

Abstract
Background. The emergence and spread of drug resistance to current antimalarial therapies remains a pressing concern, escalating the need for compounds that demonstrate novel modes of action. Diversity-Oriented Synthesis (DOS) libraries bridge the gap between conventional small molecule and natural product libraries, allowing the interrogation of more diverse chemical space in efforts to identify probes of novel parasite pathways.
Methods. We screened and optimized a probe from a DOS library using whole-cell phenotypic assays. Resistance selection and whole-genome sequencing approaches were employed to identify the cellular target of the compounds.
Results. We identified a novel macrocyclic inhibitor of Plasmodium falciparum with nanomolar potency and identified the reduction site of cytochrome b as its cellular target. Combination experiments with reduction and oxidation site inhibitors showed synergistic inhibition of the parasite.
Conclusions. The cytochrome b oxidation center is a validated antimalarial target. We show that the reduction site of cytochrome b is also a druggable target. Our results demonstrating a synergistic relationship between oxidation and reduction site inhibitors suggests a future strategy for new combination therapies in the treatment of malaria.
Keywords: cytochrome b, diversity-oriented synthesis, drug development, drug resistance, malaria, target identification
Malaria remains one of the most significant global public health burdens with over 40% of the world's population at risk for infection and approximately 800 000 annual deaths due to the disease [1]. Disease is caused by infection with protozoan parasites of the Plasmodium genus and can range from asymptomatic or uncomplicated clinical malaria to severe disease, including anemia, respiratory distress, and cerebral complications. A major challenge in the treatment of malaria has been the emergence of resistance to antimalarial drugs [2, 3]. A large proportion of the antimalarial drugs currently used as front-line therapies are combinations of structures in the aminoquinoline and artemisinin classes. With documented resistance to each class [1], the discovery of new structural classes of antimalarial molecules with potential novel mode of action is greatly needed by the malaria research community. Recent efforts by several groups have aimed to develop leads with novel mechanisms of action [4], and we believe that the exploration of new chemical space is critical to the identification of new targets and pathways.
With the goal of developing diverse, novel compounds representing little probed chemical space, the Broad Institute has synthesized approximately 100 000 complex small molecules through Diversity-Oriented Synthesis (DOS) for use in high-throughput screens [5]. This strategy aims to cover chemical space and access structural features beyond that of traditional libraries, combining the complexity of natural products and the efficiency of high-throughput synthesis [6–8]. A subset of the DOS library called the “informer set” (approximately 8000 compounds) was screened in a phenotypic whole-cell assay against both drug-sensitive (3D7) and drug-resistant (Dd2) P. falciparum strains. The molecules in this screening set were selected to maximize chemotype and stereochemical diversity. This effort identified a new series of active molecules from the “ring-closing metathesis” (RCM) collection. Analysis of the 16 stereoisomers of the hit molecule identified the SRRS configuration to be the most potent, with marginal activity observed with the RRRS stereoisomer and no activity observed for the remaining isomers. Medicinal chemistry efforts resulted in the identification of lead ML238, a Molecular Libraries Probe Production Centers Network (MLPCN) designated probe [5] (Figure 1A). Further optimization yielded BRD6323 (Figure 1A), which contains a 2,4-dimethylisoxazolyl at the urea substituent and demonstrates both increased water and buffer solubility (400 µM, water), with somewhat diminished potency (Dd2 half maximal effective concentration [EC50] = 9 nM) [9]. Both compounds are stable in plasma and demonstrate neither cellular toxicity in HepG2 cells nor erythrocyte hemolysis [5]. The challenge remained to identify the molecular target of these molecules.
Figure 1.

Selection of DOS-resistant parasites. A, Molecules used for resistance selection experiments. ML238 is an MLPCN-designated probe. B, Dose-response phenotype of representative clones from ML238-resistant (Dd2: G33A) and BRD6323-resistant (Dd2: G33V) parasites demonstrate significant shifts in EC50 (***P ≤ .0001). Solid white bars: Dd2; bars with diagonal lines: Dd2:G33A mutant; solid black bars: Dd2:G33V parasites. C, Topological representation of PfCYTb highlighting the resistance mutations identified in this study (red) and previously reported oxidation (Qo) site mutations (yellow, ATV resistance [22]; green, decoquinate resistance [21]; blue, BZT resistance [10]). EC50 values were determined using a whole-cell SYBR green assay [12]. Error bars indicate the standard deviation of 3 biological replicates, each with triplicate measurements. Significance relative to Dd2 EC50 was determined by 1-way ANOVA with Dunnett's multiple comparison post-test; n = 3. Abbreviations: ANOVA, analysis of variance; ATV, atovaquone; BZT, benzothiazepine; DOS, Diversity-Oriented Synthesis; EC50, half maximal effective concentration; MLPCN, Molecular Libraries Probe Production Centers Network; Qo, ubiquinol oxidation center; Qi, ubiquinone reduction site of cytochrome b.
METHODS
Synthesis of ML238 (a.k.a. BRD9554, IDI-5918)
ML238 was prepared following the literature procedure [5]. 1H nuclear magnetic resonance spectra matched that reported, and high-performance liquid chromatography analysis indicated >95% purity.
Synthesis of BRD6323 (a.k.a. IDI-5994) N-(((2S,8R,9R)-11-((S)-1-(Dimethylamino)Propan-2-yl)-14-(3-(3,5-dimethylisoxazol-4-yl)Ureido)-2,9-dimethyl-12-oxo-2,3,4,5,6,8,9,10,11,12-decahydrobenzo[b][1,9,5]Dioxaazacyclotetradecin-8-yl)Methyl)-4-fluoro-N-methylbenzenesulfonamide
This compound was synthesized using the same procedure used to prepare the previously reported phenyl urea [5] with the exception that 4-isocyanato-3,5-dimethylisoxazole was used instead of phenyl isocyanate during urea formation. Complete methods for the synthesis of this compound can be found in Supplementary Information.
Parasite Strains and Culture Maintenance
Parasites were obtained from the Malaria Research and Reagent Resource Repository (MR4). We used the following parasite lines from the MR4 repository of the American Type Culture Collection (ATCC): 3D7 (MRA-151), Dd2 (MRA-156), the atovaquone-resistant cell line TM90C6B (MRA-205) harboring a Y268S mutation in PfCYTb, and an atovaquone-resistant parasite line of unknown origin with a K272R mutation in PfCYTb (ATVR: K272R). Transgenic Dd2 parasites expressing a chromosomally integrated copy of the Saccharomyces cerevisiae dihydroorotate dehydrogenase (DHOD) were utilized as previously described [10]. Parasites were cultured by standard methods [11] in Roswell Park Memorial Institute (RPMI) media supplemented with 5% human O+ serum and 0.25% AlbuMAX II (Life Technologies 11021-045).
Selection for Drug Resistance
Approximately 5 × 108 mixed stage parasites were treated at 5 nM of ML238 or 50 nM BRD6323 in each of 4 independent flasks until cultures were negative for parasites by microscopy (6–8 days). After this treatment, compound pressure was removed and the cultures fed on alternate days with complete compound free-RPMI media. Once healthy parasites reappeared in the culture flasks and parasitemia reached 2%–4%, compound exposure was repeated. These steps were executed for 30–50 days until the parasites were growing in the presence of compound at a good multiplication rate. To prevent the lysis of red blood cells, 30%–40% of parasite culture was replaced with freshly washed cells once a week during the entire selection period. Selected parasites were cloned by limiting dilution in a 96-well plate in the presence of 5 nM ML238 or 50 nM BRD6323. Parasite clones were detected by light microscopy after 3 weeks of growth and expanded for cryopreservation and phenotypic analysis. The resistant mutant cell lines are available to the research community upon request.
In Vitro Drug Sensitivity and EC50 Determination
Drug assays were performed as previously described [12], with modifications for 384-well format. Briefly, synchronized ring-stage parasites were cultured in the presence of 12-point 2-fold serial dilutions of test compounds in 40 µL of RPMI supplemented with 0.5% AlbuMAX II at 1.0% hematocrit and an initial parasitemia of 1.0% in black clear-bottom plates (Greiner Bio-one 781090). Following 72 hours' incubation under standard culture conditions, SYBR Green I dye (Invitrogen S7563) was added to a dilution of 1:5000, and plates were stored at room temperature until fluorescence signal was read on a Spectramax M5 plate reader (Molecular Devices, ex 480 nm, em 530 nM). After background subtraction and normalization, EC50 values were calculated using the Levenberg-Marquardt algorithm [13, 14] as implemented in the Collaborative Drug Discovery database [15].
Isobologram experiments were performed in similar fashion, utilizing the modified fixed-ratio methodology [16]. Briefly, DOS and control compounds were mixed at multiple fixed volumetric ratios (10:0, 8:2, 6:4, 5:5, 4:6, 2:8, and 0:10) and then serially diluted in 12-point 2-fold dilutions and dispensed in triplicate to 384-well assay plates. Fractional inhibitory concentrations (FICs) were calculated for each drug combination as described [16]. Synergy was defined as Σ FIC < 1.0, additivity as Σ FIC = 1, and antagonism as Σ FIC > 1.0.
Genome Sequencing and Single-Nucleotide Polymorphisms Identification
Genomic DNA extractions from late-stage parasite cultures were performed using Qiagen DNeasy kit (Qiagen). Genomic DNA was sheared and made into a 200 bp fragment Illumina sequencing library, and sequenced with paired-end reads on an Illumina GAIIx machine. The sequenced reads were aligned against the P. falciparum 3D7 reference from PlasmoDB (version 7.1*) [17] using Burrows-Wheeler Aligner program version 0.5.7 [18]. Duplicate reads were marked using the Picard MarkDuplicates tool <http://picard.sourceforge.net/>. The consensus bases were called using the Genome Analysis Toolkit's (GATK) Unified Genotyper (version 1.0.5974) [19] and the SAMtools (version 0.1.16) [20] mpileup command. Only bases that were called as homozygous for the reference or the alternate allele with a genotype quality of at least 30 were considered.
Polymerase Chain Reaction Analysis and DNA Sequencing
Gene-specific primers were used to polymerase chain reaction (PCR) amplify the region around each single-nucleotide polymorphism (SNP) identified by whole-genome sequencing. Primer sequences are included in the online Supplementary Information file. PCR was performed using 20 pmol of each primer, genomic DNA template, and Pfusion high-fidelity DNA polymerase HF master mix (New England Biolabs). The mixture was heated to 95°C for 5 minutes and then cycled at 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute over 35 cycles, with an additional extension at 72°C for 10 minutes after the completion of all cycles. PCR products were evaluated by agarose gel electrophoresis, purified using ExoSAP-IT per the manufacturer's protocol (Affymetrix), and sequenced using the same gene-specific amplification primers. PCR sequences were aligned and SNPs identified using the SeqMan program from the Lasergene suite, version 11.2.1 (DNASTAR, Inc).
Sequence Data
Whole-genome sequence data were deposited in GenBank under accession number SRX120899, root sample ID: SM-26YXR. PCR sequences were deposited in GenBank under accession numbers KM032225-KM032247.
Statistical Analyses
EC50 data are listed as mean ± standard deviation and were analyzed by 1-way analysis of variance with Dunnett's multiple comparison post-test; n = 3 as implemented in the Mac OS X Prism 6.0c software package (GraphPad Software, Inc) and as noted in the figure legends. Differences were considered significant for P < .05.
RESULTS
To explore whether ML238 and BRD6323 targeted known antimalarial drug pathways, we screened a panel of parasite isolates representing diverse drug-resistance genotypes, including a set of recent clinical isolates. There was not an appreciable difference in EC50 between the strains tested, suggesting that this chemical series has a nonoverlapping mode of action with existing drugs (Supplementary Figure 1). To identify the target, we selected resistant parasites and used whole-genome sequencing to identify causal mutations. Selection of Dd2 parasites with ML238 resulted in a single mutant parasite line, while 2 independent lines were generated upon exposure to BRD6323. Phenotypic whole-cell dose-response assays showed a 30- to 100-fold shift in EC50 for the mutants relative to their Dd2 parental line (ML238: 5 nM to 160 nM, BRD6323: 9 nM to 900 nM) (Figure 1B); the efficacy of control compounds was unchanged between mutants and parent.
The full genome sequence of the parasite line resistant to ML238 was determined, and SNPs between the Dd2 parental genome and the resistant parasite were identified. Silent mutations and known highly polymorphic surface antigens were filtered from the dataset, and 4 nonsynonymous mutations were identified in the resistant parasite (Table 1). Sequencing of each of these loci in the BRD6323-resistant clones revealed that only the mutation in the cytb locus was consistent across all resistant lines. ML238-selected parasites had a G98C nucleotide substitution, resulting in a G33A amino acid change, while both the BRD6323-resistant parasite lines harbor a G33V amino acid substitution (G98T nucleotide change). Cytochrome b contains 2 discrete reaction sites involved in the Q cycle: a ubiquinone reduction center (Qi site) and a ubiquinol oxidation center (Qo site). These mutations map to the ubiquinone reduction (Qi) center of P. falciparum cytochrome b (PfCYTb) in a region of the protein completely distinct from known PfCYTb drug resistance mutations in the Qo center (Figure 1C) [10, 21, 22].
Table 1.
Single-Nucleotide Polymorphisms Identified in Whole-Genome Sequencing of Parasites Resistant to ML238
| Chromosome | Position | Dd2 |
ML238R |
Gene ID | Annotation | Coding Position | Protein Position | Dd2 |
ML238R |
BRD6323R |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Base | Quality | Base | Quality | Codon | a.a. | Codona | a.a. | Codonb | a.a. | ||||||
| M76611 | 3589 | G | 93 | C | 93 | mal_mito_3 | Cytb | 98 | 33 | GGA | G | GCA | A | GTA | V |
| Pf3D7_05 | 593 876 | C | 93 | G | 93 | PFE0705c | Helicase, belonging to UvrD family, putative | 1786 | 596 | CAA | Q | GAA | E | CAA | Q |
| Pf3D7_06 | 572 486 | A | 93 | T | 93 | PFF0670w | Transcription factor with AP2 domain(s), putative | 6353 | 2118 | AAA | K | ATA | I | AAA | K |
| Pf3D7_10 | 1228 345 | T | 93 | C | 93 | PF10_0294 | RNA helicase, putative | 746 | 249 | CTC | L | CCC | P | CTC | L |
SNPs were identified relative to the parental Dd2 genome sequence. Silent mutations and those occurring in highly polymorphic surface antigens were filtered from the dataset.
Abbreviations: a.a., amino acid; BRD6323R, BRD6323-resistant clone; ML238R, ML238-resistant clone; PCR, polymerase chain reaction; SNPs, single-nucleotide polymorphisms.
a Whole-genome sequencing result.
b Result from PCR resequencing of targeted region.
Cytochrome b multimerizes with a Rieske iron-sulfur protein and cytochrome c1 subunit to form Complex III of the electron transport chain located in the mitochondrial inner membrane. In P. falciparum, electron transport mediated by Complex III plays a critical role in the pyrimidine biosynthetic pathway by regenerating the ubiquinone cofactor of DHOD [23]. To further validate cytochrome b as the target of the DOS molecules, we tested a transgenic Dd2 parasite line carrying a copy of the type-1A yeast DHOD enzyme (Dd2-scDHOD), which allows the parasite to bypass the need for electron transport in the pyrimidine biosynthetic pathway [10, 23]. Electron transport inhibitors targeting complexes I-III are therefore rendered ineffective against the transgenic parasites expressing the yeast enzyme. Dose response to either selection compound in the Dd2-scDHOD cell line showed a dramatic shift in EC50 (100-fold less sensitive), confirming that parasite electron transport is involved in the primary mechanism of action of the DOS molecules (Supplementary Table 2). The relative potency of the control compound, chloroquine, which has a nonmitochondrial antimalarial mechanism, remained unchanged across these parasite strains.
The Qo site is known to be the target of atovaquone (ATV), while the Qi site appears to be an underappreciated alternate binding site for an effective antiparasitic agent. To understand the interplay between resistance to Qi and Qo site inhibitors, we tested cell lines harboring Qo site mutations along with our Qi site mutants against a panel of Qi site and Qo site inhibitors. TM90C6B, a culture-adapted clinical isolate resistant to ATV, contains a Y268S mutation in the Qo site, which has been shown to be an important contributor to ATV resistance [22]. Additionally, we tested an independent ATV-resistant cell line (ATVR: K272R) with a K272R mutation in the Qo site. Dd2: G131S is a Dd2-derived cell line resistant to a class of tetracyclic benzothiazepine (BZT) cytochrome b inhibitors and harbors a G131S mutation in the ubiquinol binding pocket of cytochrome b [10].
To evaluate the effect of Qi site inhibitors on our various mutant cell lines, we assayed them in the presence of the DOS molecules or antimycin. Antimycin is a natural product derived from Streptomyces that has modest antimalarial activity [10, 22] and has been shown to potently inhibit human and yeast cytochrome b. Resistance to antimycin in yeast has been attributed to multiple mutations in the Qi site [24], leading us to predict that our Dd2: G33A/V mutant lines would be cross-resistant to the molecule. Interestingly, only the Dd2: G33V mutant and not the Dd2: G33A mutant showed reduced sensitivity to antimycin (Table 2), suggesting that disruption of antimycin binding and/or activity requires larger steric changes than the small alteration represented by the addition of the methyl group in the G33A mutant. The ATV-resistant cell lines, TM90C6B and ATVR: K272R, were not cross-resistant with any of the Qi site inhibitors. In fact, they were hypersensitive to the DOS molecules and showed a similar trend in response to antimycin. Dd2: G131S parasites also demonstrated increased sensitivity to ML238 (Table 2). These results could imply that mutations in the Qo site (TM90C6B, ATVR: K272R, Dd2: G131S cell lines) come at a fitness cost, rendering the parasites more susceptible to inhibition at the Qi site. Indeed, reduced fitness of Qo site–mutant parasites relative to wild-type has been previously described and attributed to reduced ubiquinone binding [25].
Table 2.
Activity of Qi Site Inhibitors in PfCYTb-Mutant Cell Lines
| EC50 (nM) ± SD |
||||||
|---|---|---|---|---|---|---|
| Dd2 | Dd2: G33A | Dd2: G33V | Dd2: G131S | TM90C6B | ATVR: K272R | |
| Chloroquine | 95 ± 7 | 94 ± 12 | 92 ± 10 | 71 ± 25 | 250 ± 67** | 130 ± 20 |
| ML238 | 4.8 ± 1.0 | 160 ± 17*** | 84 ± 110*** | 2.0 ± 0.7* | 1.23 ± 0.6*** | 1.7 ± 0.7** |
| BRD6323 | 9.9 ± 1.0 | 920 ± 120*** | 1300 ± 230*** | 9.6 ± 1.2 | 5.92 ± 1.4* | 4.1 ± 0.04*** |
| Antimycin | 160 ± 22 | 110 ± 33 | 600 ± 94** | 170 ± 95 | 72.8 ± 49 | 71 ± 18 |
EC50s (nM) are mean ± SD of 3 independent assays (each run in triplicate). Significance relative to Dd2 EC50 was determined by 1-way ANOVA followed by Dunnett's multiple comparison post-test; n = 3.
Abbreviations: ANOVA, analysis of variance; ATVR, atovaquone resistant; EC50, half maximal effective concentration; PfCYTb, Plasmodium falciparum cytochrome b; Qi, ubiquinone reduction site of cytochrome b.
* P < .05; **P < .005; ***P < .0005.
We further interrogated whether Qo site inhibitors would demonstrate a similar pattern of activities and implied fitness consequences in our panel of mutant parasite lines. The Dd2: G33A/V cell lines did not show cross-resistance to any of the Qo site inhibitors tested (Table 3). Paralleling the effect seen with Qi site inhibitors in the Qo site mutant cell lines, the Dd2: G33V cell line showed increased sensitivity to ATV, suggesting that there might be reciprocal fitness costs in mutating either the Qi or Qo site.
Table 3.
Activity of Qo Site Inhibitors in PfCYTb-Mutant Cell Lines
| EC50 (nM) ± SD |
||||||
|---|---|---|---|---|---|---|
| Dd2 | Dd2: G33A | Dd2: G33V | Dd2: G131S | TM90C6B | ATVR: K272R | |
| Decoquinate | 1.3 ± 0.3 | 0.62 ± 0.13 | 0.64 ± 0.39* | 290 ± 36*** | 485 ± 73*** | 12 ± 2*** |
| Atovaquone | 0.16 ± 0.02 | 0.10 ± 0.02 | 0.072 ± 0.02* | 0.099 ± 0.02 | 6690 ± 3700*** | 5.6 ± 0.6*** |
| BZT1 | 24 ± 2 | 30 ± 23 | 21 ± 7 | 2700 ± 140*** | 342 ± 120*** | 66 ± 5* |
EC50s (nM) are mean ± standard deviation of 3 independent assays. Significance relative to Dd2 EC50 was determined by 1-way ANOVA followed by Dunnett's multiple comparison post-test; n = 3.
Abbreviations: ANOVA, analysis of variance; ATVR, atovaquone resistant; EC50, half maximal effective concentration; PfCYTb, Plasmodium falciparum cytochrome b; Qo, ubiquinol oxidation site of cytochrome b.
* P < .05, **P < .005, ***P < .0005.
Given the increased sensitivity of our mutant cell lines to inhibitors of the other site, we were interested in exploring the effect of Qi and Qo site inhibitors on the malaria parasite when administered in combination. Using a modified fixed-ratio methodology [16], isobolar analysis of ML238 in combination with either ATV or decoquinate demonstrated a synergistic relationship between the 2 classes of inhibitors (mean Σ FIC (ML238 + ATV) = 0.77, mean Σ FIC (ML238 + DEC) = 0.57) (Figure 2, Supplementary Table 2). These results are consistent with what is known about the shuttle of electrons between these 2 sites during the Q-cycle and observations that bypass mechanisms in the face of perturbation at the Qi site (by either inhibitor binding or site mutation) are not permitted [26]. Their synergistic effect also suggests the possibility of using Qo and Qi site inhibitors in combination therapies, particularly in the face of clinical resistance to ATV.
Figure 2.
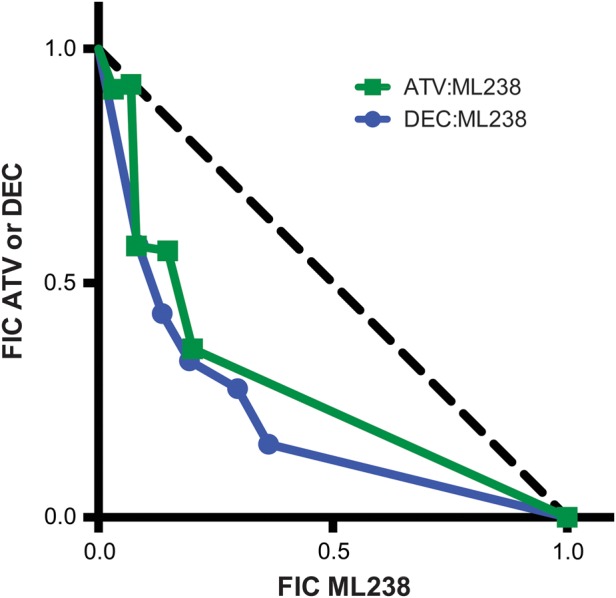
Qi and Qo site inhibitors are synergistic. ML238 was tested in combination with ATV (closed green squares and curve) and DEC (closed blue circles and curve) at multiple fixed volumetric ratios (10:0, 8:2, 6:4, 5:5, 4:6, 2:8, and 0:10) [16]. FICs for each drug were calculated and plotted. Synergy was defined as Σ FIC < 1, additivity as Σ FIC = 1, and antagonism as Σ FIC > 1. Abbreviations: ATV, atovaquone; DEC, decoquinate; FICs, fractional inhibitory concentrations; Qo, ubiquinol oxidation center; Qi, ubiquinone reduction site of cytochrome b.
DISCUSSION
Screening of chemically diverse DOS libraries coupled with advances in target identification technologies offer a powerful, integrated approach toward elucidating new pathways for drug development. Here we have presented a proof-of-concept chemogenomic approach identifying the quinolone reduction site of cytochrome bc1 as the molecular target for an antimalarial probe from a DOS library. Cytochrome b is a validated target in Apicomplexa parasites; ATV, one of the partner drugs in Malarone, inhibits the oxidation site of cytochrome b and demonstrates activity in both the erythrocytic and liver stages of the parasite lifecycle and Endochin-like quinolones have activity against both malaria and Toxoplasma gondii parasites and have been shown to target cytochrome bc1 [22, 27]. We demonstrate that the Qi site is also a druggable target in P. falciparum and lacks cross-resistance with Qo site inhibitors. This study, along with those of Vailleres et al [28] and Nilsen et al [29], offers new hope in utilizing Qi site inhibitors to thwart ATV resistance. We believe the lack of cross-resistance between Qo or Qi site mutants/inhibitors coupled with their effect in combination suggest that using targeted combination therapy toward 2 active sites in the same enzyme could represent a promising avenue for antimalarial development.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Nobutaka Kato of the Broad Therapeutics Platform and Leila Ross of the Harvard School of Public Health Department of Immunology and Infectious Diseases for valuable discussions. We also thank Justin Dick, Gilberto Ramirez, and Kate Fernandez for technical assistance and culture support.
Financial support. This work was supported by the National Institutes of Health (AI093716-01A1 to R. C. W.), the ExxonMobil Foundation (gift to D. F. W.), the National Institutes of Health–funded Molecular Libraries Probe Production Centers Network (1U54HG005032-1 to S. L. S.), and the Bill & Melinda Gates Foundation (OPP1053644 to D. F. W. and OPP1032518 to S. L. S.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Alonso PL, Brown G, Arevalo-Herrera M, et al. A research agenda to underpin malaria eradication. PLOS Med. 2011;8:e1000406. doi: 10.1371/journal.pmed.1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sibley CH, Ringwald P. A database of antimalarial drug resistance. Malar J. 2006;5:48. doi: 10.1186/1475-2875-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venture MfM. MMV Annual Report 2013. 2014.
- 5.Heidebrecht J, Richard W, Mulrooney C, Austin CP, et al. Diversity-oriented synthesis yields a novel lead for the treatment of malaria. ACS Med Chem Lett. 2012;3:112–7. doi: 10.1021/ml200244k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcaurelle LA, Comer E, Dandapani S, et al. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: discovery of macrocyclic histone deacetylase inhibitors. J Am Chem Soc. 2010;132:16962–76. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dandapani S, Marcaurelle LA. Grand challenge commentary: Accessing new chemical space for ‘undruggable’ targets. Nat Chem Biol. 2010;6:861–3. doi: 10.1038/nchembio.479. [DOI] [PubMed] [Google Scholar]
- 8.Schreiber SL. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science. 2000;287:1964–9. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 9.Comer E, Beaudoin JA, Kato N, et al. Diversity-oriented synthesis-facilitated medicinal chemistry: Toward the development of novel antimalarial agents. J Med Chem. 2014;57:8496–502. doi: 10.1021/jm500994n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong CK, Urgaonkar S, Cortese JF, et al. Identification and validation of tetracyclic benzothiazepines as Plasmodium falciparum Cytochrome bc1 Inhibitors. Chem Biol. 2011;18:1602–10. doi: 10.1016/j.chembiol.2011.09.016. Elsevier Ltd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–5. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 12.Johnson JD, Dennull RA, Gerena L, Lopez-Sanchez M, Roncal NE, Waters NC. Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrob Agents Chemother. 2007;51:1926–33. doi: 10.1128/AAC.01607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levenberg K. A method for the solution of certain non-linear problems in least squares. Q Appl Math. 1944;2:164–8. [Google Scholar]
- 14.Marquardt D. An algorithm for least-squares estimation of nonlinear parameters. SIAM J Appl Math. 1963;11:431–41. [Google Scholar]
- 15.Hohman M, Gregory K, Chibale K, Smith PJ, Ekins S, Bunin B. Novel web-based tools combining chemistry informatics, biology and social networks for drug discovery. Drug Discov Today. 2009;14:261–70. doi: 10.1016/j.drudis.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 16.Fivelman QL, Adagu IS, Warhurst DC. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother. 2004;48:4097–102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aurrecoechea C, Brestelli J, Brunk BP, et al. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37:D539–43. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nam T-G, McNamara CW, Bopp S, et al. A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem Biol. 2011;6:1214–22. doi: 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher N, Meunier B. Molecular basis of resistance to cytochrome bc1 inhibitors. FEMS Yeast Res. 2008;8:183–92. doi: 10.1111/j.1567-1364.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- 23.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 24.di Rago JP, Colson AM. Molecular basis for resistance to antimycin and diuron, Q-cycle inhibitors acting at the Qi site in the mitochondrial ubiquinol-cytochrome c reductase in Saccharomyces cerevisiae. J Biol Chem. 1988;263:12564–70. [PubMed] [Google Scholar]
- 25.Peters JM, Chen N, Gatton M, et al. Mutations in cytochrome b resulting in atovaquone resistance are associated with loss of fitness in Plasmodium falciparum. Antimicrob Agents Chemother. 2002;46:2435–41. doi: 10.1128/AAC.46.8.2435-2441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooley JW, Ohnishi T, Daldal F. Binding dynamics at the quinone reduction (Qi) site influence the equilibrium interactions of the iron sulfur protein and hydroquinone oxidation (Qo) site of the cytochrome bc1 complex. Biochemistry. 2005;44:10520–32. doi: 10.1021/bi050571+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doggett JS, Nilsen A, Forquer I, et al. Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc Natl Acad Sci USA. 2012;109:15936–41. doi: 10.1073/pnas.1208069109. National Acad Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vallieres C, Fisher N, Antoine T, et al. HDQ, a potent inhibitor of Plasmodium falciparum proliferation, binds to the quinone reduction site of the cytochrome bc1 complex. Antimicrob Agents Chemother. 2012;56:3739–47. doi: 10.1128/AAC.00486-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsen A, LaCrue AN, White KL, et al. Quinolone-3-diarylethers: A new class of antimalarial drug. Sci Transl Med. 2013;5 doi: 10.1126/scitranslmed.3005029. 177ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.