

Abstract

2,2′-Bipyridine-ligated copper complexes, in combination with TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl), are highly effective catalysts for aerobic alcohol oxidation. Considerable uncertainty and debate exist over the mechanism of alcohol oxidation mediated by CuII and TEMPO. Here, we report experimental and density functional theory (DFT) computational studies that distinguish among numerous previously proposed mechanistic pathways. Oxidation of various classes of radical-probe substrates shows that long-lived radicals are not formed in the reaction. DFT computational studies support this conclusion. A bimolecular pathway involving hydrogen-atom-transfer from a CuII–alkoxide to a nitroxyl radical is higher in energy than hydrogen transfer from a CuII–alkoxide to a coordinated nitroxyl species. The data presented here reconcile a collection of diverse and seemingly contradictory experimental and computational data reported previously in the literature. The resulting Oppenauer-like reaction pathway further explains experimental trends in the relative reactivity of different classes of alcohols (benzylic versus aliphatic and primary versus secondary), as well as the different reactivity observed between TEMPO and bicyclic nitroxyls, such as ABNO (ABNO = 9-azabicyclo[3.3.1]nonane N-oxyl).
INTRODUCTION
Aerobic alcohol oxidation mediated by homogeneous copper/nitroxyl cocatalysts has been the subject of extensive recent investigation.1 These catalyst systems achieve some of the most efficient, selective, and functional-group-tolerant means to oxidize alcohols to aldehydes and ketones. They often use ambient air as the source of oxidant and achieve complete conversion within 1 h at room temperature. Two highly effective complementary catalyst systems are shown in Scheme 1. The Cu/TEMPO (TEMPO = 2,2,6,6-tetramethylpiperidine-N-oxyl) catalyst system (Scheme 1A),2,3 which features a CuI source such as [Cu(MeCN)4]OTf in combination with 2,2′-bipyridine (bpy), TEMPO, and N-methylimidazole (NMI), exhibits very high chemoselectivity for oxidation of primary alcohols. For example, the steric sensitivity of this catalyst system enables selective oxidation of unprotected diols, wherein only the least sterically hindered primary alcohol is oxidized. The Cu/ABNO (ABNO = 9-azabicyclo[3.3.1]nonane N-oxyl) catalyst system4,5 (Scheme 1B) is similar, but uses the less sterically hindered bicyclic ABNO cocatalyst, and it enables rapid oxidation of both primary and secondary alcohols, including molecules containing diverse functional groups.
Scheme 1.

Cu/TEMPO and Cu/ABNO Aerobic Alcohol Oxidation Catalyst Systems
The synthetic utility of Cu/nitroxyl catalyst systems has led to considerable interest in the mechanism of these reactions. An early study3a emphasized the relationship between Cu/TEMPO and other TEMPO-catalyzed alcohol oxidation reactions that proceed via an oxoammonium/hydroxylamine cycle.6 The latter reactions are effective with a variety of terminal oxidants, such as NaOCl, Br2 and PhI(OAc)2 (Scheme 2). An O2-coupled CuII/CuI redox cycle was proposed to oxidize TEMPOH to the oxoammonium species in the Cu/TEMPO-catalyzed reactions;3a however, subsequent studies by us7 and others3c,8 demonstrated that a different mechanism is involved in these reactions. Electrochemical studies showed that the CuII reduction potential is not high enough to generate the oxoammonium species under the reaction conditions,7 and kinetic isotope effects (KIEs) revealed that an oxoammonium reagent and the reactive oxidant in Cu/TEMPO-catalyzed oxidation reactions exhibit different intrinsic isotope effects for alcohol oxidation.7a,8 Extensive additional kinetic and in situ spectroscopic studies support the (simplified) catalytic mechanism shown in Scheme 3.7 Molecular oxygen oxidizes CuI and the hydroxylamine (TEMPOH) to a CuII–OH species and TEMPO radical in steps i and ii, and CuII–OH and TEMPO then mediate oxidation of the alcohol to the aldehyde (steps iii and iv, Scheme 3).
Scheme 2.

Oxoammonium/Hydroxylamine Mechanism for Alcohol Oxidation with Diverse Terminal Oxidants
Scheme 3.

Catalytic Mechanism for Cu/TEMPO-Catalyzed Aerobic Alcohol Oxidation
The mechanism in Scheme 3 explains the kinetic differences observed between activated and unactivated alcohols. Benzylic, allylic, and other activated alcohols are oxidized rapidly in step iv and feature turnover-limiting oxidation of CuI by O2 (step i). Unactivated aliphatic alcohols react more slowly and exhibit turnover-limiting cleavage of the C–H bond (step iv). The latter reactions exhibit a saturation dependence on [alcohol] and a first-order dependence on [TEMPO], consistent with the sequence shown in steps iii and iv. Insights such as these provided a foundation for the discovery of the Cu/ABNO catalyst system (cf. Scheme 1B),4 which exhibits faster rates and broader scope, but many fundamental questions about the mechanism of alcohol oxidation by CuII and nitroxyl remain unanswered.
The mechanism of CuII/nitroxyl-mediated alcohol oxidation has been the subject of considerable speculation and debate. Three of the most prominent mechanistic proposals are depicted in Scheme 4. In 1966, Brackman and Gaasbeek reported the first example of Cu/nitroxyl-catalyzed alcohol oxidation in the oxidation of methanol with phenanthroline/CuII species and di-tert-butylnitroxyl.9 They observed inhibition of the reaction under more basic conditions and proposed that the nitroxyl radical abstracts a hydrogen atom from a neutral methanol ligand coordinated to CuII (Scheme 4A). Sheldon and co-workers highlighted similarities between Cu/nitroxyl catalysts and galactose oxidase,8a an enzyme that features an active site with a CuII-coordinated phenoxyl radical from a modified tyrosine side chain (Figure 1). The oxidation of different benzylic alcohols by a Cu/nitroxyl catalyst system was shown to exhibit KIEs and Hammett correlations similar to those reported for galactose oxidase, and the authors proposed a galactose oxidase-like mechanism involving hydrogen atom transfer to an η2-coordinated nitroxyl radical (Scheme 4B; cf. Figure 1). This proposal finds additional support from precedents for CuII complexes bearing η1- and η2-coordinated nitroxyl ligands.10 Finally, density functional theory (DFT) computational studies have been used to probe the nature of hydrogen transfer from an alkoxide ligand to a coordinated nitroxyl.11,12 Compelling studies by Baerends and co-workers identified a low-energy pathway involving hydrogen transfer from the alkoxide to an η1-coordinated nitroxyl (Scheme 4C).11
Scheme 4.

Mechanistic Proposals for CuII/Nitroxyl-Mediated Alcohol Oxidation
Figure 1.

Active site structure of galactose oxidase.
Each of the mechanisms in Scheme 4 is potentially consistent with experimental data; however, recent spectroscopic and kinetic studies of Cu/TEMPO-catalyzed alcohol oxidation did not provide evidence for a Cu/TEMPO adduct under catalytic conditions.3c,7,13 In addition, a first-order kinetic dependence on [Cu] and [TEMPO] observed in the oxidation of aliphatic alcohols seems most consistent with a bimolecular mechanism analogous to that in Scheme 4A. The computationally derived mechanism in Scheme 4C11 was published before the recent experimental studies and cannot be compared directly to the experimental results (e.g., activation barriers) because these studies started from a [(bpy)CuII(alkoxide)(nitroxyl)]+ complex that is not the resting state of the catalyst.
Here, we report experimental and computational data that directly assess the mechanisms in Scheme 4. Several radical-probe substrates have been used to probe whether discrete radical intermediates are involved in alcohol oxidation (cf. Scheme 4A,B). DFT computational studies of (bpy)Cu/TEMPO- and (bpy)Cu/ABNO-mediated alcohol oxidation have been carried out, starting from a [(bpy)Cu(OH)(NMI)]+ species that has been proposed as the catalyst resting state. Inclusion of the full ligand and nitroxyl (TEMPO and ABNO) structures enables quantitative comparison of the computational results with experimental data, including relative reactivity trends between benzylic/aliphatic and primary/secondary alcohols and between TEMPO/ABNO nitroxyl cocatalysts. The collective results reconcile previously reported experimental and computational data and support a closed-shell two-electron hydrogen-transfer pathway most closely resembling that in Scheme 4C. The results further explain the reactivity and chemoselectivity differences observed with TEMPO and ABNO cocatalysts.
RESULTS AND DISCUSSION
Oxidation of Radical Probe Substrates
Galactose oxidase-mediated alcohol oxidation is proposed to proceed via a ketyl radical intermediate.14 This conclusion is supported by results with radical-probe substrates, such as cyclopropyl carbinol derivatives. Cyclobutanol is another probe molecule used to distinguish between one- and two-electron alcohol oxidation pathways.15–21 For example, oxidation of cyclobutanol by permanganate leads to ring-opened products,20 while oxidation by perruthenate affords only cyclobutanone.21 These observations support stepwise one-electron and concerted two-electron pathways, respectively, with these reagents.
Five different radical-probe substrates were subjected to the previously reported Cu/TEMPO aerobic alcohol oxidation conditions (Table 1). In all cases, efficient oxidation was observed to afford the unrearranged aldehyde or ketone in nearly quantitative yield, based on 1H NMR analysis. The cyclopropyl carbinol derivatives (entries 1–3) show no ring-opening to the homoallylic products.22,23 Cyclopropyl radical ring-opening is known to be a reversible process,24 but the lack of trans–cis isomerization of 2,3-trans,trans-diphenyl-cyclopropanemethanol (entry 3) indicates that a ring-opening/ring-closure equilibrium does not occur. Cyclobutanol affords only cyclobutanone (entry 4), and no cyclizaton of the benzylic alcohol is observed under the reaction conditions (entry 5). The last substrate has been shown to rearrange via a 6-exo-trigcyclization to a chromanone derivative when subjected to alcohol oxidation conditions that proceed via one-electron steps.25
Table 1.
Cu/TEMPO-Catalyzed Aerobic Oxidation of Radical-Probe Substrates
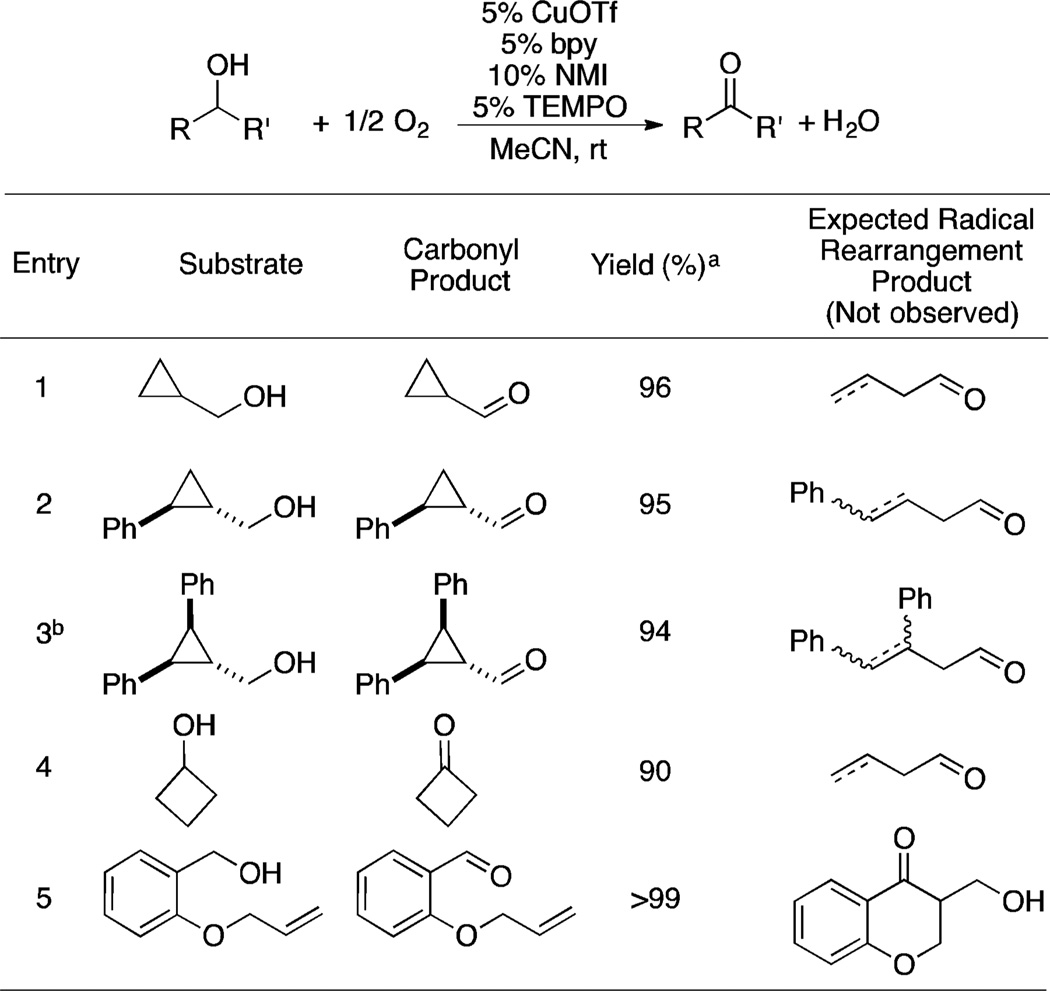 |
Yield determined by 1H NMR spectroscopy (int. std. = trimethoxybenzene).
No trans–cis isomerization observed.
To our knowledge, the rate constants for rearrangement of the ketyl radical anions derived from these substrates have not been determined, but rate constants for the corresponding neutral radicals are >108 s−1.22,26 This consideration, together with evidence for radical intermediates when these substrates have been used with galactose oxidase and other oxidants, suggests that discrete radical intermediates are not involved in Cu/nitroxyl-mediated alcohol oxidation.
Experimental Benchmarks for Computational Studies
With these results in hand, we turned our attention to DFT computational studies designed to enable direct comparison with experimental data. The following experimental observations provide important benchmarks to assess the validity of computed reaction pathways:
Cu/TEMPO-catalyzed alcohol oxidation reactions exhibit the following relative reactivity trends: primary benzylic alcohols > primary aliphatic alcohols ≫ secondary aliphatic alcohols.
The experimental activation barrier for benzyl alcohol oxidation is <15 kcal/mol (i.e., less than that associated with the turnover-frequency,27 which is limited by aerobic oxidation of CuI as the turnover-limiting step) and is 15–16 kcal/mol for the primary aliphatic alcohol cyclohexanemethanol, which was used in previous mechanistic studies.7
Spectroscopic studies provide no direct evidence for a Cu/TEMPO adduct under the catalytic reaction conditions, and the oxidation of the aliphatic primary alcohols exhibits a first-order kinetic dependence on [Cu] and [TEMPO] and a saturation dependence on [alcohol].7 These data demonstrate that the catalyst resting state cannot be a Cu/TEMPO adduct. Electron paramagnetic resonance (EPR) spectroscopic analysis of the catalytic reaction mixture supports the presence a mononuclear bpy-ligated CuII species. A (bpy)CuII–hydroxide resting state, such as [(bpy)Cu(OH)(NMI)]+, is most consistent with the data.
Alcohol oxidation does not proceed via a discrete radical intermediate.
Alcohol oxidation by the Cu/ABNO catalyst system proceeds with a much lower barrier than Cu/TEMPO, and Cu/ABNO mediates facile oxidation of secondary aliphatic alcohols.
We used the OPBE density functional in our DFT computational studies, following the lead of Baerends et al.11 Comparative calculations were carried out with the B3LYP and M06-L functionals, but both of these functionals led to poor agreement with experimental data.28 The experimental solvent, acetonitrile, was modeled using the SMD continuum solvation model.
Formation of Cu–Alkoxides Species
The experimental data implicate an equilibrium between (bpy)CuII–OH and (bpy)CuII–OCH2R species (cf. benchmark #3 in the previous section). Several four- and five-coordinate structures of this type were evaluated, with NMI and/or acetonitrile ancillary ligands, and the most stable species was found to be [(bpy)CuII(OH)(NMI)]+ (Figure 2). Equilibria for formation of CuII–alkoxide species derived from benzyl alcohol, and 1- and 2-propanol (as representative benzylic, primary aliphatic and secondary aliphatic substrates) were calculated. In each case, the energy of the CuII–alkoxide is higher than that of the CuII–hydroxide, with relative stabilities of CuII–alkoxides 2Bn, 2nPr, and 2iPr showing a qualitative correlation with the alcohol pKa values: benzyl alcohol > 1-propanol > 2-propanol.29,30 These CuII–alkoxide species served as starting points for assessment of the different oxidation pathways depicted in Scheme 4.
Figure 2.

Equilibrium for formation of Cu–alkoxide species.
Bimolecular Hydrogen Atom Abstraction Pathway
On the basis of the lack of evidence for TEMPO coordination to (bpy)CuII and the first-order rate dependence on [Cu] and [TEMPO] (cf. benchmark #3 in the previous section), we previously proposed that alcohol oxidation occurs via bimolecular abstraction of a hydrogen atom from a CuII– alkoxide by TEMPO.7a This reaction was evaluated for various (bpy)CuII–alkoxide species. The lowest energy pathway for hydrogen-atom transfer was found from [(bpy)CuII(OR)-(CH3CN)]+ species, which have a slightly higher ground-state energy than the analogous NMI-ligated species (ΔG° < 2 kcal/mol) (Scheme 5). The transition state energies for benzyl, 1-propyl, and 2-propyl alcohol derivatives were all very similar, at 20.6, 21.0, and 21.8 kcal/mol, respectively. The H atom transfer step exhibits a linear C⋯H⋯O trajectory, while the geometry of the copper center shifts from square-planar to distorted tetrahedral in the transition state. Transition states with a closed-shell electronic structure are lowest in energy, consistent with the reduction of CuII to CuI taking place in concert with the H atom transfer step.
Scheme 5.

Free Energy Surface for CuII/TEMPO-Mediated Alcohol Oxidation via Bimolecular Hydrogen Atom Transfer (cf. Scheme 4A)
These calculations show that a bimolecular hydrogen-atom transfer process is energetically accessible. It further accounts for the experimental rate law, and the lack of evidence for a radical intermediate can be explained by homolytic C–H cleavage occurring in concert with reduction of CuII to CuI, thereby avoiding formation of a carbon-centered radical. On the other hand, the calculated barrier for oxidation of benzyl alcohol (20.6 kcal/mol) is substantially higher than the 15 kcal/mol upper limit for this substrate determined experimentally,27 and the different barriers calculated for primary and secondary alcohols do not account for the dramatic reactivity differences observed with these two classes of alcohols.
Hydrogen Transfer to η1- and η2-Coordinated TEMPO Adducts of Copper(II)
Reaction pathways involving η1 and η2 CuII–nitroxyl adducts (cf. Scheme 4B,C) provide alternatives to bimolecular hydrogen-atom transfer. An η2–TEMPO adduct 6nPr was identified computationally by replacement of NMI in [(bpy)Cu(OnPr)(NMI)]+ with TEMPO (Figure 3). This complex has a closed-shell singlet electronic structure. An attempt to optimize a triplet structure resulted in the dissociation of the TEMPO ligand and optimization of an open-shell singlet produced a closed-shell singlet electronic structure. The free energy of complex 6nPr is 22.9 kcal/mol higher than that of the CuII–alkoxide complex 2nPr, (i.e., 25.0 kcal/mol higher than that of the reference CuII–hydroxide complex 1), and 4.0 kcal/mol higher than that of the transition state energy for the bimolecular hydrogen atom transfer pathway (cf. Scheme 5). The latter comparison suggests alcohol oxidation does not proceed via an η2–nitroxyl, since the transition state for H atom transfer would be even higher in energy than 6nPr.
Figure 3.

Equilibrium for formation of η2–nitroxyl complex, 6nPr.
Coordination of TEMPO as an η1 adduct is more favorable than as an η2 adduct (cf. Figures 3 and 4). This complex, 7nPr, also has a closed-shell singlet electronic structure. Displacement of NMI by TEMPO in Figure 4 remains substantially uphill energetically (+13.6 kcal/mol), but it is significantly lower in energy than the transition state for bimolecular H atom transfer, and this complex was used as a starting point for calculation of a full pathway for alcohol oxidation (Scheme 6).
Figure 4.

Equilibrium for formation of η1–nitroxyl complex, 7nPr.
Scheme 6.

Free Energy Surface for CuII/TEMPO-Mediated Alcohol Oxidation via Hydrogen Transfer to η1-Bound TEMPO
Different energies are observed for the η1–TEMPO adducts 7Bn, 7nPr, and 7iPr; however, their relative energies are compressed relative to the corresponding NMI adducts 2Bn, 2nPr, and 2iPr. The low-energy pathway involves hydrogen transfer from the alkoxide to the unbound nitrogen atom of the η1–TEMPO ligand via a six-membered transition state (8-TS), similar to that identified by Baerends et al.11 The transition-state energy exhibits a significant dependence on the identity of the alkoxide ligand, with the secondary i-propoxide complex having an energy 6.3 kcal/mol higher than that of the primary n-propoxide complex (cf. Scheme 6). In all cases, the barrier for hydrogen transfer is small (<5 kcal/mol) from the [(bpy)Cu-(OR)(η1–TEMPO)]+ intermediate (7), and the overall energy for the oxidation of the synthetically relevant primary alcohols PhCH2OH and nPrOH is <17 kcal/mol relative to the resting state [(bpy)Cu(OH)(NMI)]+ species (1).
Overall, this reaction pathway shows excellent agreement with the experimental data. The predicted rates follow the trends observed experimentally: primary benzylic alcohols > primary aliphatic alcohols ≫ secondary aliphatic alcohols. The activation energies of 15.8 and 16.9 kcal/mol calculated for benzyl alcohol and 1-propanol are very close to the ~15 kcal/mol barrier determined experimentally, and the significantly higher barrier calculated for 2-propanol concurs with the lack of reactivity observed with 2° alcohols under experimental conditions. The concerted hydrogen transfer step in 8-TS explains the lack of radical rearrangement products observed with the substrates in Table 1.
Our previous kinetic studies of Cu/TEMPO-catalyzed alcohol oxidation7 showed that the catalytic rate exhibits a first-order dependence on [TEMPO], and EPR spectroscopic analysis of the reaction mixture provided no evidence for TEMPO coordination to (bpy)CuII. These observations led us to propose a bimolecular H atom transfer mechanism for the reaction, similar to the mechanism in Scheme 4A. The results in Scheme 6, however, provide an alternative explanation for the experimental data. The first-order kinetic dependence on [TEMPO] and the lack of experimentally detectable nitroxyl coordination to CuII can be explained by an unfavorable preequilibrium for TEMPO binding to CuII (cf. Figure 4). A rate law corresponding to the mechanism in Scheme 6 (eq 1; see Supporting Information for derivation of the rate law) is consistent with the first-order dependence on [TEMPO] (as well as the saturation kinetic dependence on [RCH2OH] and first-order dependence on [Cu], observed experimentally7).
| (1) |
CuII/ABNO-Mediated Alcohol Oxidation
The only experimental result not explained by the data above is the reactivity different between TEMPO and ABNO (see benchmark #5 above). These nitroxyls have very similar redox potentials,31 but considerably different steric profiles, and the Cu/ABNO catalyst system exhibits significantly higher activity with 1° and 2° aliphatic alcohols. The η1–nitroxyl pathway in Scheme 6 was re-evaluated with CuII/ABNO for the oxidation of 1- and 2-propanol (Scheme 7). The ABNO adduct 10 and corresponding hydrogen-transfer transition state 11-TS exhibit much lower energies than the TEMPO-derived structures. The overall barriers for CuII/ABNO-mediated oxidation of 1- and 2- propanol are less than 10 kcal/mol, and there is essentially no difference between the barrier for the two different alcohol substrates. Similar results are obtained with AZADO (2-azaadamantane N-oxyl), a nitroxyl with electronic and steric profiles similar to ABNO (Supporting Information Figure S2). These results show that steric effects can have a significant influence on the stability of CuII/nitroxyl adducts and their reactivity toward alcohol oxidation.
Scheme 7.

CuII/ABNO-Mediated Oxidation of 1-Propanol and 2-Propanol
Mechanistic Analysis and Comparison to Other Alcohol Oxidation Methods
The experimental and computational studies outlined above clearly distinguish among the mechanisms that have been proposed for Cu/TEMPO-catalyzed alcohol oxidation. The elegant and compelling analogy between Cu/nitroxyl catalyst systems and galactose oxidase is undermined by the lack of evidence for radical intermediates in the Cu/TEMPO reactions (cf. Table 1). The computational results further show that a bimolecular pathway for hydrogen-atom transfer (cf. Schemes 4A and 5), which is conceptually related to the intramolecular hydrogenatom transfer step proposed for galactose oxidase, has a barrier that is too high in energy to account for the experimental results, and does not reproduce the substrate steric effects observed in the reactions.
On the other hand, the experimental data are fully explained by a mechanism involving concerted hydrogen transfer from an alkoxide ligand to an η1-coordinated nitroxyl. Structurally characterized Cu–TEMPO complexes have been reported in the literature, including examples with η1 and η2 TEMPO coordination modes (Figure 5).10,32 Each of these complexes is diamagnetic, consistent with the closed-shell singlet electronic structure of the [(bpy)Cu(OR)(nitroxyl)]+ intermediates 7 and 10 described above (cf. Schemes 6 and 7). The η2–TEMPO coordination mode is much too high in energy to participate the catalytic reaction (cf. Figure 3 and Scheme 4B), but the η1 coordination mode is energetically accessible. Nevertheless, coordination of TEMPO as an η1 adduct is sufficiently uphill energetically that the adduct cannot be detected experimentally. A previous study of the well-defined Cu–TEMPO complexes 13 and 14 (Figure 5) noted that pyridine readily displaces TEMPO from Cu,10d so perhaps it is not surprising that TEMPO coordination is disfavored in the presence of bpy and NMI as ancillary ligands.
Figure 5.

Structurally characterized Cu–nitroxyl complexes.
The six-membered transition state for hydrogen transfer in Schemes 6 and 7 shows considerable resemblance to the transition state for Oppenauer oxidation of alcohols mediated by Al(iOPr)3 (Figure 6).33 The latter mechanism involves approximately thermoneutral hydrogen transfer from an AlIII-bound alkoxide to a coordinated ketone, such as acetone, as a hydrogen acceptor. While the two transition states are structurally similar, CuII/nitroxyl-mediated alcohol oxidation features a thermodynamic driving force associated with reduction of CuII and nitroxyl to CuI and hydroxylamine, and oxidation of CuI and hyroxylamine by O2 continuously replenishes the oxidized species (Scheme 3). This driving force in the reaction expands the scope of alcohols that can be oxidized effectively.
Figure 6.

Structural similarity between the transition states for alcohol oxidation in Cu/TEMPO and Oppenauer oxidation methods.
Substantial contemporary efforts are focused on replacing noble-metal catalysts with first-row transition metals. One intrinsic challenge in achieving this goal is the tendency of first-row transition metals to undergo one-electron, rather than two-electron, redox steps. The Cu/nitroxyl catalysts show how a first-row transition metal can be combined with a one-electron redox-active organic cocatalyst to achieve a two-electron transformation, and their mechanism may be compared to palladium(II)-catalyzed alcohol oxidation reactions.34 The latter reactions feature PdII–alkoxide intermediates that undergo β-hydride elimination via a four-membered transition state (Figure 7). This step is often the turnover-limiting step of the reaction. The six-membered transition state for Cu/nitroxyl-mediated alcohol oxidation should have less strain, which may may account for the lower barrier associated with these reactions.35 The identification of new catalyst systems that exploit the redox synergy between transition metal and organic cocatalysts represents a promising target for future studies.
Figure 7.

Four-membered transition states for β-hydride elimination from a PdII-alkoxide.
CONCLUSIONS
The study described herein resolves long-standing questions about the mechanism of Cu/TEMPO-catalyzed alcohol oxidation by reconciling an array of experimental data that have provided the basis for the different mechanistic proposals. Specifically, a concerted two-electron alcohol oxidation pathway involving a closed-shell η1–nitroxyl–Cu adduct, identified by DFT computational methods, is shown to be favored over intramolecular and intermolecular homolytic mechanisms. It further rationalizes experimentally determined catalytic laws and accounts for the steric and kinetic effects of alcohol oxidations that employ TEMPO and ABNO cocatalysts. Cu/TEMPO is commonly described as a functional mimic of galactose oxidase, but the mechanistic conclusions highlight important distinctions between the two systems in light of the strong evidence for radical intermediates in the enzymatic reaction.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the DOE for financial support of this work (DE-FG02-05ER15690). The NSF provided partial support for the computational resources (CHE-0840494) and NMR instrumentation (NSF CHE-1048642 and NSF CHE-0342998) used in this work.
Footnotes
ASSOCIATED CONTENT
S Supporting Information
Experimental details and characterization data for radical-probe oxidation reactions and computational details and atomic coordinates and energies for all computed structures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.For reviews, see: Sheldon RA, Arends IWCE. Adv. Synth. Catal. 2004;346:1051–1071. Cao Q, Dornan LM, Rogan L, Hughes NL, Muldoon ML. Chem. Commun. 2014;50:4524–4543. doi: 10.1039/c3cc47081d. Ryland BL, Stahl SS. Angew. Chem, Int. Ed. 2014 doi: 10.1002/anie.201403110. Early View.
- 2.(a) Hoover JM, Stahl SS. J. Am. Chem. Soc. 2011;133:16901–16910. doi: 10.1021/ja206230h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoover JM, Steves JE, Stahl SS. Nat. Protoc. 2012;7:1161–1166. doi: 10.1038/nprot.2012.057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hoover JM, Stahl SS. Org. Synth. 2013;90:240–250. [Google Scholar]
- 3.For discussion of related catalyst systems, see the reviews in ref 1 and the following leading references: Semmelhack MF, Schmid CR, Cortés DA, Chou CS. J. Am. Chem. Soc. 1984;106:3374–3376. Gamez P, Arends IWCE, Sheldon RA, Reedijk J. Adv. Synth. Catal. 2004;346:805–811. Kumpulainen ETT, Koskinen AMP. Chem.— Eur. J. 2009;15:10901–10911. doi: 10.1002/chem.200901245.
- 4.Steves JE, Stahl SS. J. Am. Chem. Soc. 2013;135:15742–15745. doi: 10.1021/ja409241h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For discussion of related catalyst systems, see the review in ref 1 and the following leading references: Sasano Y, Nagasawa S, Yamazaki M, Shibuya M, Park J, Iwabuchi Y. Angew. Chem, Int. Ed. 2014;53:3236–3240. doi: 10.1002/anie.201309634. Rogan L, Hughes NL, Cao Q, Dornan LM, Muldoon MJ. Catal. Sci. Technol. 2014;4:1720–1725.
- 6.For a recent comprehensive reviews, see: Bobbitt JM, Brückner C, Merbouh N. Org. React. (Hoboken, NJ, U.S.) 2010;74:103–424.
- 7.(a) Hoover JM, Ryland BL, Stahl SS. J. Am. Chem. Soc. 2013;135:2357–2367. doi: 10.1021/ja3117203. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoover JM, Ryland BL, Stahl SS. ACS Catal. 2013;3:2599–2605. doi: 10.1021/cs400689a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Dijksman A, Arends IWCE, Sheldon RA. Org. Biomol. Chem. 2003;1:3232–3237. doi: 10.1039/b305941c. [DOI] [PubMed] [Google Scholar]; (b) Gamez G, Arends IWCE, Reedijk J, Sheldon RA. Chem. Commun. 2003:2414–2415. doi: 10.1039/b308668b. [DOI] [PubMed] [Google Scholar]
- 9.Brackman W, Gaasbeek C. Recl. Trav. Chim. Pays-Bas. 1966;85:221–241. [Google Scholar]
- 10.(a) Dickman MH, Doedens RJ. Inorg. Chem. 1981;20:2677–2681. [Google Scholar]; (b) Porter LC, Dickman MH, Doedens RJ. Inorg. Chem. 1986;25:678–684. [Google Scholar]; (c) Caneschi A, Grand A, Laugier J, Rey P, Subra R. J. Am. Chem. Soc. 1988;110:2307–2309. [Google Scholar]; (d) Laugier J, Latour JM, Caneschi A, Rey P. Inorg. Chem. 1991;30:4474–4477. [Google Scholar]
- 11.(a) Michel C, Belanzoni P, Gamez P, Reedijk J, Baerends EJ. Inorg. Chem. 2009;48:11909–11920. doi: 10.1021/ic902155m. [DOI] [PubMed] [Google Scholar]; (b) Belanzoni P, Michel C, Baerends EJ. Inorg. Chem. 2011;50:11896–11904. doi: 10.1021/ic200725k. [DOI] [PubMed] [Google Scholar]
- 12.Cheng L, Wang J, Wang M, Wu Z. Inorg. Chem. 2010;49:9392–9399. doi: 10.1021/ic100996b. [DOI] [PubMed] [Google Scholar]
- 13.Gamba I, Mutikainen I, Bouwman E, Reedijk J, Bonnet S. Eur. J. Inorg. Chem. 2013:115–123. [Google Scholar]
- 14.(a) Branchaud BP, Montague-Smith MP, Kosman DJ, McLaren FR. J. Am. Chem. Soc. 1993;115:798–800. [Google Scholar]; (b) Wachter RM, Branchaud BP. J. Am. Chem. Soc. 1996;118:2782–2789. [Google Scholar]; (c) Wachter RM, Montague-Smith MP, Branchaud BP. J. Am. Chem. Soc. 1997;119:7743–7749. [Google Scholar]; (d) Turner BE, Branchaud BP. Bioorg. Med. Chem. Lett. 1999;9:3341–3346. doi: 10.1016/s0960-894x(99)00611-3. [DOI] [PubMed] [Google Scholar]; (e) Himo F, Eriksson LA, Maseras F, Siegbahn PEM. J. Am. Chem. Soc. 2000;122:8031–8036. [Google Scholar]; (f) Whittaker MM, Whittaker JW. Biochemistry. 2001;40:7140–7148. doi: 10.1021/bi010303l. [DOI] [PubMed] [Google Scholar]; (g) Branchaud BP, Turner BE. Methods Enzymol. 2002;354:415–425. doi: 10.1016/s0076-6879(02)54032-5. [DOI] [PubMed] [Google Scholar]
- 15.Rocek J, Radkowsky AE. J. Am. Chem. Soc. 1968;90:2986–2988. [Google Scholar]
- 16.Rocek J, Aylward DE. J. Am. Chem. Soc. 1975;97:5452–5456. [Google Scholar]
- 17.(a) Pestovsky O, Bakac A. J. Am. Chem. Soc. 2004;126:13757–13764. doi: 10.1021/ja0457112. [DOI] [PubMed] [Google Scholar]; (b) Beach ES, Malecky RT, Gil RR, Horwitz CP, Collins TJ. Catal. Sci. Technol. 2011;1:437–443. [Google Scholar]; (c) Lee DG, Gai H. Can. J. Chem. 1993;71:1394–1400. [Google Scholar]
- 18.Meyer K, Rocek J. J. Am. Chem. Soc. 1972;94:1209–1214. [Google Scholar]
- 19.Whitesides GM, Sadowski JS, Lilburn J. J. Am. Chem. Soc. 1974;96:2829–2835. [Google Scholar]
- 20.Lee DG, Chen T. J. Org. Chem. 1991;56:5341–5345. [Google Scholar]
- 21.Lee DG, Spitzer UA, Cleland J, Olson ME. Can. J. Chem. 1976;54:2124–2126. [Google Scholar]
- 22.For a study of the rearrangement of the parent cyclopropylcarbinol radical species, see: Davies AG, Muggleton B. J. Chem. Soc., Perkin Trans. 1976;2:502–510.
- 23.For examples of the use of this as a probe for synthetic alcohol oxidation systems, see, e.g., Kretzschmar I, Levinson JA, Friend CM. J. Am. Chem. Soc. 2000;122:12395–12396. Markó IE, Gautier A, Mutonkole J-L, Dumeunier R, Ates A, Urch CJ, Brown SM. J. Organomet. Chem. 2001;624:344–347.
- 24.Tanko JM, Drumright RE. J. Am. Chem. Soc. 1992;114:1844–1854. [Google Scholar]
- 25.Astolfi P, Brandi P, Galli C, Gentili P, Gerini MF, Greci L, Lanzalunga O. New J. Chem. 2005;29:1308–1317. [Google Scholar]
- 26.Tanko JM, Phillips JP. J. Am. Chem. Soc. 1999;121:6078–6079. [Google Scholar]
- 27.The estimated experimental barriers are obtained from the initial rates measured under the reaction conditions reported in ref 7a, adjusted to account for standard states for all reagents.
- 28.See Supporting Information for details.
- 29.For pKa of benzyl alcohols see: Bordwell FG, Liu W-Z. J. Am. Chem. Soc. 1996;118:8777–8781.
- 30.Olmstead WN, Margolin Z, Bordwell FG. J. Org. Chem. 1980;45:3295–3299. [Google Scholar]
- 31.Lauber MB, Stahl SS. ACS Catal. 2013;3:2612–2616. [Google Scholar]
- 32.Numerous TEMPO complexes with metals other than Cu have been reported. For leading references, see: Dickman MH, Doedens RJ. Inorg. Chem. 1982;21:682–684. Jaitner P, Huber W, Gieren A, Betz H. J. Organomet. Chem. 1986;311:379–385. Mahanthappa MK, Huang K-W, Cole AP, Waymouth RM. Chem. Commun. 2002:502–503. doi: 10.1039/b110147a. Isrow D, Captain B. Inorg. Chem. 2011;50:5864–5866. doi: 10.1021/ic2007342. Smith JM, Mayberry DE, Margarit CG, Sutter J, Wang H, Meyer K, Bontchev RP. J. Am. Chem. Soc. 2012;134:6516–6519. doi: 10.1021/ja211882e. Scepaniak JJ, Wright AM, Lewis RA, Wu G, Hayton TW. J. Am. Chem. Soc. 2012;134:19350–19353. doi: 10.1021/ja309499h. Lomont JP, Nguyen SC, Harris CB. J. Am. Chem. Soc. 2013;135:11266–11273. doi: 10.1021/ja404476m. Wright AM, Zaman HT, Wu G, Hayton TW. Inorg. Chem. 2013;52:3207–3216. doi: 10.1021/ic302697v.
- 33.de Graauw CF, Peters JA, van Bekkum H, Huskens J. Synthesis. 1994:1007–1017. [Google Scholar]
- 34.For representative reviews, see: Sheldon RA, Arends IWCE, ten Brink G-J, Dijksman A. Acc. Chem. Res. 2002;35:774–781. doi: 10.1021/ar010075n. Stahl SS. Angew. Chem. Int. Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630. Schultz MJ, Sigman MS. Tetrahedron. 2006;62:8227–8241.
- 35.For a comparison of homogeneous Pd and Cu/TEMPO catalyst systems under continuous flow conditions, see: Ye X, Johnson MD, Diao T, Yates MH, Stahl SS. Green Chem. 2010;12:1180–1186. doi: 10.1039/c0gc00106f. Greene JF, Hoover JM, Mannel DS, Root TW, Stahl SS. Org. Process Res. Dev. 2013;17:1247–1251.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.