pathways for therapies targeting the β-cell. They provide the potential of glucose dependent enhancement of insulin secretion, combined with protection of β-cell mass. In addition, extra-pancreatic effects contribute to obtain weight loss and improved blood sugar control. These effects warrant further examination and optimization to improve the potential of therapies to derive benefits to the β-cells to treat diabetes.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH) to SR (DK075706), BW (DK085129), and LHP (DK063493 and DK092616), and by University of Chicago Diabetes Research and Training Center funding from the NIH (DK020595) to BW and LHP. CMDO was supported by a T32 NIH training grant (DK087703). JHE was supported by a Junior Diabetes Fonds Fellowship from Diabetes Fonds (2013.81.1675). Author contributions: SR, LHP and BW designed this study. SR, EM, LMD, JHE and BW conducted the experimental research and analyzed the data. SR, LHP and BW contributed to the preparation of the manuscript. BW and SR are the guarantors of this work and, as such, had full access to all the data and take responsibility for the integrity of data and the accuracy of data analysis.
Conflict of interest
The authors declare no competing interests.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Drucker D.J., Nauck M.A. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 2.Doyle M.E., Egan J.M. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacology & Therapeutics. 2007;113(3):546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Furman B., Pyne N., Flatt P., O'Harte F. Targeting beta-cell cyclic 3'5' adenosine monophosphate for the development of novel drugs for treating type 2 diabetes mellitus. A review. The Journal of Pharmacy and Pharmacology. 2004;56(12):1477–1492. doi: 10.1211/0022357044805. [DOI] [PubMed] [Google Scholar]
- 4.Zander M., Madsbad S., Madsen J.L., Holst J.J. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359(9309):824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- 5.Bunck M.C., Diamant M., Corner A., Eliasson B., Malloy J.L., Shaginian R.M. One-year treatment with exenatide improves beta-cell function, compared with insulin glargine, in metformin-treated type 2 diabetic patients: a randomized, controlled trial. Diabetes Care. 2009;32(5):762–768. doi: 10.2337/dc08-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallwitz B., Guzman J., Dotta F., Guerci B., Simo R., Basson B.R. Exenatide twice daily versus glimepiride for prevention of glycaemic deterioration in patients with type 2 diabetes with metformin failure (EUREXA): an open-label, randomised controlled trial. Lancet. 2012;379(9833):2270–2278. doi: 10.1016/S0140-6736(12)60479-6. [DOI] [PubMed] [Google Scholar]
- 7.Stumvoll M., Fritsche A., Haring H.U. Clinical characterization of insulin secretion as the basis for genetic analyses. Diabetes. 2002;51(Suppl 1):S122–S129. doi: 10.2337/diabetes.51.2007.s122. [DOI] [PubMed] [Google Scholar]
- 8.Kjems L.L., Holst J.J., Volund A., Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes. 2003;52(2):380–386. doi: 10.2337/diabetes.52.2.380. [DOI] [PubMed] [Google Scholar]
- 9.Fritsche A., Stefan N., Hardt E., Haring H., Stumvoll M. Characterisation of beta-cell dysfunction of impaired glucose tolerance: evidence for impairment of incretin-induced insulin secretion. Diabetologia. 2000;43(7):852–858. doi: 10.1007/s001250051461. [DOI] [PubMed] [Google Scholar]
- 10.Knop F.K., Vilsboll T., Hojberg P.V., Larsen S., Madsbad S., Volund A. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes. 2007;56(8):1951–1959. doi: 10.2337/db07-0100. [DOI] [PubMed] [Google Scholar]
- 11.Ahren B. Incretin dysfunction in type 2 diabetes: clinical impact and future perspectives. Diabetes & Metabolism. 2013;39(3):195–201. doi: 10.1016/j.diabet.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Shao Y., Yuan G., Feng Y., Zhang J., Guo X. Early liraglutide treatment is better in glucose control, beta-cell function improvement and mass preservation in db/db mice. Peptides. 2014;52:134–142. doi: 10.1016/j.peptides.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Hojberg P.V., Zander M., Vilsboll T., Knop F.K., Krarup T., Volund A. Near normalisation of blood glucose improves the potentiating effect of GLP-1 on glucose-induced insulin secretion in patients with type 2 diabetes. Diabetologia. 2008;51(4):632–640. doi: 10.1007/s00125-008-0943-x. [DOI] [PubMed] [Google Scholar]
- 14.Hojberg P.V., Vilsboll T., Rabol R., Knop F.K., Bache M., Krarup T. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia. 2009;52(2):199–207. doi: 10.1007/s00125-008-1195-5. [DOI] [PubMed] [Google Scholar]
- 15.Hansen K.B., Vilsboll T., Bagger J.I., Holst J.J., Knop F.K. Impaired incretin-induced amplification of insulin secretion after glucose homeostatic dysregulation in healthy subjects. The Journal of Clinical Endocrinology & Metabolism. 2012;97(4):1363–1370. doi: 10.1210/jc.2011-2594. [DOI] [PubMed] [Google Scholar]
- 16.Lynn F.C., Thompson S.A., Pospisilik J.A., Ehses J.A., Hinke S.A., Pamir N. A novel pathway for regulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. Faseb Journal. 2003;17(1):91–93. doi: 10.1096/fj.02-0243fje. [DOI] [PubMed] [Google Scholar]
- 17.Xu G., Kaneto H., Laybutt D.R., Duvivier-Kali V.F., Trivedi N., Suzuma K. Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes. Diabetes. 2007;56(6):1551–1558. doi: 10.2337/db06-1033. [DOI] [PubMed] [Google Scholar]
- 18.Biden T.J., Schmitz-Peiffer C., Burchfield J.G., Gurisik E., Cantley J., Mitchell C.J. The diverse roles of protein kinase C in pancreatic beta-cell function. Biochemical Society Transactions. 2008;36(Pt 5):916–919. doi: 10.1042/BST0360916. [DOI] [PubMed] [Google Scholar]
- 19.Widmann C., Dolci W., Thorens B. Desensitization and phosphorylation of the glucagon-like peptide-1 (GLP-1) receptor by GLP-1 and 4-phorbol 12-myristate 13-acetate. Molecular Endocrinology. 1996;10(1):62–75. doi: 10.1210/mend.10.1.8838146. [DOI] [PubMed] [Google Scholar]
- 20.Widmann C., Dolci W., Thorens B. Heterologous desensitization of the glucagon-like peptide-1 receptor by phorbol esters requires phosphorylation of the cytoplasmic tail at four different sites. The Journal of Biological Chemistry. 1996;271(33):19957–19963. doi: 10.1074/jbc.271.33.19957. [DOI] [PubMed] [Google Scholar]
- 21.Baggio L.L., Kim J.G., Drucker D.J. Chronic exposure to GLP-1R agonists promotes homologous GLP-1 receptor desensitization in vitro but does not attenuate GLP-1R-dependent glucose homeostasis in vivo. Diabetes. 2004;53(Suppl 3):S205–S214. doi: 10.2337/diabetes.53.suppl_3.s205. [DOI] [PubMed] [Google Scholar]
- 22.Roed S.N., Wismann P., Underwood C.R., Kulahin N., Iversen H., Cappelen K.A. Real-time trafficking and signaling of the glucagon-like peptide-1 receptor. Molecular and Cellular Endocrinology. 2013;382(2):938–949. doi: 10.1016/j.mce.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Tian Y., Laychock S.G. Protein kinase C and calcium regulation of adenylyl cyclase in isolated rat pancreatic islets. Diabetes. 2001;50(11):2505–2513. doi: 10.2337/diabetes.50.11.2505. [DOI] [PubMed] [Google Scholar]
- 24.Ramos L.S., Zippin J.H., Kamenetsky M., Buck J., Levin L.R. Glucose and GLP-1 stimulate cAMP production via distinct adenylyl cyclases in INS-1E insulinoma cells. The Journal of General Physiology. 2008;132(3):329–338. doi: 10.1085/jgp.200810044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zippin J.H., Chen Y., Straub S.G., Hess K.C., Diaz A., Lee D. CO2/HCO3(-)- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. The Journal of Biological Chemistry. 2013;288(46):33283–33291. doi: 10.1074/jbc.M113.510073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajan S., Torres J., Thompson M.S., Philipson L.H. SUMO downregulates GLP-1-stimulated cAMP generation and insulin secretion. American Journal of Physiology. Endocrinology and Metabolism. 2012;302(6):E714–E723. doi: 10.1152/ajpendo.00486.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orellana S.A., Amieux P.S., Zhao X., McKnight G.S. Mutations in the catalytic subunit of the cAMP-dependent protein kinase interfere with holoenzyme formation without disrupting inhibition by protein kinase inhibitor. The Journal of Biological Chemistry. 1993;268(10):6843–6846. [PubMed] [Google Scholar]
- 28.Woodford T.A., Correll L.A., McKnight G.S., Corbin J.D. Expression and characterization of mutant forms of the type I regulatory subunit of cAMP-dependent protein kinase. The effect of defective cAMP binding on holoenzyme activation. The Journal of Biological Chemistry. 1989;264(22):13321–13328. [PubMed] [Google Scholar]
- 29.Dickson L.M., Lingohr M.K., McCuaig J., Hugl S.R., Snow L., Kahn B.B. Differential activation of protein kinase B and p70(S6)K by glucose and insulin-like growth factor 1 in pancreatic beta-cells (INS-1) The Journal of Biological Chemistry. 2001;276(24):21110–21120. doi: 10.1074/jbc.M101257200. [DOI] [PubMed] [Google Scholar]
- 30.Landa L.R., Jr., Harbeck M., Kaihara K., Chepurny O., Kitiphongspattana K., Graf O. Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. The Journal of Biological Chemistry. 2005;280(35):31294–31302. doi: 10.1074/jbc.M505657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clegg C.H., Correll L.A., Cadd G.G., McKnight G.S. Inhibition of intracellular cAMP-dependent protein kinase using mutant genes of the regulatory type I subunit. The Journal of Biological Chemistry. 1987;262(27):13111–13119. [PubMed] [Google Scholar]
- 32.Permutt A., Chirgwin J., Giddings S., Kakita K., Rotwein P. Insulin biosynthesis and diabetes mellitus. Clinical Biochemistry. 1981;14(5):230–236. doi: 10.1016/s0009-9120(81)90940-1. [DOI] [PubMed] [Google Scholar]
- 33.Newgard C.B., McGarry J.D. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annual Review of Biochemistry. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 34.Goodge K.A., Hutton J.C. Translational regulation of proinsulin biosynthesis and proinsulin conversion in the pancreatic beta-cell. Seminars in Cell & Developmental Biology. 2000;11(4):235–242. doi: 10.1006/scdb.2000.0172. [DOI] [PubMed] [Google Scholar]
- 35.Delmeire D., Flamez D., Hinke S.A., Cali J.J., Pipeleers D., Schuit F. Type VIII adenylyl cyclase in rat beta cells: coincidence signal detector/generator for glucose and GLP-1. Diabetologia. 2003;46(10):1383–1393. doi: 10.1007/s00125-003-1203-8. [DOI] [PubMed] [Google Scholar]
- 36.Dou H., Wang C., Wu X., Yao L., Zhang X., Teng S. Calcium influx activates adenylyl cyclase 8 for sustained insulin secretion in rat pancreatic beta cells. Diabetologia. 2014;58(2):324–333. doi: 10.1007/s00125-014-3437-z. [DOI] [PubMed] [Google Scholar]
- 37.Klinger S., Poussin C., Debril M.B., Dolci W., Halban P.A., Thorens B. Increasing GLP-1-induced beta-cell proliferation by silencing the negative regulators of signaling cAMP response element modulator-alpha and DUSP14. Diabetes. 2008;57(3):584–593. doi: 10.2337/db07-1414. [DOI] [PubMed] [Google Scholar]
- 38.Drucker D.J. Incretin action in the pancreas: potential promise, possible perils, and pathological pitfalls. Diabetes. 2013;62(10):3316–3323. doi: 10.2337/db13-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chepurny O.G., Kelley G.G., Dzhura I., Leech C.A., Roe M.W., Dzhura E. PKA-dependent potentiation of glucose-stimulated insulin secretion by Epac activator 8-pCPT-2'-O-Me-cAMP-AM in human islets of Langerhans. American Journal of Physiology. Endocrinology and Metabolism. 2010;298(3):E622–E633. doi: 10.1152/ajpendo.00630.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fehmann H.C., Jiang J., Schweinfurth J., Wheeler M.B., Boyd A.E., 3rd, Goke B. Stable expression of the rat GLP-I receptor in CHO cells: activation and binding characteristics utilizing GLP-I(7-36)-amide, oxyntomodulin, exendin-4, and exendin(9-39) Peptides. 1994;15(3):453–456. doi: 10.1016/0196-9781(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 41.Agbor T.A., Cheong A., Comerford K.M., Scholz C.C., Bruning U., Clarke A. Small ubiquitin-related modifier (SUMO)-1 promotes glycolysis in hypoxia. The Journal of Biological Chemistry. 2011;286(6):4718–4726. doi: 10.1074/jbc.M110.115931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giri R., Yeh H.H., Wu C.H., Liu H.S. SUMO-1 overexpression increases RbAp46 protein stability and suppresses cell growth. Anticancer Research. 2008;28(6A):3749–3756. [PubMed] [Google Scholar]
- 43.Cox B., Briscoe J., Ulloa F. SUMOylation by Pias1 regulates the activity of the hedgehog dependent gli transcription factors. PLoS One. 2010;5(8):e11996. doi: 10.1371/journal.pone.0011996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han L., Pan Y., Wang B. Small ubiquitin-like modifier (SUMO) modification inhibits GLI2 protein transcriptional activity in vitro and in vivo. The Journal of Biological Chemistry. 2012;287(24):20483–20489. doi: 10.1074/jbc.M112.359299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X., Vadrevu S., Dunlop A., Day J., Advant N., Troeger J. Selective SUMO modification of cAMP-specific phosphodiesterase-4D5 (PDE4D5) regulates the functional consequences of phosphorylation by PKA and ERK. Biochemical Journal. 2010;428(1):55–65. doi: 10.1042/BJ20091672. [DOI] [PubMed] [Google Scholar]
- 46.Fehmann H.C., Jiang J., Pitt D., Schweinfurth J., Goke B. Ligand-induced regulation of glucagon-like peptide-I receptor function and expression in insulin-secreting beta cells. Pancreas. 1996;13(3):273–282. doi: 10.1097/00006676-199610000-00010. [DOI] [PubMed] [Google Scholar]
- 47.Larsen J., Hylleberg B., Ng K., Damsbo P. Glucagon-like peptide-1 infusion must be maintained for 24 h/day to obtain acceptable glycemia in type 2 diabetic patients who are poorly controlled on sulphonylurea treatment. Diabetes Care. 2001;24(8):1416–1421. doi: 10.2337/diacare.24.8.1416. [DOI] [PubMed] [Google Scholar]
- 48.Gromada J., Dissing S., Rorsman P. Desensitization of glucagon-like peptide 1 receptors in insulin-secreting beta TC3 cells: role of PKA-independent mechanisms. British Journal of Pharmacology. 1996;118(3):769–775. doi: 10.1111/j.1476-5381.1996.tb15466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alarcon C., Wicksteed B., Rhodes C.J. Exendin 4 controls insulin production in rat islet beta cells predominantly by potentiation of glucose-stimulated proinsulin biosynthesis at the translational level. Diabetologia. 2006;49(12):2920–2929. doi: 10.1007/s00125-006-0433-y. [DOI] [PubMed] [Google Scholar]
- 50.Wong W., Scott J.D. AKAP signalling complexes: focal points in space and time. Nature Reviews. Molecular Cell Biology. 2004;5(12):959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 51.Herberg F.W., Maleszka A., Eide T., Vossebein L., Tasken K. Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. Journal of Molecular Biology. 2000;298(2):329–339. doi: 10.1006/jmbi.2000.3662. [DOI] [PubMed] [Google Scholar]
- 52.Alto N.M., Soderling S.H., Hoshi N., Langeberg L.K., Fayos R., Jennings P.A. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4445–4450. doi: 10.1073/pnas.0330734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song W.J., Seshadri M., Ashraf U., Mdluli T., Mondal P., Keil M. Snapin mediates incretin action and augments glucose-dependent insulin secretion. Cell Metabolism. 2011;13(3):308–319. doi: 10.1016/j.cmet.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.D'Alessio D.A. Taking aim at islet hormones with GLP-1: is insulin or glucagon the better target? Diabetes. 2010;59(7):1572–1574. doi: 10.2337/db10-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dupre J., Behme M.T., McDonald T.J. Exendin-4 normalized postcibal glycemic excursions in type 1 diabetes. The Journal of Clinical Endocrinology & Metabolism. 2004;89(7):3469–3473. doi: 10.1210/jc.2003-032001. [DOI] [PubMed] [Google Scholar]
- 56.Kielgast U., Holst J.J., Madsbad S. Antidiabetic actions of endogenous and exogenous GLP-1 in type 1 diabetic patients with and without residual beta-cell function. Diabetes. 2011;60(5):1599–1607. doi: 10.2337/db10-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.