

Abstract
Objective
Disruption of TCF7L2 in mouse pancreatic β-cells has generated different outcomes in several investigations. Here we aim to clarify role of β-cell TCF7L2 and Wnt signaling using a functional-knockdown approach.
Methods
Adenovirus-mediated dominant negative TCF7L2 (TCF7L2DN) expression was conducted in Ins-1 cells. The fusion gene in which TCF7L2DN expression is driven by PTRE3G was utilized to generate the transgenic mouse line TCF7L2DNTet. The double transgenic line was created by mating TCF7L2DNTet with Ins2-rtTA, designated as βTCFDN. β-cell specific TCF7L2DN expression was induced in βTCFDN by doxycycline feeding.
Results
TCF7L2DN expression in Ins-1 cells reduced GSIS, cell proliferation and expression of a battery of genes including incretin receptors and β-cell transcription factors. Inducing TCF7L2DN expression in βTCFDN during adulthood or immediately after weaning generated no or very modest metabolic defect, while its expression during embryonic development by doxycycline feeding in pregnant mothers resulted in significant glucose intolerance associated with altered β-cell gene expression and reduced β-cell mass.
Conclusions
Our observations support a cell autonomous role for TCF7L2 in pancreatic β-cells suggested by most, though not all, investigations. βTCFDN is a novel model for further exploring the role of TCF7L2 in β-cell genesis and metabolic homeostasis.
Keywords: GLP-1, GSIS, Pancreatic β-cells, TCF7L2, TCF7L2DN, Wnt, Incretin receptors
1. Introduction
Genome wide association studies (GWAS) have led to the recognition of a number of Wnt signaling pathway components, including Wnt ligand [1], the co-receptor LRP-5 [2], and the Wnt pathway effector TCF7L2, as risk genes of type 2 diabetes (T2D) and other metabolic disorders [3]. In addition, the expression of a few known diabetes risk or obesity related genes was shown to be positively or negatively regulated by the Wnt signaling pathway effector β-catenin (β-cat)/TCF (cat/TCF) [4]. Among these diabetes risk genes, TCF7L2, also known as TCF-4 [5], has drawn our attention the most as its association with T2D susceptibility has been reproducibly observed globally in different ethnic groups [3,6–12].
After the discovery of the association between TCF7L2 and T2D in 2006 [3], enormous effort has been made to explore mechanisms underlying the function of this Wnt pathway effector in pancreatic β-cells and elsewhere [13–26]. Evidence has been collected, indicating the beneficial effect of TCF7L2 in pancreatic β-cell for cell proliferation, insulin gene expression and insulin secretion [16,19,20,27–29]. Interestingly, disruption of Tcf7l2 in mouse β-cells in developing pancreas led to the development of glucose intolerance and β-cell dysfunction [27,29], while inducing Tcf7l2 disruption in adulthood in another Tcf7l2−/− mouse line generated no metabolic defect [30]. Very recently, Takamoto et al. have demonstrated that the expression of dominant negative Tcf7l2 (Tcf7l2DN, the 55 kDa short form) in mouse β-cells led to the development of impaired glucose homeostasis, associated with the alteration of β-cell gene expression [22]. By utilizing RNA-sequencing in rodent and human pancreatic islets, Zhou et al. have identified the Tcf7l2-Isl-1 regulatory transcriptional network responsible for Tcf7l2 in exerting its pancreatic function, including the regulation of insulin secretion [31].
Here we generated a novel mouse model βTCFDN, in which human β-cell TCF7L2DN (the 75 kDa long form) expression is inducible upon doxycycline administration. The induction of β-cell TCF7L2DN expression during adulthood or immediately after weaning generated a very minor metabolic defect, without affecting Isl-1 expression level. When the pregnant mothers were fed with doxycycline, the βTCFDN offspring showed significant intolerance to glucose, associated with reduced β-cell mass and altered expression of certain β-cell specific genes, including Isl-1. Thus, our observations support a cell autonomous role for Tcf7l2 in pancreatic β-cells from a novel angle.
2. Materials and methods
2.1. Cell cultures and reagents
The INS-1 832/13 (INS-1) cell line and mouse islets were cultured in RPMI 1640 as described previously [32]. GLP-1 (7–37) was provided by Abcam Inc. (Toronto, Canada). Antibodies for Tcf7l2 and β-actin were purchased from Cell Signaling Technology (Beverly, MA). The anti-HA antibody (clone 16B12) was the product of Covance Research Products Inc. (USA, NJ). 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) was the product of Sigma–Aldrich (Oakville, Ontario, Canada).
2.2. Animals and doxycycline feeding
The human TCF7L2DN cDNA, provided by Eric Fearon [33], was inserted into PTRE3G (GSL Biotech LLC, Chicago, IL). Following DNA sequencing verification, it was utilized to generate the transgenic mice by FVB mouse zygote pronuclear microinjection and the implantation into pseudopregnant recipients, performed by the Toronto Centre for Phenogenomics Transgenic Core [34]. The mouse lines were designated as TCF7L2DNTet. The double transgenic line, designated as βTCFDN, was generated by mating TCF7L2DNTet with Ins2-rtTA. Doxycycline diet was provided by Harlan (625 mg/kg doxycycline, TD.08434). In each experiment, sex and age match Ins2-rtTA littermates were utilized as the control. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University Health Network.
2.3. Adenovirus experiments
The generation of Ad-TCF7L2WT and Ad-TCF7L2DN adenoviruses is detailed in Supplemental Experimental Procedures [35].
2.4. Metabolic tolerance tests and metabolic measurement
Oral glucose tolerance test (OGTT) and intraperitoneal insulin tolerance test (IPITT) were performed as previously described [34].
Serum insulin levels were measured using an insulin immunoassay kit (Cat 32392, AIS). Methods for glucose stimulated insulin secretion (GSIS) and GLP-1 stimulated insulin secretion are detailed in Supplemental Experimental Procedures.
2.5. Measurement of β-cell and α-cell mass and immunohistochemistry study
β-cell and α-cell mass were measured as we have previously described [36], detailed in Supplemental Experimental Procedures. Pdx-1 and Nkx-6.1 expression in mouse pancreatic islets were assessed by immunohistochemistry staining, detailed in Supplemental Experimental Procedures.
2.6. Cell proliferation assay
Cell proliferation on Ad-TCF7L2WT, or Ad-TCF7L2DN, or control virus infected Ins-1 cells was determined with the MTT method [37], detailed in Supplemental Experimental Procedures.
2.7. Western blotting
Methods for Western blotting against either whole cell lysate or isolated mouse islets have been described in our previous studies [37,38].
2.8. RNA isolation, reverse transcription, and quantitative RT-PCR
RNA isolation, cDNA synthesis, and real-time RT-PCR (qPCR) were performed as previously described [39]. All nucleotide primers for qPCR are listed in Supplementary Table 1.
2.9. Statistical analysis
Data are presented as means ± SEM. Significance was determined using the Student's t-test or one-way ANOVA followed by Bonferroni post hoc test as appropriate for single or multiple comparisons respectively. Differences were considered statistically significant when p < 0.05.
3. Results
3.1. TCF7L2DN attenuates GSIS and alters β-cell gene expression in the Ins-1 cell line
Considering that other Tcfs, including Tcf7 and Tcf7l1, are also expressed in pancreatic β-cells [40], which may exert redundant function with Tcf7l2, we tested the effect of functional knockdown of Tcfs or cat/Tcf with TCF7L2DN. The full-length human TCF7L2 cDNA (encoding the 78 kDa isoform) and one that lacks the first 117 bp nucleotide sequence (Figure 1A) were utilized to generate adenoviruses, designated as Ad-TCF7L2WT and Ad-TCF7L2DN. We reported very recently that TCF7L2DN is still able to bind to the consensus TCF binding motif but cannot recruit β-cat [35]. When Ins-1 cells were infected with the GFP control virus (Ad-GFP), Ad-TCF7L2WT, or Ad-TCF7L2DN, the infection efficiency reached more than 95%, based on the detection of GFP expression (data not shown). The expression of endogenous Tcf7l2 and exogenous TCF7L2 can be detected by Western blotting with the TCF7L2 antibody, while TCF7L2 and TCF7L2DN can be differentiated based on their sizes (78 kDa versus 75 kDa; Figure 1B). The expression of exogenous TCF7L2 can be detected by the HA-tag antibody (Figure 1B). Ad-TCF7L2DN infection, but not Ad-TCF7L2WT infection, completely blocked GSIS (Figure 1C) and attenuated cell growth (Figure 1D). Wild type TCF7L2-stimulated growth of Ins-1 cells was observed 48 h and 72 h after the infection (Figure 1D).
Figure 1.
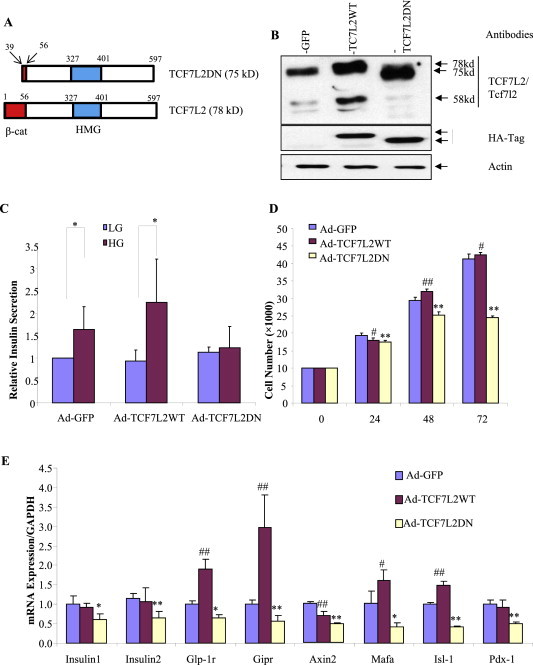
TCF7L2DN expression in the Ins-1 cell line attenuates GSIS, inhibits cell growth and represses the expression of a battery of β-cell specific genes. A) A schematic of TCF7L2DN (75 kDa) and wild type TCF7L2 (78 kDa). β-cat, the β-cat interaction domain; HMG, the HMG-box DNA binding domain. B) Detection of TCF7L2 and TCF7L2DN in virus infected Ins-1 cells by Western blotting. GFP, the control virus. C) The expression of TCF7L2DN but not wild type TCF7L2 inhibited GSIS. LG, low glucose (2.8 mM), HG, high glucose (16.7 mM). Ad-GFP, the control virus. n = 4 for each virus infection. D) TCF7L2DN expression inhibited Ins-1 cell growth, assessed by MTT. n = 8. E) qPCR assessment shows that TCF7L2DN suppressed the expression of a panel of β-cell specific genes.
We then assessed the effect of Ad-TCF7L2WT and Ad-TCF7L2DN on the expression of two pro-insulin genes, genes that encode the two incretin hormone receptors (Gipr and Glp-1r), the Wnt pathway downstream target Axin2, and the β-cell specific genes Mafa, Isl-1 and Pdx-1. As shown in Figure 1E, the expression of these genes was significantly inhibited by TCF7L2DN expression. Ad-TCF7L2WT infection generated no significant effect on the two pro-insulin genes and Pdx-1, increased the expression of the two incretin hormone receptors (Glp-1r and Gipr) and elevated the expression of Mafa and Isl-1. Furthermore, Ad-TCF7L2WT infection also repressed the expression of the Wnt pathway target Axin2, supporting the notion for the bi-directional effect of TCFs [35,41].
3.2. Inducing β-cell TCF7L2DN expression in βTCFDN during adulthood or immediately after weaning generates very modest defect on glucose tolerance
The fusion gene construct in which TCF7L2DN expression is driven by PTRE3G was then created (Supplementary Figure 1A). When it was co-transfected with Tet-On3G (i.e. rtTA) into Ins1 cells, the addition of doxycycline resulted in mCherry and TCF7L2DN expression (Supplementary Figure 1B and C). We then generated the transgenic mice with this construct. Among the five founders (Supplementary Figure 1D), founder 4 showed consistent germ line transmission, which was utilized for further investigation and designated as TCF7L2DNTet. After TCF7L2DNTet was mated with Ins2-rtTA, the double transgenic mouse line was designated as βTCFDN. We found that doxycycline feeding at the age of 6 wk for 2 wks allowed us to visualize mCherry fluorescence in the pancreatic islets (Supplementary Figure 1E) co-localized with the insulin-producing cells (Supplementary Figure 1F).
We then assessed the effect of TCF7L2DN expression on metabolic homeostasis either in adult mice or in mice immediately after weaning. As shown in Supplementary Figures 2 and 3, we found a tendency for impaired OGTT and reduced expression of certain β-cell specific genes (Supplementary Figure 2D and I) or reduced insulin secretion after OGTT (Supplementary Figure 3D). Mice fed with doxycycline during adulthood also showed reduced expression of pro-insulin 2, Pdx-1 and Mafa. However, mRNA levels of other genes, including Isl-1 and Insulin 1, were comparable in βTCFDN and the control Ins2-rtTA mice (Supplementary Figure 2I). It is worth pointing out that TCF7L2DN expression repressed endogenous Tcf7l2 expression in vitro (Figure 1B) and in vivo (Supplementary Figure 2C). This supports our previous observation that TCF7L2 expression can be auto-regulated [42]. This may represent another mechanism for this dominant negative molecule to exert its inhibitory effect on Wnt signaling. Together, the defect generated by inducing TCF7L2DN expression in adulthood or immediately after weaning is modest. This is likely due to complicated compensatory mechanisms in response to Wnt functional knockdown during adulthood in the absence of a challenge.
3.3. Inducing TCF7L2DN expression during the embryonic developmental stage causes impaired glucose tolerance
Male Ins2-rtTA mice were then mated with TCF7L2DNTet female mice. After female mice became pregnant, they were fed with doxycycline throughout the pregnancy until the weaning of the offspring at 21 days. The offspring were then fed with doxycycline diet. Doxycycline feeding did not alter the body weight of the mice (Figure 2A). Importantly, the βTCFDN offspring showed impaired tolerance to oral glucose (Figure 2B) but comparable tolerance to insulin challenge (Figure 2C). Figure 2D shows that while prior to glucose gavage βTCFDN mouse plasma glucose levels were comparable with that of Ins2-rtTA controls, 15 min after oral glucose challenge, βTCFDN mice showed even higher plasma glucose levels. Consistently, Figure 2E shows that in βTCFDN mice, oral glucose challenge-stimulated insulin secretion was significantly attenuated.
Figure 2.
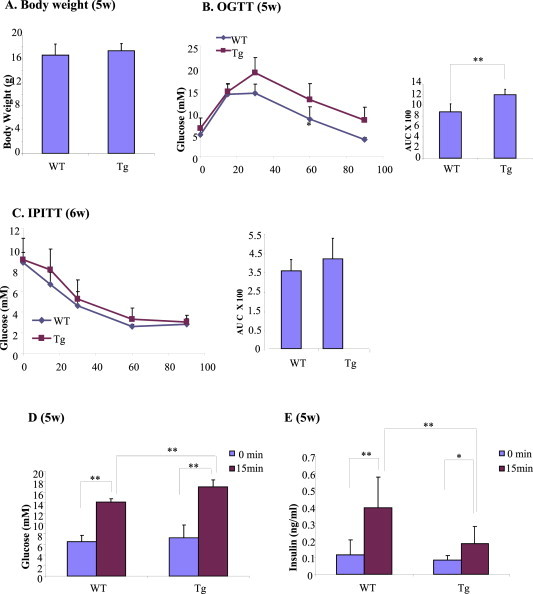
Impaired glucose homeostasis in βTCFDN offspring when the pregnant mothers were fed with doxycycline to induce TCF7L2DN expression. A) Body weight comparison. B–C) Impaired response to OGTT (B) but normal response to IPITT (C). D–E) Increased blood glucose levels after oral glucose gavage D) and attenuated plasma insulin levels after glucose gavage in βTCFDN mice. n = 4 to 5 for panels A–E.
3.4. Inducing TCF7L2DN expression during the embryonic developmental stage causes reduced β-cell mass
Mice were sacrificed at 6 wk of age for β-cell and α-cell mass measurement. βTCFDN mice had significantly reduced β-cell mass compared with Ins2-rtTA controls (Figure 3A). α-cell mass, however, was not altered (Figure 3B). Since β-cell numbers within the islets appeared reduced, we assessed the expression of two β-cell specific homeobox-gene transcription factors Pdx-1 and Nkx6-1, which were both detectable (Figure 3C, D). We then counted Pdx-1 and Nkx6.1 positive cells in mouse islets. The percentage of Pdx-1 positive islet cells was 51.7% in the Ins2-rtTA mouse but dropped to 36.0% in the βTCFDN mouse (Figure 3C, right panel). Similarly, the percentage of Nkx6.1 positive islet cells dropped from 57.5% in the Ins2-rtTA mouse to 34.9% in the βTCF7DN mouse (Figure 3D, right panel). Thus, β-cell TCF7L2DN expression starting from the embryonic stage reduces the genesis of Pdx-1 and Nkx6.1 positive islet cells.
Figure 3.
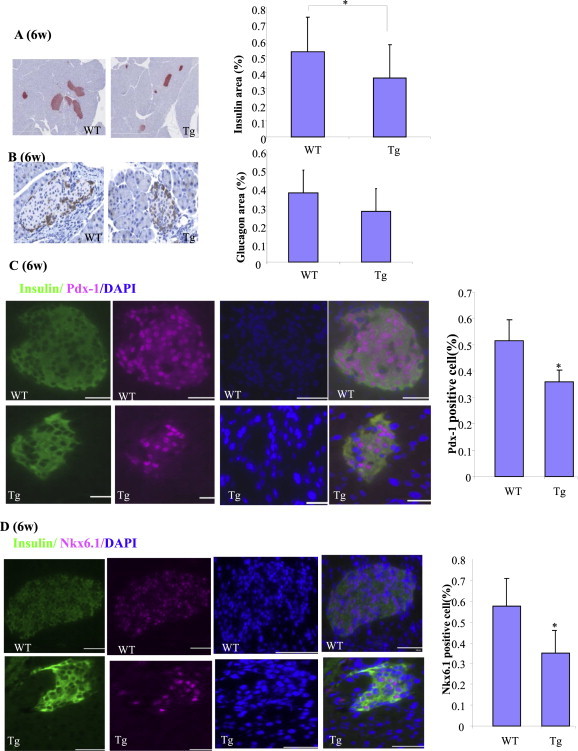
Reduced β-cell mass, Pdx-1 and Nkx6.1 positive islet cells in βTCFDN offspring when pregnant mothers were fed with doxycycline to induce TCF7L2DN expression. A–B) Assessment of β-cell mass (A) and α-cell (B) mass. n = 3. C–D) Immunostaining shows reduced percentage of Pdx-1 positive islet cells in βTCFDN mice. Immunostaining shows reduced percentage of Nkx6.1 positive islet cells in βTCFDN mice. The percentages were determined by counting 2,200–3,000 islet cells from βTCFDN mice and the Ins2-rtTA control mice (n = 3 for each group).
3.5. Inducing TCF7L2DN expression during the embryonic developmental stage altered β-cell gene expression and the ex vivo response to GLP-1
Another set of βTCFDN and control Ins2-rtTA mice were generated by doxycycline feeding of the pregnant mothers followed by doxycycline feeding of the offspring after weaning. We then assessed alterations of β-cell specific gene expression in mice at the age of 6 wks. As shown in Figure 4A, the expression of pro-insulin genes, two incretin receptor genes, Axin2, as well as Pdx-1 and Isl-1 was reduced in the βTCF7DN mouse islets. The expression levels of Mafa and Ngn3, however, were not altered.
Figure 4.
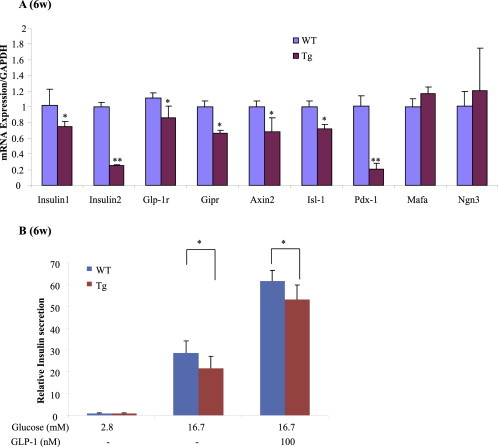
βTCFDN mouse islets show reduced β-cell specific gene expression and attenuated response to GLP-1 treatment. A) qPCR assessment for β-cell specific gene expression. n = 3 for both βTCFDN and control ins2-rtTA mice. B) Assessment of insulin secretion by GLP-1 treatment in isolated βTCFDN mouse islets. n = 3 for both βTCFDN and control ins2-rtTA mice.
Another set of βTCF7L2DN and Ins2-rtTA control mice were generated by doxycycline feeding of the pregnant mothers, followed by doxycycline feeding of the offspring after weaning. At the age of 5 wks impaired tolerance to OGTT was detected in the βTCFDN offspring (data not shown). At the age of 6 wk, mouse islets were isolated for GLP-1 treatment. As shown in Figure 4B, islets isolated from the βTCFDN offspring showed attenuated response to both GSIS and GLP-1-potentiated GSIS.
4. Discussion
Here we examined the effect of Wnt pathway functional knockdown in pancreatic β-cells with TCF7L2DN. TCF7L2DN expression in Ins-1 cells attenuated the expression of the Wnt pathway downstream target Axin2, inhibited cell growth, and suppressed the expression of two pro-insulin genes as well as genes that encode the receptors for the incretin hormones. More importantly, the functional knockdown inhibited GSIS. Although the induction of TCF7L2DN expression in β-cells in adult mice generated a tendency towards metabolic defect and resulted in the inhibition of several β-cell specific genes, the expression of a number of other β-cell specific genes, including Isl-1 was not affected. This could be due to the existence of complicated compensatory mechanism, which deserves further investigation. The induction of β-cell TCF7L2DN expression during embryonic development, by feeding doxycycline to pregnant mothers, however, revealed a fundamental role of TCFs on the generation of Pdx-1 and Nkx6.1 positive islet cells as well as the maintenance of glucose homeostasis.
As a key component of the Wnt signaling pathway, TCF7L2 may not exert its function on its own. A TCF member, including TCF7L2, forms the bipartite transcription factor cat/TCF with a free β-cat molecule. Different TCFs may exert certain redundant functions [43]. To explore the function of Wnt pathway in pancreatic β-cells, several investigations have assessed the effect of β-cat deletion in pancreatic β-cells, even before the recognition of TCF7L2 as the diabetes risk gene [44–47]. An early report showed that β-cat deletion did not significantly perturb pancreatic islet endocrine cell mass or function, although β-cat is evidently essential for acinar cell differentiation [44]. When Pdx-1 promoter was utilized to deliver the Cre enzyme to delete β-cat, Papadopoulou and Edlund found that β-cat deleted cells had a competitive disadvantage during pancreas development [45]. Interestingly, although they observed a reduction in islet numbers during the early embryonic development stage, the mice recovered from the pancreatitis at a later stage and regenerated normal pancreas and duodenal villi from cells that escaped β-cat deletion [45]. Heiser and colleagues took advantage of the S33Y β-cat mutant. The transgenic mice generated with this constitutive mutant β-cat revealed a temporal/spatial role for β-cat signaling in the regulation of pancreas organ growth [46]. Finally, the metabolic function of Wnt signaling in pancreatic β-cells has been demonstrated with the utilization of Wnt ligands [47,48]. Thus, although Wnt signaling does play an important role in β-cell genesis and function, the defect caused by β-cat genetic deletion may not be easily detected due to the existence of complicated, redundant, and compensatory mechanisms. This can be attributed to the redundant function of TCF members, the bi-directional and temporal/spatial feature of β-cat molecules [49,50], as well as the fact that the knockout approach for β-cat cannot reach 100%. TCF7L2DN utilized in this as well as a few previous studies serves as a unique tool in assessing Wnt signaling function [5,22,33,34].
Following the milestone discovery of the association between TCF7L2 polymorphisms and the risk of T2D [3], studies have been conducted to explore the function of TCF7L2 in pancreatic β-cells and elsewhere. A considerable amount of effort has been made on pancreatic β-cells, as Schafer and colleagues demonstrated that impaired GLP-1-induced insulin secretion occurred in the carriers of T2D risk TCF7L2 SNPs [15]. Utilizing both the over-expression and knockdown approaches in isolated human islets, Shu et al. demonstrated the role of TCF7L2 in protecting cells from cytokine-induced apoptosis, stimulating β-cell proliferation and mediating GSIS [20]. The same group then conducted further exploration and showed that siRNA-mediated TCF7L2 knockdown, resulting in reduced Glp-1r and Gipr expression and attenuated incretin response [19]. The beneficial effect of TCF7L2 in human islets observed by Shu and colleagues was also demonstrated in mouse islets by da Silva Xavier and colleagues by various means [16]. In the current study, we have verified that TCF7L2DN reduced Glp-1r and Gipr expression in vitro and in vivo, and attenuated the response to GLP-1. We observed the stimulation of the expression of Glp-1r and Gipr by WT TCF7L2 in the Ins-1 cell line (Figure 1E). However, the additive effect of wild type TCF7L2 on GSIS was not appreciable (Figure 1C). This might be due to the difference among the species or the expression level in these two studies, which requires further examinations.
Tcf7l2−/− mice die 8–10 days after birth [51]. Several transgenic mouse models have been generated, exploring the metabolic function of TCF7L2. Savic et al. found that the 92-kb genomic interval associated with the risk of T2D contains enhancer elements that regulate the temporal/spatial expression patterns of Tcf7l2 in metabolic tissues. By deleting this interval, they establish a critical role for these enhancer elements in regulating Tcf7l2 expression. They then developed a TCF7l2 copy-number allelic series in mice. The TCF7l2 null mice displayed enhanced glucose tolerance coupled to significantly lowered insulin levels, while transgenic mice harboring multiple TCF7L2 copies and overexpressing this gene display reciprocal phenotypes, including glucose intolerance [13]. These observations suggest that Tcf7l2 has a deleterious metabolic effect, which is in disagreement with the beneficial effect of pancreatic TCF7L2 observed by Shu et al. [19,20], as well as by Da Silva Xavier et al. [16], but is in agreement with the observations that TCF7L2 expression in human islets was increased in T2D, particularly in TCF7L2 T2D risk allele carriers, while its over-expression in human islets reduced GSIS [14].
The pancreatic β-cell specific knockout of Tcf7l2 has been performed recently. Boj et al. demonstrated that the deletion of Tcf7l2 in β-cells generated no deleterious effects on metabolic homeostasis [30] while da Silva Xavier et al. found that TCF7L2 deletion in β-cells resulted in an impairment of glucose homeostasis and β-cell function [27]. The discrepancy was partially attributed to the use of Pdx-1 versus RIP (insulin) promoters to drive Cre recombinase-mediated excision of LoxP-flanked TCF7L2 sequences. However, a very recent study by Mitchell et al. demonstrates that a β-cell specific TCF7L2−/− mouse line generated with the utilization of the Ins-1 promoter also shows a defect in glucose intolerance and insulin secretion [29]. It is worth pointing out that Boj and colleagues did not examine mice beyond 12 wk of age while the other two teams did [30]. Furthermore, Boj et al. did not apply the alternative OGTT approach to assess glucose intolerance in their model, potentially underestimating the effects of changes in GLP-1 (or other incretin) receptor expression in their Tcf7l2-null β-cells [30]. More importantly, the approach by Boj and colleagues may only expect to see effects of TCF7L2 loss at post-embryonic stage, as they started to inject tamoxifen to initiate the cleavage at weaning [30], in contrast to the deletion during embryonic development stages utilized by the other groups [27,29]. Here, we demonstrate that Wnt pathway functional knockdown during embryonic development resulted in impaired glucose homeostasis and reduced β-cell mass. In contrast, Wnt pathway functional knockdown in adulthood did not significantly affect glucose homeostasis or β-cell mass. Our observations revealed the temporal/spatial role of β-cat/Tcf and Tcf7l2 in pancreatic β-cells from a novel angle. However, we cannot eliminate the possibility that differences in the genetic background of different animal models, including the one in this study, play a role in the subtle difference in metabolic homeostasis. In addition, the use of Cre system in β-cell gene disruption may cause impairment of β-cell function, as suggested recently [52]. Cre per se may generate no effect on β-cell function in the case of the Ins-1Cre delete strain [53], as utilized by Mitchell et al. [29]. The use of Pdx-1-Cre, RIP-Cre and MIP-Cre, however, may cause complications, in part due to the inclusion of a second cistron which leads to the local production of human growth hormone, demonstrated recently by Brouwers and colleagues [54].
It is worth emphasizing that TCF7L2DN expression leads to impaired Glp-1r expression and impaired GLP-1 action on insulin secretion, which were also observed very recently by Mitchell et al. in their β-cell specific Tcf7l2−/− mouse model [29]. These observations, along with the study in human islets by Shu and colleagues [19], support a β-cell autonomous effect of TCF7L2 in controlling the expression of incretin receptors.
Although Tcf7l2 has multiple alternatively spliced isoforms, in metabolic organs, the two major isoforms encode the two proteins in sizes of 78 kDa and 58 kDa, respectively. Both isoforms share the common HMG and β-cat interaction domains. Very recently, Takamoto et al. generated a transgenic mouse model in which the 55 kDa short isoform of Tcf7l2DN was driven by the Ins2 promoter. These transgenic mice showed impaired glucose homeostasis, reduced β-cell mass, and insulin secretion [22]. It is yet to be determined whether eithervof these two major isoforms exerts a unique function in pancreatic β-cells.
Very recently, Zhou et al. have identified a TCF7L2-regulated transcriptional network that is responsible for its effect on human and rodent islets insulin secretion [31]. A key component of the network is the LIM homeodomain protein Isl-1, which is a Wnt downstream target in cardiovascular progenitors as well [55]. We observed reduced Isl-1 expression in response to TCF7L2DN expression in Ins-1 cells and in βTCFDN mice. Our βTCFDN mouse model as well as the TCF7L2 adenovirus will serve as unique tools to further explore the TCF7L2/Isl-1 regulatory network in the genesis of pancreatic β-cells and their function in health and diseases.
5. Conclusions
In summary, TCF7L2DN expression in Ins-1 cells attenuates cell growth, GSIS and β-cell specific gene expression. Its expression during embryonic development significantly impairs the generation of Pdx-1 and Nkx6.1 positive islet cells, β-cell gene expression and the function of pancreatic islets in response to GLP-1. Our new findings provide further support for a cell autonomous role for Tcf7l2 in β-cells, as previously indicated by some, though not all Tcf7l2 knockout mouse models.
Author contributions
W.S., X.X., M.H., M.C.N., and T.J. designed and conducted experiments, and analyzed the data. W.S. and T.J. wrote the manuscript. W.I., F.X., Z.S., K.Z. and T.L. conducted experiments; H.G., D.M., and A.N. provided research material. H.G. and J.W. assisted the experimental design, and edited the manuscript. All authors approved the submission of the manuscript.
Acknowledgments
We thank Dr. Douglas Melton (Harvard University) for the permission of using the transgenic mouse line Ins2-rtTA, and Dr. Andras Nagy (Mount Sinai Hospital Lunenfeld-Tanenbaum Research Institute) for providing the Ins2-rtTA mice. This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) to T.J. (MOP-89987 and MOP-97790). W.I. is a recipient of the CIHR Doctoral Canada Graduate Scholarship, Ontario Graduate Scholarship, and Banting and Best Diabetes Centre – University Health Network Graduate Award. Tianru Jin is the guarantor of the manuscript.
Conflict of interest
The authors have declared that no conflict of interest exists.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Kanazawa A., Tsukada S., Sekine A., Tsunoda T., Takahashi A., Kashiwagi A. Association of the gene encoding wingless-type mammary tumor virus integration-site family member 5B (WNT5B) with type 2 diabetes. The American Journal of Human Genetics. 2004;75:832–843. doi: 10.1086/425340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Twells R.C., Mein C.A., Payne F., Veijola R., Gilbey M., Bright M. Linkage and association mapping of the LRP5 locus on chromosome 11q13 in type 1 diabetes. Human Genetics. 2003;113:99–105. doi: 10.1007/s00439-003-0940-6. [DOI] [PubMed] [Google Scholar]
- 3.Grant S.F., Thorleifsson G., Reynisdottir I., Benediktsson R., Manolescu A., Sainz J. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z., Habener J.F. Wnt signaling in pancreatic islets. Advances in Experimental Medicine and Biology. 2010;654:391–419. doi: 10.1007/978-90-481-3271-3_17. [DOI] [PubMed] [Google Scholar]
- 5.Yi F., Brubaker P.L., Jin T. TCF-4 mediates cell type-specific regulation of proglucagon gene expression by beta-catenin and glycogen synthase kinase-3beta. The Journal of Biological Chemistry. 2005;280:1457–1464. doi: 10.1074/jbc.M411487200. [DOI] [PubMed] [Google Scholar]
- 6.Florez J.C. The new type 2 diabetes gene TCF7L2. Current Opinion in Clinical Nutrition and Metabolic Care. 2007;10:391–396. doi: 10.1097/MCO.0b013e3281e2c9be. [DOI] [PubMed] [Google Scholar]
- 7.Diabetes Genetics Initiative of Broad Institute of H, Mit LU, Novartis Institutes of BioMedical R, Saxena R., Voight B.F., Chen H. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 8.Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 9.Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groves C.J., Zeggini E., Minton J., Frayling T.M., Weedon M.N., Rayner N.W. Association analysis of 6,736 U.K. subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes. 2006;55:2640–2644. doi: 10.2337/db06-0355. [DOI] [PubMed] [Google Scholar]
- 11.Cauchi S., El Achhab Y., Choquet H., Dina C., Krempler F., Weitgasser R. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. Journal of Molecular Medicine. 2007;85:777–782. doi: 10.1007/s00109-007-0203-4. [DOI] [PubMed] [Google Scholar]
- 12.Chang Y.C., Chang T.J., Jiang Y.D., Kuo S.S., Lee K.C., Chiu K.C. Association study of the genetic polymorphisms of the transcription factor 7-like 2 (TCF7L2) gene and type 2 diabetes in the Chinese population. Diabetes. 2007;56:2631–2637. doi: 10.2337/db07-0421. [DOI] [PubMed] [Google Scholar]
- 13.Savic D., Ye H., Aneas I., Park S.Y., Bell G.I., Nobrega M.A. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Research. 2011;21:1417–1425. doi: 10.1101/gr.123745.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyssenko V., Lupi R., Marchetti P., Del Guerra S., Orho-Melander M., Almgren P. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. Journal of Clinical Investigation. 2007;117:2155–2163. doi: 10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schafer S.A., Tschritter O., Machicao F., Thamer C., Stefan N., Gallwitz B. Impaired glucagon-like peptide-1-induced insulin secretion in carriers of transcription factor 7-like 2 (TCF7L2) gene polymorphisms. Diabetologia. 2007;50:2443–2450. doi: 10.1007/s00125-007-0753-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.da Silva Xavier G., Loder M.K., McDonald A., Tarasov A.I., Carzaniga R., Kronenberger K. TCF7L2 regulates late events in insulin secretion from pancreatic islet beta-cells. Diabetes. 2009;58:894–905. doi: 10.2337/db08-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ip W., Shao W., Chiang Y.T., Jin T. The Wnt signaling pathway effector TCF7L2 is upregulated by insulin and represses hepatic gluconeogenesis. American Journal of Physiology Endocrinology and Metabolism. 2012;303:E1166–E1176. doi: 10.1152/ajpendo.00249.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norton L., Fourcaudot M., Abdul-Ghani M.A., Winnier D., Mehta F.F., Jenkinson C.P. Chromatin occupancy of transcription factor 7-like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia. 2011;54:3132–3142. doi: 10.1007/s00125-011-2289-z. [DOI] [PubMed] [Google Scholar]
- 19.Shu L., Matveyenko A.V., Kerr-Conte J., Cho J.H., McIntosh C.H., Maedler K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Human Molecular Genetics. 2009;18:2388–2399. doi: 10.1093/hmg/ddp178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shu L., Sauter N.S., Schulthess F.T., Matveyenko A.V., Oberholzer J., Maedler K. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes. 2008;57:645–653. doi: 10.2337/db07-0847. [DOI] [PubMed] [Google Scholar]
- 21.Shu L., Zien K., Gutjahr G., Oberholzer J., Pattou F., Kerr-Conte J. TCF7L2 promotes beta cell regeneration in human and mouse pancreas. Diabetologia. 2012;55:3296–3307. doi: 10.1007/s00125-012-2693-z. [DOI] [PubMed] [Google Scholar]
- 22.Takamoto I., Kubota N., Nakaya K., Kumagai K., Hashimoto S., Kubota T. TCF7L2 in mouse pancreatic beta cells plays a crucial role in glucose homeostasis by regulating beta cell mass. Diabetologia. 2014;57:542–553. doi: 10.1007/s00125-013-3131-6. [DOI] [PubMed] [Google Scholar]
- 23.Chen Z., Shao W., Xu F., Liu L., Lin B., Wei X.H. Acute Wnt pathway activation positively regulates leptin gene expression in mature adipocytes. Cellular Signalling. 2015 doi: 10.1016/j.cellsig.2014.12.012. in press. [DOI] [PubMed] [Google Scholar]
- 24.Bailey K.A., Savic D., Zielinski M., Park S.Y., Wang L.J., Witkowski P. Evidence of non-pancreatic beta cell-dependent roles of Tcf7l2 in the regulation of glucose metabolism in mice. Human Molecular Genetics. 2015 doi: 10.1093/hmg/ddu577. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locke J.M., Da Silva Xavier G., Rutter G.A., Harries L.W. An alternative polyadenylation signal in TCF7L2 generates isoforms that inhibit T cell factor/lymphoid-enhancer factor (TCF/LEF)-dependent target genes. Diabetologia. 2011;54:3078–3082. doi: 10.1007/s00125-011-2290-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norton L., Chen X., Fourcaudot M., Acharya N.K., DeFronzo R.A., Heikkinen S. The mechanisms of genome-wide target gene regulation by TCF7L2 in liver cells. Nucleic Acids Research. 2014;42:13646–13661. doi: 10.1093/nar/gku1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.da Silva Xavier G., Mondragon A., Sun G., Chen L., McGinty J.A., French P.M. Abnormal glucose tolerance and insulin secretion in pancreas-specific Tcf7l2-null mice. Diabetologia. 2012;55:2667–2676. doi: 10.1007/s00125-012-2600-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutter G.A. Dorothy Hodgkin lecture 2014. Understanding genes identified by genome-wide association studies for type 2 diabetes. Diabetic Medicine. 2014;31:1480–1487. doi: 10.1111/dme.12579. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell R.K., Mondragon A., Chen L., McGinty J.A., French P.M., Ferrer J. Selective disruption of Tcf7l2 in the pancreatic beta cell impairs secretory function and lowers beta cell mass. Human Molecular Genetics. 2015 doi: 10.1093/hmg/ddu553. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boj S.F., van Es J.H., Huch M., Li V.S., Jose A., Hatzis P. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell. 2012;151:1595–1607. doi: 10.1016/j.cell.2012.10.053. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y., Park S.Y., Su J., Bailey K., Ottosson-Laakso E., Shcherbina L. TCF7L2 is a master regulator of insulin production and processing. Human Molecular Genetics. 2014;23:6419–6431. doi: 10.1093/hmg/ddu359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shao W., Yu Z., Fantus I.G., Jin T. Cyclic AMP signaling stimulates proteasome degradation of thioredoxin interacting protein (TxNIP) in pancreatic beta-cells. Cellular Signalling. 2010;22:1240–1246. doi: 10.1016/j.cellsig.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Kolligs F.T., Hu G., Dang C.V., Fearon E.R. Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Molecular and Cellular Biology. 1999;19:5696–5706. doi: 10.1128/mcb.19.8.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao W., Wang D., Chiang Y.T., Ip W., Zhu L., Xu F. The Wnt signaling pathway effector TCF7L2 controls gut and brain proglucagon gene expression and glucose homeostasis. Diabetes. 2013;62:789–800. doi: 10.2337/db12-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ip W., Shao W., Song Z., Chen Z., Wheeler M.B., Jin T. Liver-specific expression of dominant negative transcription factor 7-like 2 causes progressive impairment in glucose homeostasis. Diabetes. 2015 doi: 10.2337/db14-1329. in press. [DOI] [PubMed] [Google Scholar]
- 36.Shao W., Wang Z., Ip W., Chiang Y.T., Xiong X., Chai T. GLP-1(28-36) improves beta-cell mass and glucose disposal in streptozotocin-induced diabetic mice and activates cAMP/PKA/beta-catenin signaling in beta-cells in vitro. The American Journal of Physiology Endocrinology and Metabolism. 2013;304:E1263–E1272. doi: 10.1152/ajpendo.00600.2012. [DOI] [PubMed] [Google Scholar]
- 37.Sun J., Khalid S., Rozakis-Adcock M., Fantus I.G., Jin T. P-21-activated protein kinase-1 functions as a linker between insulin and Wnt signaling pathways in the intestine. Oncogene. 2009;28:3132–3144. doi: 10.1038/onc.2009.167. [DOI] [PubMed] [Google Scholar]
- 38.Yi F., Sun J., Lim G.E., Fantus I.G., Brubaker P.L., Jin T. Cross talk between the insulin and Wnt signaling pathways: evidence from intestinal endocrine L cells. Endocrinology. 2008;149:2341–2351. doi: 10.1210/en.2007-1142. [DOI] [PubMed] [Google Scholar]
- 39.Ip W., Shao W., Chiang Y.T., Jin T. GLP-1-derived nonapeptide GLP-1(28–36)amide represses hepatic gluconeogenic gene expression and improves pyruvate tolerance in high fat diet fed mice. American Journal of Physiology Endocrinology and Metabolism. 2013;305:E1348–E1358. doi: 10.1152/ajpendo.00376.2013. [DOI] [PubMed] [Google Scholar]
- 40.Columbus J., Chiang Y., Shao W., Zhang N., Wang D., Gaisano H.Y. Insulin treatment and high-fat diet feeding reduces the expression of three Tcf genes in rodent pancreas. Journal of Endocrinology. 2010;207:77–86. doi: 10.1677/JOE-10-0044. [DOI] [PubMed] [Google Scholar]
- 41.Jin T. The WNT signalling pathway and diabetes mellitus. Diabetologia. 2008;51:1771–1780. doi: 10.1007/s00125-008-1084-y. [DOI] [PubMed] [Google Scholar]
- 42.Sun J., Wang D., Jin T. Insulin alters the expression of components of the Wnt signaling pathway including TCF-4 in the intestinal cells. Biochimica et Biophysica Acta. 2010;1800:344–351. doi: 10.1016/j.bbagen.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Okamura R.M., Sigvardsson M., Galceran J., Verbeek S., Clevers H., Grosschedl R. Redundant regulation of T cell differentiation and TCRalpha gene expression by the transcription factors LEF-1 and TCF-1. Immunity. 1998;8:11–20. doi: 10.1016/s1074-7613(00)80454-9. [DOI] [PubMed] [Google Scholar]
- 44.Murtaugh L.C., Law A.C., Dor Y., Melton D.A. Beta-catenin is essential for pancreatic acinar but not islet development. Development. 2005;132:4663–4674. doi: 10.1242/dev.02063. [DOI] [PubMed] [Google Scholar]
- 45.Papadopoulou S., Edlund H. Attenuated Wnt signaling perturbs pancreatic growth but not pancreatic function. Diabetes. 2005;54:2844–2851. doi: 10.2337/diabetes.54.10.2844. [DOI] [PubMed] [Google Scholar]
- 46.Heiser P.W., Lau J., Taketo M.M., Herrera P.L., Hebrok M. Stabilization of beta-catenin impacts pancreas growth. Development. 2006;133:2023–2032. doi: 10.1242/dev.02366. [DOI] [PubMed] [Google Scholar]
- 47.Rulifson I.C., Karnik S.K., Heiser P.W., ten Berge D., Chen H., Gu X. Wnt signaling regulates pancreatic beta cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6247–6252. doi: 10.1073/pnas.0701509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schinner S., Ulgen F., Papewalis C., Schott M., Woelk A., Vidal-Puig A. Regulation of insulin secretion, glucokinase gene transcription and beta cell proliferation by adipocyte-derived Wnt signalling molecules. Diabetologia. 2008;51:147–154. doi: 10.1007/s00125-007-0848-0. [DOI] [PubMed] [Google Scholar]
- 49.Jin T., George Fantus I., Sun J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of beta-catenin. Cellular Signalling. 2008;20:1697–1704. doi: 10.1016/j.cellsig.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 50.Ip W., Chiang Y.T., Jin T. The involvement of the wnt signaling pathway and TCF7L2 in diabetes mellitus: the current understanding, dispute, and perspective. Cell & Bioscience. 2012;2:28. doi: 10.1186/2045-3701-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korinek V., Barker N., Moerer P., van Donselaar E., Huls G., Peters P.J. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nature Genetics. 1998;19:379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 52.Teitelman G., Kedees M. Mouse insulin cells expressing an inducible RIPCre transgene are functionally impaired. The Journal of Biological Chemistry. 2015 doi: 10.1074/jbc.M114.615484. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kone M., Pullen T.J., Sun G., Ibberson M., Martinez-Sanchez A., Sayers S. LKB1 and AMPK differentially regulate pancreatic beta-cell identity. FASEB Journal. 2014;28:4972–4985. doi: 10.1096/fj.14-257667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brouwers B., de Faudeur G., Osipovich A.B., Goyvaerts L., Lemaire K., Boesmans L. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metabolism. 2014;20:979–990. doi: 10.1016/j.cmet.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin L., Cui L., Zhou W., Dufort D., Zhang X., Cai C.L. Beta-catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9313–9318. doi: 10.1073/pnas.0700923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.