

Abstract
Despite more than three decades of intensive effort, no effective pharmacologic inhibitors of the Ras oncoproteins have reached the clinic, prompting the widely held perception that Ras proteins are “undruggable”. However, there is renewed hope that this is not the case. In this review, we summarize the progress and promise of five key directions. First, we focus on the prospects of direct inhibitors of Ras. Second, we revisit the issue of whether blocking Ras membrane association is a viable approach. Third, we assess the status of targeting Ras downstream effector signalling, arguably the most favourable current direction. Fourth, we address whether the search for synthetic lethal interactors of mutant RAS still holds promise. Finally, Ras-mediated changes in cell metabolism have recently been described. Can these changes be exploited for new therapeutic directions? We conclude with perspectives on how additional complexities, not yet fully understood, may impact each of these approaches.
Introduction
In 1982 mutationally activated RAS genes were detected in human cancers, marking the first discovery of mutated genes in this disease1. Subsequent intensive sequencing of the cancer genome has revealed that, despite the identification of over 500 validated cancer genes2 (COSMIC), the three RAS genes (HRAS, NRAS and KRAS) still comprise the most frequently mutated oncogene family in human cancers (TABLE 1; SUPPLEMENTARY TABLES 1 and 2). The frequent mutation of RAS in three of the top four cancer killers in the US (lung, colon, pancreatic) has spurred intense interest and effort in developing Ras inhibitors. However, despite more than three decades of effort by academia and industry, no effective anti-Ras inhibitors have reached the clinic, prompting a widely held perception that Ras oncoproteins are an “undruggable” cancer target. Although past failures dampened enthusiasm for anti-Ras drug discovery, mutated Ras proteins clearly merit continued attention. Given that the greatest success in signal transduction-based therapies has been achieved against mutationally activated targets, there is now renewed hope that recent advances in understanding Ras function, together with new approaches and technology, may finally have brought the holy grail of cancer research within reach3.
Table 1.
Frequency of RAS mutations in human cancers
| Cancer | % KRAS | % NRAS | % HRAS | % All RAS |
|---|---|---|---|---|
| Pancreatic ductal adenocarcinoma | 97.7 | 0.0 | 0.0 | 97.7 |
| Colorectal adenocarcinoma | 44.7 | 7.5 | 0.0 | 52.2 |
| Multiple myeloma | 22.8 | 19.9 | 0.0 | 42.6 |
| Lung adenocarcinoma | 30.9 | 0.9 | 0.3 | 32.2 |
| Skin cutaneous melanoma | 0.8 | 27.6 | 1.0 | 29.4 |
| Uterine corpus endometrioid carcinoma | 21.4 | 3.6 | 0.4 | 24.6 |
| Uterine carcinosarcoma | 12.3 | 1.8 | 0.0 | 14.0 |
| Thyroid carcinoma | 1.0 | 8.5 | 3.5 | 13.0 |
| Stomach adenocarcinoma | 11.4 | 0.9 | 0.0 | 12.3 |
| Acute myeloid leukaemia | 3.1 | 6.7 | 1.6 | 11.4 |
| Bladder urothelial carcinoma | 3.1 | 1.4 | 5.9 | 10.6 |
| Cervical adenocarcinoma | 8.3 | 0.0 | 0.0 | 8.3 |
| Head and neck squamous cell carcinoma | 0.5 | 0.3 | 4.7 | 5.5 |
| Gastric carcinoma | 4.0 | 1.0 | 0.0 | 5.0 |
| Esophageal adenocarcinoma | 4.1 | 0.0 | 0.7 | 4.8 |
| Chronic lymphocytic leukaemia | 1.9 | 2.5 | 0.0 | 4.4 |
| Lung squamous cell carcinoma | 2.2 | 0.0 | 2.2 | 4.4 |
| Small cell lung carcinoma | 1.4 | 0.0 | 1.4 | 2.8 |
| Renal papillary cell carcinoma | 1.8 | 0.0 | 0.0 | 1.8 |
| Adenoid cystic carcinoma | 0.0 | 0.0 | 1.7 | 1.7 |
| Medulloblastoma & pilocytic astrocytoma | 1.0 | 0.5 | 0.0 | 1.6 |
| Breast invasive carcinoma | 0.7 | 0.4 | 0.3 | 1.4 |
| Hepatocellular carcinoma | 0.8 | 0.4 | 0.0 | 1.4 |
| Cervical squamous cell carcinoma | 1.3 | 0.0 | 0.0 | 1.3 |
| Ovarian serous adenocarcinoma | 0.6 | 0.6 | 0.0 | 1.3 |
| Adrenocortical carcinoma | 1.1 | 0.0 | 0.0 | 1.1 |
| Neuroblastoma | 0.0 | 0.8 | 0.0 | 0.8 |
| Brain lower grade glioma | 0.5 | 0.5 | 0.0 | 0.7 |
| Prostate adenocarcinoma | 0.3 | 0.0 | 0.3 | 0.7 |
| Glioblastoma multiforme | 0.7 | 0.0 | 0.0 | 0.7 |
| Medulloblastoma | 0.0 | 0.4 | 0.0 | 0.4 |
| Kidney renal clear cell carcinoma | 0.2 | 0.0 | 0.2 | 0.4 |
| Esophageal adenocarcinoma | 0.0 | 0.0 | 0.0 | 0.0 |
| Osteosarcoma (paediatric) | 0.0 | 0.0 | 0.0 | 0.0 |
| Rhabdoid tumours | 0.0 | 0.0 | 0.0 | 0.0 |
| Sarcoma | 0.0 | 0.0 | 0.0 | 0.0 |
| Small intestine neuroendocrine tumours | 0.0 | 0.0 | 0.0 | 0.0 |
| T-cell prolymphocytic leukemia | 0.0 | 0.0 | 0.0 | 0.0 |
Compiled from references or databases referred to in Supplementary Table 1
In this review, we provide a critical assessment of past efforts and discuss the most promising directions for future success (FIG. 1). First, we begin with what was once thought impossible: direct inhibition of Ras. Second, we discuss the prospects for blocking Ras membrane association, a direction that had lost favour with the disappointing outcome of farnesyltransferase inhibitors4. Third, we provide a status report on inhibitors of Ras downstream effector signalling, in particular through Raf and phosphoinositide 3-kinase (PI3K). Fourth, we summarize the efforts in the search for synthetic lethal interactors with mutant RAS and discuss whether this direction might yield alternative targets. We next evaluate the prospect that Ras-mediated changes in cell metabolism can be exploited for drug discovery. We conclude with a discussion of unresolved issues that will likely add complexity and further challenges to anti-Ras drug discovery.
Figure 1. Approaches to discover and develop pharmacologic inhibitors of mutant Ras.

Past and ongoing approaches to inhibitors of mutationally activated Ras include Ras-binding small molecules that disrupt a key function(s) of Ras, inhibition of the CAAX motif-targeted enzymes that promote Ras membrane association, inhibitors of effector signalling function, unbiased interfering RNA, genetic or chemical screens for synthetic lethal interactors and inhibitors of metabolism.
RAS mutations and human cancer
RAS mutations are early genetic events in tumour progression. Numerous genetically engineered mouse models of RAS-driven cancers have demonstrated the potent cancer-inducing activity of mutant RAS. Consistent with the need for additional genetic alterations to cooperate with mutant RAS for full transforming activity, loss of tumour suppressor function (e.g., TP53, LKB1, APC) together with RAS activation results in enhanced tumour formation and progression5–7. Despite the early onset of RAS mutations, there is considerable experimental evidence that continued expression of mutant RAS is necessary for tumour maintenance. Suppression of RAS by RNA interference impaired the in vitro and in vivo growth of RAS-mutant human cancer cell lines8–10. Similarly, in mouse models driven by inducible mutant RAS, withdrawal of Ras expression leads to tumour regression11–15. Hence, there is little doubt that mutant Ras is a therapeutically useful drug target even in advanced metastatic tumours. Until recently, RAS-mutant cancers were discussed as if they comprised a homogeneous subset of all cancers. Instead, there is now an emerging understanding that not all mutant RAS genes are created equal. Both the frequency with which each RAS isoform is mutated and the specific mutations thereof vary strikingly in different cancer types (BOX 1), and these may need to be addressed differently. Thus, there may not be one single anti-Ras therapy that fits all RAS-mutant cancers.
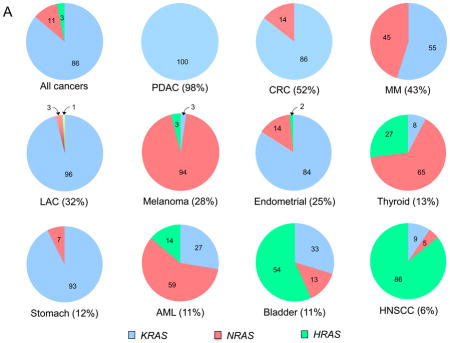
Box 1. RAS mutations in human cancer.
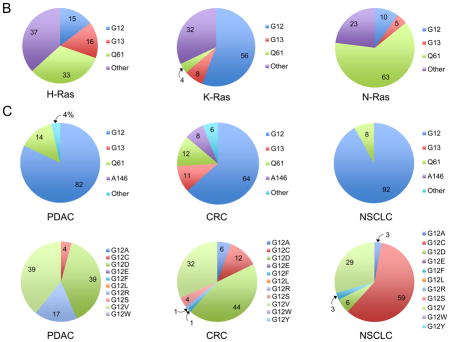
The frequency and distribution of RAS gene mutations are not uniform1,191. KRAS is the isoform most frequently mutated (86%), followed by NRAS (11%), and, infrequently, HRAS (3%) (COSMIC) (SUPPLEMENTARY TABLE 1). Overall, RAS mutations have been detected in 9–30% of all tumour samples sequenced (depending on the database utilized), with the specific RAS isoform generally differing according to cancer type. In pancreatic ductal adenocarcinoma (PDAC; ~90% of all pancreatic cancers) and lung adenocarcinoma (LAC; 30–35% of all lung cancers) there is a near 100% frequency of KRAS mutations. In colon and rectal carcinomas (CRC), KRAS is also the predominant mutated isoform (86%), whereas NRAS mutations are infrequent (14%) and HRAS mutations have not been detected. Conversely, KRAS and NRAS are seen at equivalent frequencies in multiple myeloma (MM), and NRAS is the predominant isoform mutated in cutaneous melanomas (94%) and acute myelogenous leukaemias (AML; 59%). Although rare overall, HRAS mutations are predominant in bladder (57%) and in head and neck squamous cell carcinomas (HNSCC; 86%).
Cancer-associated RAS genes are characterized by single base missense mutations, 99% of which are found at residues G12, G13 or Q61. There are also cancer-type differences in the relative frequency of mutations at these positions. In PDAC and NSCLC, KRAS mutations are found predominantly at G12. In CRC, G12 is also the predominant position (78%), but additionally there is a significant frequency of G13 mutations (20%), of mutations at A146, a position rarely mutated in other cancers, and, to a lesser frequency, at K117. There are also cancer-type differences in the substitutions seen at a given residue. For example, at G12, in PDAC and CRC the predominant substitution is G12D, followed by G12V. In contrast, in NSCLC, the major substitution is G12C, which is rare in PDAC.
The basics: in search of the Achilles’ heel of Ras
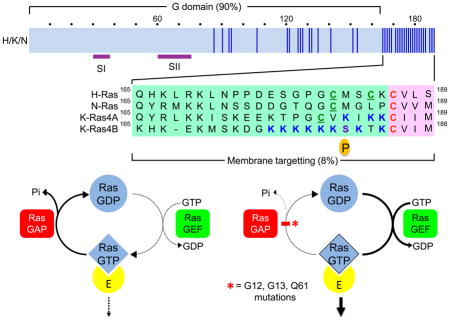
In order to develop antagonists of Ras, it is essential to understand the requirements for Ras function. The canonical property of Ras is that of a small GTPase which normally cycles between a GTP-bound active state and a GDP-bound inactive state, facilitated in part by GTPase activating protein (GAP) stimulation of GTP hydrolysis. However, when Ras proteins are mutationally activated, impaired GAP stimulation favours the formation of persistently GTP-bound Ras16 (BOX 2). This critical biochemical defect prompted the earliest efforts to target mutant Ras. By analogy to the ATP-competitive inhibitors that are effective antagonists of protein kinases, identification of GTP-competitive inhibitors of Ras has been attempted. However, whereas ATP binds protein kinases with low micromolar affinity, GTP binds Ras proteins with picomolar affinity, preventing discovery of effective inhibitors. Another early effort searched for small molecules that could act as GAPs for mutant Ras. To date, these efforts have not succeeded17.
Box 2. Regulation of the Ras GDP-GTP cycle.
The three RAS genes encode four 188–189 amino acid proteins that share 82–90% overall sequence identity (blue vertical line indicates amino nonidentity). KRAS encodes two splice variants that result from alternative exon 4 utilization, encoding divergent C-terminal sequences. Exons 4A and 4B encode 39 and 38 amino acids, respectively, with 19 identical and 4 conserved substitutions. K-Ras4A is most similar to the original retroviral K-Ras while K-Ras4B is the predominant isoform expressed in human cells. Residues in Switch I (SI; aa 30–38) and II (SII; aa 60–76) change in conformation during GDP-GTP cycling. Wild type Ras proteins bind GDP and GTP with low picomolar affinities. With cellular levels of GDP and GTP at millimolar concentrations, Ras is persistently nucleotide-bound. Ras proteins possess low intrinsic GTP hydrolysis and guanine nucleotide exchange activities, which are regulated and accelerated by GTPase activating proteins (RasGAPs) and guanine nucleotide exchange factors (RasGEFs), respectively. In resting cells, Ras is predominantly GDP-bound. Upon growth factor stimulation and activation of RasGEFs, nucleotide binding is destabilized and the nucleotide is released. Since the cellular concentrations of GTP are 10-fold that of GDP, RasGEFs promote transient formation of Ras-GTP, with RasGAPs returning the protein to the inactive GDP-bound state. Mutant Ras is impaired in intrinsic and GAP-stimulated GTP hydrolysis activity, favouring persistent formation of Ras-GTP.
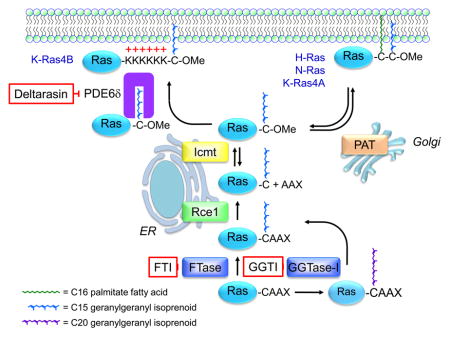
Another key requirement for Ras function is its association with the inner face of the plasma membrane18 (BOX 3), for which it is obligatory that Ras be modified by addition of a C15 farnesyl isoprenoid lipid to its C-terminal CAAX motif. The identification of farnesyltransferase (FTase) and the determination that the C-terminal CAAX tetrapeptide sequence alone was sufficient to inhibit FTase activity19 prompted an intensive effort to develop FTase inhibitors (FTIs)4. Many potent and selective FTIs were shown in preclinical mouse studies to effectively block the growth of Hras-driven tumours. These remarkable results led to clinical trials, with two FTIs (lonafarnib and tipifarnib) advancing to Phase III. However, these FTIs showed no anti-tumour activity in clinical trials focused on pancreatic and colon cancers, which are associated with mutations in KRAS or NRAS rather than HRAS (TABLE 1, BOX 1). One basis for this failure was predicted years earlier when it was found unexpectedly that, when FTase activity is blocked by FTI treatment, K-Ras4B and N-Ras proteins are no longer farnesylated but instead become substrates for the related prenyltransferase geranylgeranyltransferase-I (GGTase I) that catalyses addition of a C20 geranylgeranyl isoprenoid group20–22. Since this alternate lipid can also support their membrane association and oncogenic activities, K-Ras4B and N-Ras escape FTI-mediated inhibition of their function. The failure of this massive effort to develop FTIs as anti-Ras drugs created a stigma towards other ideas for blocking Ras membrane association. Worse, the failure of FTIs to act as anti-Ras drugs also prompted a surprisingly widely held misconception that Ras is not a useful therapeutic target. It did create large clues that conflating all the Ras isoforms together would be an unfortunate oversimplification.
Box 3. Posttranslational modifications that regulate Ras membrane association and subcellular localization.
Ras proteins are synthesized initially as cytosolic and inactive proteins. All four Ras proteins terminate in a C-terminal CAAX tetrapeptide motif comprised of an invariant cysteine residue to which the lipid is attached, followed by two typically aliphatic residues (AA), and the C-terminal residue (X) that contributes to prenyltransferase substrate specificity. The CAAX motif is necessary and sufficient to signal a series of posttranslational modifications that enhance the hydrophobicity of Ras, facilitating membrane association. The first step is catalysed by cytosolic farnesyltransferase (FTase)-mediated covalent addition of a C15 farnesyl isoprenoid to the cysteine of the CAAX motif, followed by Ras converting enzyme 1 (Rce1)-catalysed proteolytic removal of the AAX peptide, and finally isoprenylcysteine methyltransferase (ICMT)-catalysed carboxylmethylation of the now terminal farnesylated cysteine. FTase inhibitors (FTIs) prevent all three modifications. All Ras proteins are modified by FTase in cells. However, upon FTase inhibition by FTI treatment, K-Ras4B and N-Ras can be modified instead by addition of a C20 geranylgeranyl isoprenoid by the related enzyme geranylgeranyltransferase I (GGTase I). The CAAX-signalled modifications alone are not sufficient to promote Ras trafficking and association with the inner face of the plasma membrane. Ras proteins possess a second membrane targetting element comprised of either a polybasic amino acid stretch (K-Ras4B) or cysteine residues that undergo reversible acylation by the fatty acid palmitate.
The third key requirement for Ras function is the engagement of effector proteins that transmit signals downstream. The GTP-bound form of Ras is the active form by virtue of its increased affinity for effectors. Presently, at least 11 distinct classes of Ras effectors have been identified. The majority of these possess Ras-binding (RBD) or Ras-association (RA) domains23. Association with Ras-GTP promotes effector activation by promoting concentration at the plasma membrane, where additional events facilitate activation and/or enhance intrinsic catalytic activity. Genes encoding members of the two canonical Ras effector families, BRAF and PIK3CA, are also frequently mutationally activated in human cancers (20% and 12%, respectively; COSMIC) (SUPPLEMENTARY TABLE 1), supporting the importance of these pathways in driving cancer growth. Additionally, cell culture and mouse model studies have established driver roles for at least four other Ras effectors1. In light of the critical requirement for effector signalling in mutant Ras function, most current efforts to develop anti-Ras therapies have centered on pharmacologic inhibitors of effector signalling, as described below.
In the following sections, we summarize progress made in five key approaches to target Ras. We note that, since almost none of these approaches is selective for mutant Ras, additional future strategies that do not rely on selective Ras-dependency may be required to enhance the therapeutic index for cancer cells.
Direct inhibitors of Ras – druggable after all?
Direct targeting of Ras has been thought to be a very challenging task, if not downright impossible. Although some potential binding sites have been identified using computational approaches24,25, there do not appear to be any deep hydrophobic pockets on the surface of K-Ras that would allow tight binding of small molecules. Nevertheless, there have been several attempts to discover small molecules that bind directly to Ras26. This section describes these efforts.
Low-affinity inhibitors
One of the first compounds reported (SCH-53239, FIG. 2) was designed to compete with GDP for the nucleotide binding site of Ras27. Instead of binding in this fashion, a water-soluble analog (SCH-54292, FIG. 2) was found by NMR spectroscopy to bind to a hydrophobic pocket near the switch II (SII) region of Ras (BOX 2), with the hydroxylamine located near the Mg2+ cation on the basis of the intermolecular NOEs involving I100 and R68 of Ras27. Additional compounds were prepared in which the sugar of SCH-54292 was replaced with a bicyclic moiety based on molecular modeling (bicyclic analog, FIG. 2) that inhibited nucleotide exchange similarly to the originally described molecules within this series28. Two of these analogs also inhibited Ras-dependent cell growth. However, all of the compounds from this series contain a hydroxylamine, which appears to be critical for their activity but is not an ideal functional group in a drug molecule due to its toxicity and poor metabolic stability. Furthermore, these compounds are not very potent.
Figure 2. Compounds that have been reported to bind to Ras.

SCH-53239 was designed to inhibit guanine nucleotide exchange27. Structure-activity relationship studies led to the development of a derivative with greater water solubility, SCH-54292. Subsequently, another group used molecular modeling to design a series of sugar-derived bicyclic analogs28. Based on earlier observations that the NSAID sulindac showed anti-tumour activity in Hras-mutant rat mammary carcinomas224, the active metabolite sulindac sulphide was evaluated and found to bind to H-Ras29. IND12 is a sulindac derivative that blocks the growth of Ras-transformed cells31,32. MCP1 was identified in a yeast two-hybrid screen for inhibitors of H-Ras binding to full length Raf-134. Zn-cyclen selectively binds and stabilizes the weak effector binding affinity conformational state of Ras35. The HBS3 peptide is a mimic of the Sos1 αH helix that interacts with H-Ras36. DCAI and VU0460081 were identified in fragment-based library screens for K-Ras4B-binding molecules35,36. Kobe0065 was identified in a computer docking screen of a virtual compound library and selected for its ability to inhibit H-Ras-GTP binding to Raf-RBD40. Kobe2602 was identified in a subsequent computer-assisted similarity search of 160,000 compounds. A K-Ras G12C inhibitor was identified using a disulphide-fragment-based screening approach with GDP-bound K-Ras G12C. SML-8-73-1 covalently binds to K-Ras G12C and occupies the nucleotide binding site44. The Nucleotide exchange activator (compound 4) stimulates Ras-GTP formation, yet disrupts ERK and PI3K signalling45.
Another series of compounds that have been reported to noncovalently bind to Ras and to inhibit Ras/Raf complex formation was based on the non-steroidal anti-inflammatory drug, sulindac sulphide29 (FIG. 2). Sulindac analogs were synthesized, and the most potent was shown to bind to Ras at the Raf binding site when complexed with the nonhydrolysable GTP analog, GppNHp30. In addition, this compound inhibited the Ras-Raf interaction with an IC50 of 30 μM and decreased downstream phosphorylation of ERK (see FIG. 4 for pathway). The sulindac derivative IND12 selectively inhibited the proliferation of Ras-transformed cells31, induced the expression of E-cadherin, and increased the level of E-cadherin-bound β-catenin, thereby decreasing invasion32. However, none of the sulindac series of compounds are thus far potent enough to serve as useful drug molecules, and it is unclear whether the other off-target activities of sulindac and its derivatives would prove to be problematic.
Figure 4. Inhibitors of RAS effector signalling under clinical evaluation.

Compiled from clinicaltrials.gov. Ras proteins bind to the Ras-binding domain (RBD) of the p110 catalytic subunit of class I PI3Ks (α, γ, and δ). Unless indicated otherwise, PI3K inhibitors are pan-class I. Ras binds to the RBD of A-Raf, B-Raf and C-Raf. mTOR exists as two distinct complexes, mTORC1 (Raptor) and mTORC2 (Rictor). Rapamycin/sirolimus and its analogs (rapalogs: everolimus, ridaforolimus, and temsirolimus) are selective for mTORC1, forming a complex with mTOR and FKBP12. Second-generation mTOR inhibitors are ATP-competitive inhibitors of mTOR kinase activity.
In a yeast two-hybrid screen for other compounds that would inhibit the Ras-Raf interaction, small molecules (e.g., MCP1, FIG. 2, and later derivatives) were discovered that inhibited Raf activation in cancer cells and reversed Ras-transformed phenotypes33,34. Again, the compounds were not very potent. Structural information on how they bind has not yet been obtained but could be useful for improving their potency if it becomes available. For Zn2+ cyclen (FIG. 2), structural information has been reported on how it binds to Ras and blocks the Ras-Raf interaction35, but due to its lack of selectivity for binding proteins and chelating different metals, Zn2+ cyclen also does not represent a good starting point for the design of drug-like molecules. Nevertheless, this work revealed the existence of at least two distinct conformational states of Ras when bound to GTP analogs and demonstrated the possibility of stabilizing one of these conformations to inhibit effector interactions.
GEF inhibitors
The Ras GTP/GDP cycle is regulated negatively by GAPs and positively by GEFs, or guanine nucleotide exchange factors, that facilitate the dissociation of GDP and thereby promote binding of the more abundant GTP moiety (BOX 2). The most prominent RasGEF is Sos1, which has multiple binding sites for Ras. In addition to the small organic molecules that bind to Ras, a peptide based on the αH helix of the Sos1 RasGEF, the region that forms contacts with Ras (HBS3 peptide; FIG. 2), was found to bind directly to Ras in a cleft near the SI and SII regions36. The new peptide was designed to be more water soluble than the Sos αH peptide and to have improved helical character. To do this, the hydrogen bond surrogate approach was used, in which the N-terminal hydrogen bond between the carbonyl of the ith amino acid and the amine of the i+4th amino acid was replaced with a carbon-carbon bond. This peptide bound to GDP-bound Ras with a Kd of 158 μM, inhibited Ras activation, and regulated ERK-MAPK signalling in cells. Although the reported peptide was not very potent, further optimization of the helical peptide could lead to drug-like molecules, as was recently demonstrated for stapled helix peptides targeting challenging protein-protein interactions37.
In 2012, two groups reported on K-Ras4B binders that were discovered from fragment-based screening38,39. The group from Genentech identified a compound called DCAI (FIG. 2) which was shown to bind to a pocket located between the α2 helix and β sheet of K-Ras, as revealed by an X-ray structure of the co-complex (FIG. 3a). DCAI weakly inhibited Sos-mediated nucleotide exchange (IC50 = 340 μM) by blocking the interaction between Ras and Sos, and inhibited Ras activation in cells. Independently, the Vanderbilt group discovered several series of fragment hits in an NMR-based screen of 11,000 compounds that bind weakly to GDP-bound K-Ras. The X-ray structures of several of these hits and of their water soluble analogs bound to K-Ras indicated that they all bind to the same pocket: the same site where DCAI binds to the protein (FIG. 3b). This pocket is not readily observed in the ligand-free form of K-Ras. Instead, compound binding induces a conformational change in the protein in which Tyr71 moves out of the way, along with Met67 and α2 of the SII region, to create a primary binding pocket and a second nearby cleft39. From the structural information obtained from the X-ray data, additional compounds were designed and synthesized with improved affinity (e.g., VU0460081, Kd = 240 μM) compared to the initial fragment hits, but were still not very potent. Nevertheless, these compounds inhibited Sos-mediated nucleotide exchange by blocking Ras-Sos complex formation, as evidenced by a series of NMR experiments. Importantly, detailed structural information on how multiple small molecules bind to K-Ras were obtained from these studies. However, like many other previously reported compounds, the small molecules reported in these two papers bind only weakly to K-Ras, and the discovery of analogs with large improvements in affinity is likely to be a very challenging task. Also, even if this can be accomplished, whether compounds with such a mechanism of action would be advantageous in the setting of mutationally active Ras proteins is currently controversial.
Figure 3. Three-dimensional structures of Ras/ligand complexes.

Ras is represented as a molecular surface, the ligands are in stick models with yellow carbon atoms, and the nucleotide is in stick models with tan carbon atoms. The switch I region is in sky blue, and the switch II region is in purple. a | X-ray structure of a compound covalently linked to K-Ras G12D (PDB 4M22). GCP bound to DCAI (PDB 4DST)38. b | X-ray structure of K-Ras G12V. GDP bound to VU046000939. c | NMR-derived structure of K-Ras T35S. GNP bound to Kobe 2601 (PDB 2LWI)40. d | X-ray structure of K-Ras G12C. GDP bound to Shokat compound (PDB 4M22)41.
Inhibitors of Raf-1 binding
In another study, using a computer docking screen of a virtual library of compounds, several small molecules were selected for their ability to inhibit Ras-GTP binding to the Ras binding domain of Raf-140. One compound (Kobe 0065, FIG. 2) was identified that inhibited this interaction with a Ki = 46 μM, and another (Kobe 2602, FIG. 2) with a Ki = 149 μM was obtained from a subsequent similarity search. In cells, these compounds decreased the phosphorylation of downstream proteins in the Ras pathway, including MEK, ERK, and AKT, and inhibited the allosteric Ras binding site of Sos. In addition, the compounds inhibited colony formation of NIH 3T3 mouse fibroblasts and several different human cancer cell lines in soft agar, blocked the proliferation of H-Ras G12V-transformed NIH 3T3 cells, and inhibited tumour growth in a xenograft tumour model in nude mice. On the basis of NMR data, the compounds were postulated to bind to a site near the SII region of Ras in a similar but not identical pocket as the Genentech and Vanderbilt compounds (FIG. 3c). The Kobe compounds bind onto the surface on the side of the α2 helix. Although the compounds are not very potent, it was suggested that they may serve as a scaffold for the discovery of analogs with higher potency and selectivity. However, to be useful starting points for lead optimization, a suitable replacement for the toxic thiosemicarbazide scaffold would have to be found.
Mutant-specific inhibitors
Another interesting study recently described a very novel approach for targeting Ras41. Shokat and colleagues reported on small molecules that covalently bind selectively to the G12C mutant form of K-Ras, which is the most frequent RAS mutation found in non-small cell lung cancer (BOX 1). These compounds were discovered by screening a library of small molecules with GDP-bound K-Ras G12C using a tethering approach, followed by the design and synthesis of analogs guided by X-ray structures of co-complexes (FIG. 3d). The compounds were found to bind to a pocket between the α2 and α3 helices, on the other side of the SII region from the Genentech and Vanderbilt compounds, but at a site similar to where SCH-54292 was postulated to bind to K-Ras based on NMR data. As predicted, these compounds blocked Sos-mediated nucleotide exchange and decreased the binding of Ras to both B-Raf and C-Raf. Interestingly, they also appeared to selectively kill cancer cells harbouring the G12C mutation (K-Ras (G12C) inhibitor, FIG 2). Lung cancer aside, the G12C mutation is found in only a small subset of cancers compared to other K-Ras mutations (BOX 1), and it may be difficult to selectively target other commonly found mutations. However, this work demonstrates that it is possible to selectively target the G12C mutant form of Ras over the wild type protein. This could result in compounds with low toxicity, providing that small molecules could be discovered that do not bind to other cysteine-containing proteins. In other studies aimed at covalent attachment to cysteine-containing proteins, compounds lacking the electrophile that forms the irreversible bond typically bind very tightly to the protein42,43. For the K-Ras inhibitors in this study, however, fragments lacking the electrophile do not appear to bind to K-Ras G12C, suggesting that it might be difficult to achieve the needed selectivity.
Gray and co-workers have also targeted K-Ras G12C. They prepared a GDP analog with an attached electrophile (SML-8-73-1) that could covalently bind to the cysteine of the G12C mutant form of K-Ras44. Although small molecules targeting the guanine nucleotide binding site have been disregarded due to the picomolar affinity of GTP and GDP to K-Ras and their high intracellular concentration, SML-8-73-1 was shown to covalently bind K-Ras G12C even in the presence of 1 mM concentrations of GDP and GTP. In addition, a cell-permeable analog was shown to attenuate the downstream phosphorylation of AKT and ERK and to exhibit antiproliferative effects in several cell lines44.
Despite the many attempts to directly target K-Ras that have been published thus far, K-Ras still remains a very challenging target. Although some progress has been made in the identification of different compounds that weakly bind to Ras, the binding affinity of these early stage compounds would have to be markedly improved for them to be useful as drugs. In principle, this might be accomplished by linking these molecules together, providing that the linked compounds could possess drug-like properties. Alternatively, a different approach may be required, such as the targeting of a Ras/protein interface in which more suitable binding pockets may be present. In this regard, small molecules have been identified that bind to a hydrophobic pocket in the Ras:Sos1:Ras complex that is formed by the CDC25 homology (RasGEF) catalytic domain of Sos1 adjacent to the SII region of Ras45 (FIG. 2; Nucleotide exchange activator/compound 4). These compounds were shown to increase the rate of SOS-catalyzed nucleotide exchange and, as expected, increase the levels of Ras-GTP. However, paradoxically, they cause a biphasic response in the MAPK pathway with inhibition of ERK phosphorylation at high compound concentration, and they inhibit PI3K signalling as evidenced by a decrease in AKT phosphorylation. These compounds decrease cell proliferation and anchorage-independent growth, and suggest another possible approach to target Ras signalling for the treatment of Ras-driven tumours.
Inhibitors of membrane association/subcellular localization – back to the future?
Farnesyl transferase inhibitors
Although FTIs are not effective against the Ras isoforms most commonly mutated in human cancers, there is continued discussion as to whether FTIs can still be an effective therapeutic approach for HRAS-mutant cancers46 such as thyroid, bladder, head and neck, and skin cancers (TABLE 1 and BOX 1). However, even in these cancers, the HRAS mutation frequency is low and the driver function of HRAS and hence the potential therapeutic value of anti-HRAS drugs remains to be vigorously evaluated. Since the future of cancer treatment is personalized medicine and full exome sequencing, if HRAS is ultimately validated as a cancer driver, then a rigorous assessment of whether HRAS-mutant cancers are responsive to FTIs should be done.
The disappointing clinical outcome of FTIs in KRAS-mutant cancers extinguished interest in the further pursuit of anti-Ras strategies targeting Ras membrane association4. Thus, while observations from genetic studies in a KrasG12D-driven lung cancer mouse model suggested that combination treatment strategies to concurrently block both FTase and GGTase-I can be effective and might not be overtly deleterious47, there remain considerable caution and lack of interest in this approach. This lack of interest is based in part on the perception that concurrent inhibition of both enzymes, that collectively may have up to 300 substrates4, will be limited by serious toxicity in normal tissues overall, a point not addressed by the genetic studies cited above.
Another therapeutic approach that targets the farnesyl modification is salirasib/S-farnesylthiosalicylic acid (FTS), which acts as a mimic of the C-terminal S-farnesyl cysteine modification of Ras. The specific consequences of salirasib treatment on Ras function are varied and include decreased plasma membrane association, decreased Ras-GTP levels and protein stability, and decreased effector signalling48–50. Salirasib can also impair the activities of other farnesylated proteins (e.g., Rheb)51 and mTOR, through a mechanism that is not clearly understood52,53. Based on positive preclinical results in pancreatic cancer mouse models54,55, a clinical trial was performed in pancreatic cancer patients56. Although tantalizingly there were some longterm survivors, the main conclusion that could be reached definitively was that the treatment was well-tolerated. Unfortunately, the patient numbers were too small to determine if a significant clinical benefit was gained and if K-Ras function was effectively perturbed. Further clinical evaluation is merited, but better understanding of the key mechanism(s) of FTS action and of accurate biomarkers to monitor target inhibition are also needed.
Inhibitors of Rce1 and ICMT
While there has been some interest in developing inhibitors of the two other CAAX-signalled Ras processing enzymes, Rce1 and ICMT (BOX 3), concern that they are likely also required for the function of other proteins with critical roles in normal cell physiology has limited enthusiasm for discovery efforts. For example, the 19 ICMT substrates in the CXC-terminating Rab family of small GTPases are critical for regulating vesicular transport. Thus, pharmacologic inhibition of either Rce1 or ICMT activity will almost certainly impact the functions of many proteins beyond Ras, and consequently normal tissue toxicity may well be a limitation for these two targets.
In addition, although earlier studies in Rce1- or Icmt-deficient mouse embryo fibroblasts (MEFs) revealed impairment in H-Ras-mediated transforming activity, subsequent studies found more complex consequences of the loss of these enzymes. For example, Rce1 ablation concurrent with KrasG12D activation in mouse hematopoietic cells enhanced myeloproliferative disease57. In contrast, Icmt ablation in the same mouse model reduced myeloproliferative disease58. However, Icmt ablation concurrent with KrasG12D activation in the pancreas exhibited increased PanIN formation and accelerated neoplastic progression59. One logical explanation for these results is that both Rce1 and ICMT may possess up to 300 CAAX-terminating substrates in addition to Ras, and that these substrates may be differentially sensitive to enzyme inhibition, depending on context. Furthermore, while cell-active small molecules have been identified and characterized60–62, potent and selective pharmacologic inhibitors of Rce1 and ICMT suitable for in vivo analyses have yet to be identified.
Inhibiting palmitoylation/depalmitoylation
Another lipid modification that has attracted some interest is palmitoylation. Modification by the fatty acid palmitate is essential for the plasma membrane association and function of H-Ras and N-Ras, and likely K-Ras4A. These Ras isoforms are reversibly and covalently modified by one or two palmitoyl fatty acids on cysteines immediately upstream of the CAAX motif. Studies with nonpalmitoylated variants of H-Ras and N-Ras support the importance of this modification in Ras-driven oncogenesis63,64. One human palmitoyl acyltransferase (DHHC9/GCP16) has been described as having activity towards H-Ras and N-Ras65. However, since K-Ras4B is not palmitoylated, this approach will not be useful for the majority (85%) of RAS-mutant cancers. Another approach is suggested by the dynamic acylation/reacylation cycle of Ras, wherein depalmitoylation, perhaps counterinteruitively, promotes Ras redistribution to membrane sites where it is active66–68. Thus, depalmitoylation inhibitors have been proposed as a means of inhibiting Ras membrane association and function69. Given that there are currently 24 known mammalian palmitoyl acyltransferases70, it is unclear whether DHHC9/GCP16 is the only member with activity towards Ras proteins, how selective it is for Ras, and whether DHHC9/GCP16-selective inhibitors can be developed. It has also not yet been shown definitively whether the observed cellular consequences of depalmitoylation inhibitors are due to inhibiting Ras depalmitoylation. These issues, together with the potential for off-target activities of these inhibitors on non-Ras substrates, are concerns that currently diminish enthusiasm for this direction.
Inhibitors of other posttranslational modifications
Other posttranslational modifications that can regulate Ras subcellular localization and/or effector interactions have also attracted interest as possible directions for anti-Ras drug discovery. In particular, a classical protein kinase C, PKCα, catalyses phosphorylation of K-Ras4B at S181 within the C-terminal polybasic sequence. This phosphorylation causes Ras to dissociate from the plasma membrane and move to endomembranes71, where it interacts with inositol triphosphate receptors (InsP3) on the endoplasmic reticulum. This converts K-Ras4B from a growth-promoting to growth-suppressing protein, in a Bcl-xL-dependent fashion72. The InsP3 interaction blocks the ability of Bcl-xL to potentiate the InsP3-regulated flux of calcium from ER to mitochondria, which is required for efficient cell survival. Whether this process can be exploited for anti-cancer therapy by promoting K-Ras4B phosphorylation on this residue is an intriguing but unrealized possibility. The goal is supported by findings that the PKC agonist bryostatin-1 can cause K-Ras4B phosphorylation and translocation to endomembranes where it drives growth suppression, and that mouse tumour xenograft growth was impaired by bryostatin-1 treatment in an S181-dependent manner71. Bryostatins have been shown to be well-tolerated in vivo and anti-tumour activity has been described73. One obvious concern is that PKCs have numerous substrates and therefore pharmacologic approaches to activate PKC will very likely have significant consequences unrelated to Ras phosphorylation. Surprisingly, in contrast to these studies, fundamentally similar analyses by another group reached the opposite conclusions, proposing instead that S181 phosphorylation is required for K-Ras4B-mediated cancer growth74. A possible basis for these opposing conclusions has not been addressed.
Another intriguing target is endothelial nitric oxide synthase (eNOS)-catalysed nitrosylation. This posttranslational modification at Cys118 causes activation of wild type Ras proteins, and may promote activities required for cancer growth driven by mutant K-Ras75. Evidence supporting eNOS as a possible therapeutic target comes from mouse model studies in which genetic ablation of eNOS in the KrasG12D/Tp53 (KPC) mouse model of pancreatic cancer prolonged survival76. Treatment with the eNOS-preferential small molecule inhibitor N(G)-nitro-L-arginine methyl ester (L-NAME) also impaired the growth of human pancreatic cell line xenografts and somewhat increased survival in KPC mice76.
A different approach to impairing Ras membrane association was suggested by the discovery of proteins that selectively recognize the farnesylated forms of Ras. Of these, phosphodiesterase 6 delta (PDE6δ) has attracted particular interest. PDE6δ, which plays a role in photoreceptor signalling, interacts with and regulates the trafficking of the PDE6 complex, which is comprised of both farnesylated and geranylgeranylated subunits77. PDE6δ can also recognize K-Ras4B and other prenylated proteins and facilitate their trafficking to membrane compartments78. Palmitoylation of H-Ras, N-Ras and K-Ras4A prevents their recognition by PDE6δ78. Recently, a small molecule that inhibits the binding of PDE6δ to K-Ras4B was shown to impair accumulation of K-Ras4B on the plasma membrane and reduce the growth of KRAS-mutant tumour cells79. While these findings are intriguing, discrepancies regarding the degree of K-Ras4B dependency on PDE6δ to drive cancer growth and whether PDE6δ function is critical for CAAX-terminating proteins modified by geranylgeranylation will need to be resolved in order to determine the potential therapeutic value of anti-PDE6δ inhibitors80.
Finally, additional posttranslational modifications of Ras that modulate Ras function include mono-ubiquitination of H-Ras81 and of K-Ras82, polyubiquitination of K-Ras83 and acetylation of K-Ras4B84. However, the enzymology of these modifications, the significance of their low stoichiometry, and their potential value as therapeutic targets remain unresolved.
Inhibitors of Ras effector signalling – currently our best bet?
There are at least 11 Ras effector families identified to date, with mutation data and functional studies validating a contribution of at least six of them to Ras-dependent cancer initiation and/or maintenance23,85. In turn, with evidence for a key driver role in Ras-mediated oncogenesis86,87 the Raf serine/threonine kinases (A-Raf, B-Raf and C-Raf/Raf-1) are arguably the most important effectors of mutant RAS-dependent cancer growth. The frequent mutational activation of BRAF (SUPPLEMENTARY TABLE 1, and its infrequent co-occurrence with RAS mutations, also provides compelling evidence for a driver role for Raf in RAS-mutant cancers.
Raf-MEK-ERK inhibitors
The only well-validated substrates of Raf are the highly related MEK1 and MEK2 dual specificity protein kinases (80% identity), and the only well-validated MEK1/2 substrates are the highly related ERK1 and ERK2 serine/threonine kinases (86% identity). These findings gave rise to the early perception that this protein kinase cascade is a simple linear and unidirectional signalling pathway. However, it is now well-established that the Raf-MEK-ERK cascade is the core of a complex signalling network with multiple inputs and outputs, with feed-forward and feedback mechanisms, and with multiple scaffold proteins that dynamically regulate signalling and ERK activity88. The more than 200 substrates that have been defined for ERK1/2 alone88 add further complexity to this effector pathway. Perhaps not surprisingly then, the earlier assumptions that pharmacologic inhibition of ERK activation at the level of either Raf or MEK would be equivalent, and would be effective to block ERK activation driven by mutant Ras, have proven incorrect.
Presently, at least 11 pharmacologic inhibitors of Raf kinases have reached clinical evaluation, with four approved for use by the US Food and Drug Administration (FDA) (FIG. 4). Although sorafenib was developed originally as a Raf inhibitor89, the anti-tumour efficacy of this multikinase inhibitor is more likely based on its ability to block tyrosine kinases involved in tumour angiogenesis, such as members of the vascular endothelial growth factor receptor (VEGFR) family90. Two ATP-competitive Raf inhibitors, vemurafenib and dabrafenib, are approved for use in BRAF-mutant metastatic melanoma91. When these Raf inhibitors were evaluated in RAS-mutant cancer cells, paradoxically, activation rather than inactivation of ERK was seen92–94, as was also documented years earlier with a preclinical agent95. Raf dimerization, mediated by active Ras, causes activation of Raf, and these first generation Raf inhibitors promote Raf dimerization96,97. This accounts for the Raf inhibitor-induced development of benign skin tumours including keratoacanthomas and of squamous cell carcinomas of the skin when mutant RAS is present. Both second generation Raf inhibitors that exhibit pan-Raf activities and/or do not promote Raf dimerization, as well as inhibitors designed to block dimerization, are now under consideration; these may prove more efficacious in the long run98.
At least 15 MEK inhibitors have reached clinical evaluation (FIG. 4), with trametinib (GSK112021) recently approved by the US FDA for BRAF-mutant metastatic melanoma. Generally, the majority of these inhibitors acts as allosteric non-ATP competitive inhibitors of MEK1 and MEK2 and consequently are highly selective. Although MEK inhibitors have been effective against BRAF-mutant melanoma, they have been only partially effective in RAS-mutant cancers and human tumour cell lines 99 and in mutant RAS-driven mouse models of cancer100. Interestingly, the activation state of MEK and the mechanisms by which it is activated in RAS- versus BRAF-mutant cancers are distinct, and one recent study suggested that this in turn influences the response to specific MEK inhibitors101. For example, MEK inhibitors that act similarly to GDC-0623 and G-573 are likely to be more effective in RAS-mutant cancers, because they form a strong hydrogen bond with MEK S212 and block MEK feedback phosphorylation by Raf, whereas those that act similarly to cobimetinib (GDC-0973) are better at inhibiting active, phosphorylated MEK101. The dual Raf/MEK inhibitor RO5126766 can act in a similar manner102. Innate or acquired resistance of RAS-mutant cancer cells to MEK inhibitors can occur by upregulation of receptor tyrosine kinase activity103 or by amplification of upstream activators104 that enhance flux through the pathway to elevate ERK activity above the >80% suppression threshold needed to achieve a therapeutic response105.
Since innate/de novo or acquired mechanisms of resistance to Raf or MEK inhibitors are often due to reactivation of ERK, one obvious approach to address this limitation is to use an ERK inhibitor. Currently, three ERK inhibitors have entered clinical evaluation (FIG. 4) and an analog of one (MK-8353/SCH9000353) has characterized in preclinical models106. However, like MEK inhibitors, ERK inhibitors also block ERK feedback phosphorylation and inactivation of Raf, leading to enhanced MEK activation. Combination inhibition of the Raf-MEK-ERK cascade at multiple nodes may provide more effective inhibition while reducing toxicity.
PI3K-AKT-mTOR inhibitors
The second best validated class of Ras effectors is comprised of the p110 catalytic subunits (α, γ and δ) of class I phosphoinositide 3-kinases (PI3Ks). The frequent mutational activation of PIK3CA (encoding p110α; 12%, COSMIC) and inactivation of the PTEN tumour suppressor support a driver role for PI3K in Ras-dependent cancer growth. Since PI3K activity can also be modulated by non-Ras mechanisms, Downward and colleagues applied an innovative approach to assess the importance of Ras-dependent PI3K activation alone. They generated mice harboring a mutant germline Pik3ca allele encoding a p110α variant with point mutations in the Ras-binding domain that prevent Ras-mediated activation but retain non-Ras-dependent activities107,108. Near-complete ablation of tumour initiation and partial regression of tumour growth was observed in a Kras-driven mouse model of lung cancer, indicating that K-Ras requires p110α in this setting. A similar partial reduction in tumour growth was seen upon pharmacologic inhibition of p110α. The importance of continued PI3K activity in tumour maintenance was also demonstrated in KRAS-mutant human tumour cells, where activated p110α was able to substitute for the loss of mutant KRAS to maintain tumour growth9. While the results from mouse model analyses provide a compelling argument that PI3K inhibition may be an effective anti-Ras strategy in some settings, there is also evidence that PI3K is not always a key Ras effector. Pharmacologic inhibition of this pathway in Kras-driven mouse lung cancers did not effectively block tumour growth100. Furthermore, KRAS silencing in KRAS-mutant colorectal cancer cell lines did not reduce activation of the PI3K effector AKT, although it effectively reduced ERK activation109; instead, increased receptor tyrosine kinase signalling accounted for the maintenance of AKT activation109. Finally, in contrast to the nonoverlapping BRAF and RAS mutations seen in human cancers, PIK3CA and RAS mutations often are seen together. Although correlative, this pattern suggests that mutated Ras alone may not potently activate PI3K signalling. Nevertheless, targeting PI3K-AKT signaling in RAS-mutant cancers, particularly in combination with other pathways, may have therapeutic value.
Currently, at least 53 inhibitors of PI3K-AKT-mTOR signaling are under clinical evaluation (FIG. 4). As monotherapy, these have shown disappointing activity against RAS-mutant cancers in preclinical and clinical evaluation. However, in mouse models, potent synergistic activity with inhibitors of ERK-MAPK signalling has been seen100. Currently, there are numerous clinical trials evaluating combined inhibition of Raf and PI3K effector signalling110.
Ral inhibitors
RalGEFs comprise the third best validated effectors of Ras-driven cancer growth111,112. Mice deficient in RalGDS, one of the four RA domain-containing RalGEFs, were viable but displayed reduced carcinogen-induced mutant Hras-driven skin tumour formation113. Another RalGEF, Rgl2, was found overexpressed in KRAS-mutant colorectal cancer cell lines, and shRNA-mediated suppression of Rgl2 reduced Ral-GTP levels and impaired cell growth in vitro114. RalGEFs are activators of two related isoforms, RalA and RalB, and shRNA-mediated silencing of RalA and/or RalB in KRAS-mutant pancreatic cancer cell lines impaired growth. Interestingly, in the majority of cell culture studies, RalA and RalB exhibit very distinct roles in cancer cell behaviour. For example, RalA but not RalB was found to be necessary for the tumourigenic growth of pancreatic cancer cell lines, whereas RalB was important for invasion in vitro and experimental metastasis in vivo115. In contrast, in conditional tissue-specific genetic ablation studies, loss of both RalA and RalB was required to reduce Ras-driven lung or skin tumour growth116. These findings may reflect different roles for Ral GTPases in tumour initiation and progression versus maintenance, or cancer-type differences.
Like Ras, the Ral GTPases are not considered tractable drug targets. Therefore, current directions for therapeutic inhibition of Ral involve protein kinases that either modulate Ral subcellular localization and effector utilization or that act as downstream effectors. Analogous to PKCα phosphorylation of K-Ras4B, Aurora A and PKCα phosphorylate C-terminal serine residues in RalA and RalB, respectively, which promotes their driver functions in cancer growth117–120. RalB effector signalling can lead to activation of the TBK1 serine/threonine kinase or the mTORC1 serine/threonine protein kinase complex121,122. Thus, protein kinase inhibitors may provide promising directions for blocking aberrant Ral GTPase signalling downstream of Ras.
Rac1 inhibitors
The fourth best validated effector of Ras-driven cancer growth is the Rac1 small GTPase123. Ras can activate RacGEFs and subsequently Rac1 through direct effector binding (e.g., Tiam1)124 or indirectly through PI3K activation (e.g., P-Rex)125. The importance of Rac1 as a critical driver of cancer growth is supported by the recent identification of activated RAC1 mutations in cutaneous melanomas126,127. That inhibition of Rac1 may have a therapeutic benefit in RAS-mutant cancers is supported by mouse model studies in which genetic ablation of Rac1 function impaired initiation of mutant KRAS-driven lung or pancreatic cancers128,129. However, like Ras, Rac1 is a small GTPase, and therefore whether direct Rac1 inhibitors can be developed is still unclear. One small molecule inhibitor of Rac1 activation by RacGEFs has been described130,131, suggesting that it may be feasible to develop inhibitors of Rac1 activation. Alternatively, analogous to current approaches for Ras, inhibitors of Rac1 effector signalling may be more tractable. However, of the many Rac1 effectors identified, which of them are essential drivers of Rac1-dependent growth remains unanswered. The PAK1 serine/threonine kinase, found overexpressed in many cancers, is one possibility. Genetic ablation of Pak1 suppressed Kras-induced lung tumour formation in mice, and pharmacologic inhibition of PAK1 phenocopied loss of Pak1 function132. In addition, the p110β isoform of PI3K was recently found to serve as an effector for Rac1 rather than for Ras133, providing another candidate effector of Rac1-dependent cancer growth.
While other Ras effectors (e.g., phospholipase C ε, RASSF1) have demonstrated roles in cancer cell proliferation, these may not act as drivers of Ras-dependent cancer growth and hence their therapeutic value is presently unclear134.
In summary, while effector inhibition appears to be the most promising anti-Ras strategy to date, significant challenges remain. First, inhibition of any one effector pathway is complicated by compensatory mechanisms, necessitating inhibition at multiple points. Second, because multiple effectors have driver roles, concurrent inhibition of multiple pathways will be needed. This has prompted both preclinical and clinical evaluation of combining inhibitors of components of the Raf and/or PI3K effector networks. However, as the combinations are expanded to achieve more effective blockage of effector signalling, the offsetting increase in normal cell toxicity may tip the balance and cause loss of the therapeutic window. How to properly strike this balance is another provocative question for the future.
Synthetic lethal interactors of mutant RAS – needles in the haystack?
One possible approach to increasing therapeutic selectivity for cancer cells would be to identify targets that have synthetic lethal interactions with the RAS oncogene; that is, genes whose loss of function would be lethal only in the presence of mutated RAS (FIG. 5). Widespread observations of synthetic lethality in lower organisms suggested that genetic buffering, where one or more genes can functionally compensate for mutations in another gene, may be common135, and that synthetic lethal interactions might be exploitable for cancer treatment136. This approach could be particularly useful in situations where pharmacological action cannot be taken against the oncogenic mutations themselves, as in the case of RAS mutations.
Figure 5. Synthetic lethal interactors in RAS-mutant cancers.

Functional screens for mutant RAS synthetic lethal interactors utilize chemically synthesized siRNA libraries or viral vector-based shRNA libraries to identify genes whose knockdown causes selective impairment of the growth of RAS mutant but not RAS WT cell lines. The libraries may be either genome-wide or target a selected set of genes. The library may be delivered well-by-well, as shown in Panel A, or as pooled viruses. RAS synthetic lethal partners may operate in different pathways to support the viability of RAS mutant cells (Panel B). These include co-operating signalling and transcriptional programs, maintenance of genomic stability, and suppression of oncogenic stress.
The existence of oncogene-specific synthetic lethal interactions is supported by the fact that oncogenic mutations lead to prominent phenotypic changes in cancer cells, collectively known as the ‘hallmarks of cancer’137–139, where normal cellular pathways are co-opted, either directly or indirectly, to support the malignant growth of cancer cells. This often manifests in one of two ways. First, oncogenic transformation leads to elevated cellular stress (oncogenic stress), which requires the cancer cell to activate cellular stress-relief pathways for survival. Second, cancer cells often adapt their signalling pathways and alter their metabolic flux to support proliferation, and this requires compensatory changes in pathways not directly downstream of the driver oncogene. Collectively these “non-oncogene addictions” constitute potential synthetic lethal interactions (FIG. 5) that may be exploitable for therapeutic intervention139. The potent effect of PARP inhibitors in BRCA-mutant cancers is an example of exploiting synthetic lethality; BRCA1/2 mutations result in deficiencies in DNA homologous recombination repair and render the cell particularly dependent on PARP-mediated non-homologous end-joining DNA repair for genomic stability.
Screen results
A number of studies have applied RNAi screens in human cancer cell lines to identify synthetic lethal interactors with the KRAS oncogene (reviewed in Ref. 140). Screens done with a variety of siRNA and shRNA libraries, whether in a well-by-well or a pooled format, have confirmed that many KRAS-mutant cell lines are indeed functionally addicted to the KRAS oncogene. These screens have identified a wide array of synthetic lethal interactions with mutant KRAS (TABLE 2) that encode proteins that can be thematically categorized into several cellular processes: cell cycle and mitosis (Survivin/BIRC5, TPX2, PLK1, APC/C), cell survival (WT1, Bcl-XL1), collaborative transcriptional programs (GATA2, SNAIL2), and parallel growth and survival signals (TBK1, TAK1) (FIG. 5). Whereas the roles of mutant KRAS in cell proliferation, genomic instability and apoptosis have been described previously141, the findings of KRAS dependency on kinases such as TBK1129 and transcription factors such as GATA2142 indicate that additional cellular pathways are involved in supporting the viability of KRAS-transformed cells. Thus far none of the synthetic lethal interactors has been found to be equal or superior to KRAS itself at discriminating KRAS-mutant and -WT cells.
Table 2.
KRAS synthetic lethal genes
| Library | Primary cell line screen – assay and format | Synthetic lethal genes | Refs |
|---|---|---|---|
| siRNA library targeting ~4,000 genes (protein kinases, G protein-coupled receptors, ubiquitin E3 ligases, transporters, ion channels and peptidases) | Isogenic KRAS G13D/WT human DLD-1 CRC cell lines – cell death (well-by-well) | BIRC5a, CDC2/ CDK1, C20ORF18/RBCK1 | 225 |
| siRNA library targeting ~3,700 genes (protein kinases, G protein-coupled receptors, ubiquitin E3 ligases, transporters, ion channels, peptidases, phosphatases, dehydrogenases, nuclear receptors, lipid-modifying enzymes, integrins, chemokines and receptors) | Human NSCLC NCI-H1299 cell line (NRAS Q61K) – cell death (well-by-well) | RAN, TPX2, SCD1 | 226 |
| Lentiviral shRNA library (5,024) targeting 1,011 genes (encoding kinases, phosphatases and cancer-related genes) | Panel of KRAS mutant (4) or WT (4) human AML, breast, CRC, glioblastoma and prostate tumour cell lines, and two nontransformed human lines – NHF and hTERT-HMEC – proliferation (well-by-well) | STK33, AKT3, CPNE1, CAMK1, MLKL, FLT3LG, DGKZ | 227 |
| Retroviral shRNA library (74,905) targeting 32,293 transcripts) | Isogenic KRAS G13D/WT DLD-1 CRC cell lines – proliferation (Pooled) | PLK1, APC/C, THOC1, Ubc9 | 143 |
| Lentiviral shRNA library targeting 957 genes (including kinases, phosphatases and oncogenes) | Panel of KRAS mutant (7) and WT (10) human breast, CRC, glioblastoma, myeloma, NSCLC, prostate, renal tumour lines, and two nontransformed human cell lines - NHF, hTERT-HMEC – proliferation (well-by- well) | TBK1, PSKH2, PSMD14, PTCH2, CPNE1, MAPKP3K8 | 144 |
| Lentiviral shRNA library (631) targeting 162 genes, including part of a Kras gene expression signature (n = 89), transcriptional regulators (n = 35) K-Ras effectors (n = 47), genes differentially expressed in human NSCLC samples (n = 20) or cell lines (n = 23)] | LKR10 and LKR13 Kras/Tp53 mutant mouse lung tumour-derived cell lines - proliferation in vitro and subcutaneous tumour growth in vivo in immunodeficient mice (pooled) | WT1 | 228 |
| Retroviral shRNA library targeting 2,500 genes (protein kinases, cancer-related genes) | Isogenic KRAS G13D/WT HCT-116 CRC cell lines – proliferation (pooled) | SNAI2b | 229 |
| siRNA library targeting 7,000 genes of the druggable genome | Isogenic KRAS G13D/WT HCT-116 CRC cell lines – cell death (well-by-well) | CDC6, GATA2 | 145, 161 |
| Lentivirus shRNA library targeting 17 protein kinases encoded by KRAS-dependent genes | KRAS-dependent (SW620) and -independent (SW837) human CRC cell lines (both KRAS mutant) – proliferation (well-by-well) | MAP3K7 c | 230 |
| Lentivirus shRNA library targeting 1,200 druggable genes, such as kinases and regulators of cell proliferation and survival | KRAS mutant CRC cell lines with different sensitivities to MEK/PI3K inhibition: HCT116 (sensitive) and SW620 (insensitive) – synergistic cell death with MEK inhibitor selumetinib treatment (pooled) | BCL2L1d | 165 |
| Lentiviral shRNA library targeting ~16,000 genes | 72 RAS mutant or WT human breast, pancreatic, and ovarian tumour cell lines (pooled) | ARHGEF2e | 152,231 |
AML, acute myelogenous leukaemia; CRC; colorectal carcinoma; HMEC, immortalized human mammary epithelial cells; NHF, normal human fibroblasts; NSCLC, non-small cell lung carcinoma;
Encodes Survivin;
Encodes Snail2;
Encodes TAK1;
Encodes Bcl-xL;
Encodes GEF-H1
In general the overlap among the hits from different KRAS synthetic lethal screens has been small, although proteasome subunits have been identified in three screens143–145. The lack of significant overlap can be attributed to several factors. The patterns of non-oncogene addictions in the presence of the common KRAS driver oncogene – and therefore the synthetic lethal interactions identified – may be context-dependent. Cancer cells harbour multiple oncogenic mutations2, and secondary mutations could alter the patterns of genetic dependency146,147. In addition, tumours arising from different organs display distinct lineage phenotypes despite harbouring similar oncogenic mutations148; thus, KRAS mutations could drive genetic dependencies in a tissue-specific fashion. All of these factors could cooperate with KRAS in determining the landscape of non-oncogene addictions in cancer cells. If the landscape is indeed this fragmented, “universal” KRAS synthetic lethal interactors might be difficult to identify.
Improving the screen
Although the first generation of KRAS synthetic lethal screens has revealed interesting biological information about KRAS-mutant cancers, the findings have suffered from a number of technical limitations that have reduced confidence in the applicability of their results going forward. The most prominent of these limitations is the lack of library validation, which could contribute to high false-negative rates in the screen. In addition, library penetrance (i.e., the number of siRNA/shRNAs that effectively knock down a given gene) is highly variable among the different libraries, which could also lead to high and variable false-negative rates. A new generation of libraries with multiple validated si/shRNAs per gene would significantly improve the penetrance of the screen and would also make it easier to rule out off-target effects, thereby decreasing the false-positive rate. Furthermore, a validated library is likely to be more widely adopted by the research community, thus making cross-study comparisons easier. Finally, due to cost and feasibility issues, first-generation synthetic lethal screens were typically carried out using either KRAS-isogenic cell lines or a small panel of KRAS-mutant and -WT cell lines. While isogenic cell lines can useful for isolating the effect of individual oncogenes, care should be taken to recognize their limitations. For example, introduction of mutant KRAS into WT cells may not necessarily confer KRAS oncogene addiction. Conversely, loss of mutant KRAS might force any surviving cells that are now KRAS WT to compensate by upregulating other pathways to which they then become addicted, such as the transcriptional coactivator Yap1149,150.
Efforts to screen large panels of cancer cell lines are underway, which should more thoroughly take into account the heterogeneity among KRAS-mutant cell lines due to different lineages and co-existing mutations151,152. Indeed, a recent siRNA analysis of a large number of lung cancer cell lines revealed that the majority of genetic dependencies are “private” to individual cell lines and only a minority of genetic dependencies are shared “publicly” among many cell lines147. Screening a very large cell line collection such as the Cancer Cell Line Encyclopedia153 would also be useful to determine the landscape of KRAS synthetic lethality in a context-dependent fashion. Finally, all currently described synthetic lethal screens were conducted in vitro using anchorage-dependent proliferation or viability as the readout. Second generation synthetic lethal screens could benefit from other assay formats such as in vitro anchorage-independent colony formation assays or in vivo xenograft tumour assays that better interrogate the tumourigenic capability of the KRAS oncogene. Both of these types of assays have been successfully adapted for high-throughput screening154–157. However, these assays are likely to be less robust and their execution requires considerable effort; hence, knockdown-validated shRNA libraries would be particularly useful in these screens.
Compared to in vitro viability assays of cells cultured on plastic, in vivo screening has the benefit of providing a more physiological setting and the potential to reveal genes that are required only for tumor growth but not for cancer cell proliferation in vitro. Pooled shRNA libraries have been successfully applied in vivo for both enrichment screens to identifiy tumor suppressor genes158–160 and for dropout screens to identify tumour-essential genes155,157. By using syngeneic mouse cancer cells155,160, in vivo screening also has the potential to address the roles of the microenvironment and immune interactions in tumour growth.
From a therapeutic point of view, targeting synthetic lethality should provide a work-around solution to an undruggable oncogene. However, KRAS synthetic lethal interactors from the first generation of screens may not serve as ideal candidates for drug discovery: targeting the cell cycle machinery could lead to significant toxicity in normal proliferating tissues, whereas transcription factors such as GATA2 are themselves undruggable and require indirect targeting strategies161. Although the protein kinase STK33 was initially proposed to be a druggable target in KRAS-mutant cells144, subsequent studies found that neither genetic or pharmacologic inhibition of STK33 selectively inhibited the growth of such cells162. Genetic and pharmacologic evaluations of TBK1 found no consistent requirement for TBK1 in the growth of KRAS-mutant tumour cell lines in vitro163, although combined TBK1 and MEK inhibition led to partial regression of Kras;p53 mutant lung tumours in mice164. This result highlights a limitation of high throughput screening - hits must be rigorously validated. Further effort is therefore necessary to identify better synthetic lethal candidates for drug discovery. However, given the uncertain success of current strategies for targeting K-Ras effector signalling, synthetic lethal screening may still be a valuable approach to broaden the target space for discovering therapeutic approaches that are orthogonal to Ras pathway inhibitors. One recent study determined that combined targeting of both MEK and BCL-XL led to synthetic lethality and improved therapeutic efficacy over loss of either alone, with tumour regressions obtained in both KRAS-driven xenografts and a genetically engineered mouse model of lung cancer165.
An exciting recent development in genetic screening is the adaptation of the bacterial Type II CRISPR-Cas9 system for genome editing in mammalian cells166. In this method, a short, 20-nucleotide single guide RNA (sgRNA) guide sequence directs the RNA-dependent DNase Cas9 to cleave any target gene bearing sequence homology to the sgRNA. Imprecise double-strand DNA repair through non-homologous end-joining subsequently leads to insertion or deletion mutations that knock out gene expression. Like RNAi, CRISPR relies on short-length sequence homology and therefore could suffer from similar off-target effects. However, unlike RNAi, which produces various degrees of hypomorphic phenotypes, CRISPR generates true nulls when designed properly, thus vastly improving library penetrance. Pooled CRISPR libraries have been deployed for genetic screens in human cell lines167,168 and this approach is likely to provide an orthogonal, and potentially superior, alternative to RNAi screens in the search for mutant RAS synthetic lethal interactions. A recent CRISPR screen in a melanoma model identified both previously validated and novel genes whose loss promoted resistance to the B-Raf inhibitor vemurafenib167.
Ras-driven changes in metabolism – ready for prime-time?
In order to meet the increased biosynthetic demands of a growing tumour, cancer cells alter their metabolism. Metabolic changes were first observed in pioneering research in the 1920s by Otto Warburg, when he demonstrated that tumour cells take up excess glucose and produce lactate even in the presence of oxygen (aerobic glycolysis, a.k.a. the Warburg effect)169. Today, these metabolic adaptations, including the Warburg effect, are now recognized as one of the hallmarks of cancer138,139. One of the major requirements of tumour cells, given their deregulated proliferation, is the need to increase biomass. Thus, there is a shift towards anabolic metabolic processes to produce building blocks such as amino acids, nucleic acids, lipids and cofactors such as NADPH for redox balance and reductive biosynthesis170–172. While much is known about the altered metabolism of tumour cells, more recent work has shown that many of the metabolic changes seen in these cells are brought about on a molecular level by oncogenes and tumour suppressor genes173–175. Indeed, oncogenic Ras has been shown to be a key player in promoting such metabolic rewiring, although the specifics may differ depending on tumour type and genetic context15,176. Importantly, many of the metabolic changes driven by oncogenic Ras become critical for tumour maintenance and thus become attractive therapeutic targets in their own right. Additionally, because they are not dependent on oncogenic Ras, normal tissues often do not have the same reliance on such pathways and therefore there is potential for a tractable therapeutic index. The fact that metabolic reactions are enzyme-mediated also presents the opportunity to inhibit their catalytic activity with small molecules. Here we review the metabolic changes driven by oncogenic Ras and explore the therapeutic possibilities of targeting these pathways in various tumour types.
Metabolic recycling
One of the convergent themes in the metabolism of Ras-driven cancers is that they have developed a reliance on several mechanisms to either recycle intracellular fuel sources or scavenge extracellular constituents to meet their metabolic needs177. One such process is macroautophagy (hereafter referred to as autophagy), a process of self-eating, whereby intracellular substrates are sequestered in double-membrane vesicles known as autophagosomes178 (FIG. 6). These autophagosomes can fuse to lysosomes, forming autophagolysosomes, where the cargo is degraded by lysosomal hydrolases and then recycled back into the cytoplasm to be used in various metabolic reactions178. In cancer the role of autophagy is complex and context-dependent179–181; however, growing evidence has shown that in certain tumour types such as those driven by oncogenic Ras, autophagy is required for tumour maintenance in large part to fuel the metabolism of these aggressive cancers182–185. Indeed, inhibition of autophagy in this context either genetically or pharmacologically causes metabolic dysfunction and a decrease in tumour growth. There have been suggestions that the tumour suppressor gene background may dictate the role of autophagy in a given tumour type harbouring activating KRAS mutations. However, there are conflicting data in this regard that could be due to differences in the various models used as well as to other factors186–188.
Figure 6. Ras-driven alterations in metabolism.

RAS-mutant cancer cells are characterized by increased macropinocytosis and uptake of albumen, leading to lysosomal degradation and release of amino acids. RAS-mutant cancer cells also exhibit altered autophagy, leading to degradation of organelles and proteins, and to production of amino acids and other components that support metabolism. Oncogenic K-Ras directs glucose metabolism into biosynthetic pathways in PDAC by upregulating many key enzymes in glycolysis. Oncogenic K-Ras induces nonoxidative PPP flux to fuel increased nucleic acid biosynthesis and activates the hexosamine biosynthesis and glycosylation pathways. PDAC cells also utilize a non-canonical pathway to process glutamine and use it to maintain redox status and support growth. Blue text indicates Ras-dependent gene and/or protein expression, with arrows indicating increased (green) or decreased (red) expression. Enzymes are indicated in italics. Abbreviations used are: Glut1, glucose transporter 1; Hk 1/2, hexokinase 1/2; Pfk1, phosphofructokinase 1; Eno1, enolase 1; Pkm, pyruvate kinase; Ldha, lactate dehydrogenase A; Gfpt1, glucosamine-fructose-6-phosphate aminotransferase-1; GlcN, glucosamine; GlcNAc, N-acetylglucosamine; Rpe, ribulose-5-phosphate-3-epimerase; Rpia, ribulose-5-phosphate isomerase; GLUD1, glutamate dehydrogenase 1; GLS1, glutaminase 1; GOT1/2, aspartate transaminase 1/2; MDH1, malate dehydrogenase 1; ME1, malic enzyme; GSH, glutathione; GSSG, glutathione disulfide; ROS, reactive oxygen species.
Chloroquine and its derivative hydroxychloroquine (HCQ) can act as autophagy inhibitors, given their ability to inhibit lysosomal acidification189,190. Since they are already approved for use in patients for other indications and have been used safely for several decades, these drugs have now made their way into multiple clinical trials in various cancer types. Ras-driven cancers such as pancreatic cancer, in which nearly 100% of tumours possess activating KRAS mutations191, are highly represented in these trials. While HCQ has been shown to be effective in the preclinical setting, there are many questions regarding its use in patients, including its pharmacological properties (a long period of time is required to reach therapeutic levels) as well as its low potency189. Furthermore, as these drugs work at the level of the lysosome to inhibit the degradation of autophagosomal contents, they likely will interfere with other cellular processes. However, this may have anti-tumour benefits in that other processes such as macropinocytosis (discussed below) converge at the level of the lysosome. Whether HCQ is potent enough to inhibit autophagy in human tumours remains to be seen, and more potent autophagy inhibitors are in development. Given the multiple kinases involved in the process, as well as key components analogous to the ubiquitin conjugation system, there are several potential opportunities for drug development to inhibit this pathway192.
In addition to autophagy, which can generate metabolic substrates through intracellular degradation, Ras-transformed cells have developed other adaptations to essentially engulf extracellular proteins. It has been known since the 1980s that cells expressing Ras oncogenes undergo a process known as macropinocytosis, whereby a membrane process envelopes extracellular contents193. This is then internalized and ultimately fuses to the lysosome, where, in a process similar to autophagy, the contents of the macropinosome are degraded. Unlike autophagy, where much of the machinery has been identified, much less is known about the proteins that are critical for macropinocytosis194. Regardless, recent work has shown that, in the context of KRAS-mutant cancer cells, macropinocytosis may be playing a key metabolic role. Indeed, K-Ras-transformed cells, including pancreatic cancer cells, utilize macropinocytosis to uptake extracellular albumin which is then degraded into amino acids and used to fuel the TCA cycle195. In addition to proteins, Ras-transformed cells take up extracellular lipids and use these as their major source of fatty acids196. Together, these data show that Ras-driven cancers utilize multiple mechanisms to scavenge various metabolites and that there may be therapeutic opportunities in this regard as normal cells are not likely to be as reliant on these metabolic adaptations. In particular, as autophagy and macropinocytosis converge at the level of the lysosome, targeting degradation using HCQ or related inhibitors may have clinical utility and such trials are underway.
Targeting metabolic changes
Several recent studies have shown that one of the major mechanisms by which oncogenic K-Ras promotes tumour growth is by rewiring key aspects of metabolism in tumours. Mutant K-Ras has been shown to increase glucose uptake, and conversely low glucose conditions can select for tumour cells with KRAS mutations197. Work in pancreatic cancer systems has shown that oncogenic K-Ras systematically controls glucose metabolism by promoting a transcriptional program that leads to alterations of key rate-limiting enzymes of anabolic glucose metabolism15 (FIG. 6). The net result of these changes is increased flux of glycolytic intermediates through pathways such as the hexosamine biosynthesis pathway (HBP) and the non-oxidative arm of the pentose phosphate pathway (PPP). This leads to increased production of precursors used for glycosylation (HBP) and ribose used for DNA and RNA biosynthesis (non-oxidative PPP). Importantly, inhibition of either of these pathways using shRNAs to the K-Ras-regulated enzymes inhibits pancreatic cancer growth in vitro and in xenografts15. While no inhibitors of these enzymes are currently available, studies of K-Ras signalling pathways that mediate the transcriptional and metabolic changes in anabolic glucose metabolism have provided a potential therapeutic approach. Indeed, the Raf-MEK-ERK pathway was shown to be critical for the metabolic shift caused by oncogenic K-Ras15. Clinical grade inhibitors of MEK are being tested in a variety of cancers and it may be possible to attenuate anabolic glucose metabolism in K-Ras mutant cancers with these compounds. Other studies have supported the approach of inhibiting various aspects of glucose metabolism in KRAS–driven tumours, including inhibition of lactate dehydrogenase198 and hexokinase 2199.
In addition to glucose metabolism, oncogenic K-Ras appears to orchestrate a shift in glutamine metabolism in pancreatic and other cancer types176,200,201. In pancreatic cancers, K-Ras appears to coordinate transcriptional changes that result in a net increase in flux through a novel pathway, whereby glutamine-derived aspartate is converted to oxaloacetate by the cytosolic aspartate aminotransferase (GOT1), then to malate by malate dehydrogenase (MDH1), and finally to pyruvate and NADPH by the cytosolic malic enzyme (ME1) (FIG. 6). The NADPH generated from this series of reactions is critical for growth because of its role in maintaining reduced glutathione pools for redox balance176. Importantly, this pathway was shown to be required for pancreatic cancer redox balance and growth in vitro and in vivo, while being dispensable in normal cells, thus implying that a therapeutic index may exist. Currently, there are no clinical grade inhibitors of GOT1/2, MDH1, or ME1 available. However, inhibitors of glutaminase (GLS), the enzyme that catalyzes the conversion of glutamine to glutamate, are available and are making their way into early phase clinical trials202. GLS inhibitors alter the redox balance in pancreatic cancer cells and synergize with treatments that increase reactive oxygen species (ROS) to inhibit pancreatic cancer growth176. Thus, there is potential to combine GLS inhibitors with treatments such as radiotherapy and chemotherapies that increase ROS. An important question remains as to whether these K-Ras-regulated glucose and glutamine pathways are specific to pancreatic cancers or are a common feature of all Ras-driven tumours. Such studies are ongoing. In this regard it has been reported that inhibition of the aspartate aminotransferase in a breast cancer cell line harboring a KRAS mutation has anti-tumour effects203.
An interesting theme emerging from studies of the metabolic changes driven by oncogenic KRAS is that this oncogene also has a prominent role in maintaining redox balance. This may be through the control of metabolic pathways that lead to increased NADPH production as mentioned above176,200 or alternatively through other mechanisms such as the upregulation of NRF2, a master regulator of redox defense204. Indeed, it was demonstrated that oncogenic K-Ras, when expressed at endogenous levels, leads to decreased ROS levels in multiple cell types through the induction of the NRF2 antioxidant program205. However, there is also evidence suggesting that KRAS-driven tumourigenesis requires ROS201. Therefore, it is likely that keeping a controlled level of ROS is beneficial for tumour growth whereas disrupting redox homeostasis in K-Ras-driven tumours may be an effective therapeutic strategy.
In conclusion, mounting evidence demonstrates that one of the mechanisms by which oncogenic Ras supports tumour growth is through altering the metabolism of cancers to suit its needs. In many cases, these pathways remain critical to tumour growth and therefore may provide tractable therapeutic opportunities for future drug development initiatives. While we await the development of novel inhibitors to various metabolic enzymes, some, such as GLS inhibitors, are currently available for use. Additionally, understanding how available compounds perturb metabolic pathways may allow a more rapid translation of preclinical findings to the clinic. Examples of new uses for older drugs include HCQ to inhibit autophagy and macropinocytosis, phenformin/metformin to inhibit mitochondrial complex I, and MEK inhibitors to globally inhibit anabolic glucose metabolism in pancreatic cancers.
Conclusions and Future Prospects
As the general and military strategist Sun Tzu wrote in The Art of War, an influential ancient Chinese book on military strategy, success in battle is better achieved when we “know the enemy”. The more than three decades of intense research focussed on Ras have provided an ever-increasingly comprehensive description of the target — we have repeatedly thought that we knew our enemy, Ras. Nevertheless, we have repeatedly failed to realize that our understanding has been far from complete, and this failure has contributed to missteps and misdirections in anti-Ras drug development. Currently, we can at least recognize that issues once thought to be understood are clearly still works in progress. In particular, our view of Ras effector signalling, once thought to be a simple, linear unidirectional protein kinase cascade, has evolved to recognize a complex and highly dynamic signalling network that can adapt and rewire in response to pharmacologic inhibitors. Other issues that remain poorly understood, yet will have significant impact on successful anti-Ras drug development, also need resolution. One likely basis for the failure of anti-Ras drug discovery efforts is the still-prevalent assumption that activated Ras comes in one simple flavour. There is now at least a greater appreciation that the three RAS isoforms are not functionally equivalent, and that each will require distinct therapeutic approaches, resulting in the intense focus now on KRAS.
Along the lines of RAS isoform differences, there remain the issues of why KRAS is mutated far more frequently than the other RAS isoforms and why some types of cancer almost exclusively harbor mutations in KRAS but not in the other RAS isoforms. In one study, this issue was addressed using a mouse model where carcinogen-induced lung tumour formation is associated with a high frequency of Kras mutations206. In a mouse where the genomic Kras locus was genetically engineered to express the H-Ras protein instead, lung tumours arose at an even higher incidence and had Hras mutations. This result argues that it is not the Ras protein itself, but rather the Kras regulatory locus, that favours KRAS mutations in human lung tumours. In contrast to this suggestion, a recent provocative study found that KRAS contains a high frequency of rare codons relative to HRAS, resulting in reduced protein translation207. When the KRAS gene was modified with more commonly used codons, K-Ras protein expression was significantly higher and exhibited more potent transforming activity. This result suggests that, since mutant Ras expression in normal cells lacking other genetic alterations causes senescence, low K-Ras protein expression favours the persistence of histologically normal cells harbouring mutant KRAS, thereby allowing subsequent genetic events that then drive cancer progression. Then, once progression is initiated, higher mutant K-Ras expression is needed to drive cancer growth; this may explain the KRAS gene amplification often seen in cancers. A critical validation of this hypothesis would be a reduced incidence of tumour formation induced by mutant Kras in a mouse harbouring an endogenous codon-optimized Kras locus.
With the predominant frequency of KRAS mutations in human cancers, in particular in cancers in dire need of improved therapies, the focus is now strongly on K-Ras. Since earlier studies had found that the KRAS4B splice variant is preferentially expressed, this focus is currently most centered on the K-Ras4B protein. However, whether K-Ras4A should still be considered a key player in cancer remains a neglected issue208. KRAS was originally discovered to be an oncogene when Werner Kirsten identified a murine leukaemia virus that transduced the rat cellular Kras gene. That this gene encodes K-Ras4A is perhaps a reminder that this splice variant should not be ignored completely. Indeed, new unpublished data indicates that K-Ras4A is more widely expressed in human cancers than previously appreciated and could also contribute to the transformed phenotype (Philips, Cox et al.).
Another less-appreciated possibility is that different KRAS mutations have distinct oncogenic properties and hence may be differentially responsive to targeted therapies. This possibility is suggested by the finding that patients with colorectal cancer whose tumours harbor KRAS G13D mutations, in contrast to G12 mutations, showed a clinical benefit from cetuximab anti-EGFR therapy209,210. Similarly, in cell culture and mouse model analyses, G13D but not G12V colorectal cancer cells were sensitive to cetuximab. In a study of NSCLC, it was found that tumours with either KRAS G12C or G12V mutations correlated with worse progression-free survival compared to tumours with WT or other KRAS mutations211. A possible functional basis for this correlation was suggested by the finding that NSCLC cell lines with KRAS G12D preferentially activated the Raf and PI3K effector pathways, whereas those with KRAS G12C or G12V preferentially activated the Ral effector pathway. These and other studies have stimulated an awareness that we may need to pursue mutation-specific anti-Ras strategies, and this awareness is reflected in the fact that the NCI-directed Ras Initiative has identified KRAS G12D, G12V, G12C and G13D as the four mutant proteins to target. However, any additional genetic alterations that co-occur with one specific KRAS mutation will also diversify the therapies required in order to be effective. WT Ras activation by indirect mechanisms, whether by upstream receptor tyrosine kinase activity or by the loss of RasGAP function will also likely require very distinct therapeutic approaches. Hence, one therapy will not fit all flavours of activated Ras, and there will not be one simple anti-Ras drug for all situations.
Perhaps one of the more perplexing issues that remains far from understood today is the role of wild type Ras in the setting of mutant Ras. This issue has two seemingly conflicting camps. On one hand, there is evidence that wild type KRAS can act as a tumour suppressor and antagonize mutant KRAS oncogenicity212,213–215. Consistently, a significant subset of KRAS-mutant tumour cell lines is homozygous for the mutant allele. But how might wild type KRAS antagonize mutationally activated KRAS – is the GDP-bound form of K-Ras not simply an inactive protein? There are clues that Ras proteins may dimerize216; perhaps a WT:mutant K-Ras dimer is inactive. However, this tumour suppressor function may not hold true for WT NRAS217. In contrast to the above noted support for WT KRAS as a tumour suppressor, other studies have found instead that WT RAS isoforms serve a supporting role for the mutated RAS isoform75,218,219. Here, there is strong experimental evidence to support the necessity of the WT isoform in the context of the mutated isoform. However, the mechanistic relationship between them and what activated wild type Ras does that cannot be fulfilled by the mutated Ras isoform remain unclear.
There are other challenges: even if an inhibitor can be developed that effectively blocks mutant Ras function and has a significant therapeutic response, compensatory mechanisms will likely be triggered to overcome the RAS addiction of cancer cells, and such newly RAS-independent cancer cells will likely drive the return of cancer growth149,150. This notion is supported by the fact that mutant Ras-driven cancer growth involves effector signalling networks that can also be activated by Ras-independent mechanisms. A clear understanding of the mechanistic basis for such resistance will be required to successfully overcome it, perhaps by prompting effective rational combinations of therapies, whether these be conventional and targeted drugs or targeted inhibitors and synthetic lethal interactors165.
An emerging and exciting new direction may come from recent advances in our understanding of the immunological underpinnings of the activities of mutant RAS220. For example, mutant K-Ras induces granulocyte-macrophage colony-stimulating factor (GM-CSF) secretion in mouse pancreatic tumour cells, stimulating the expansion of immunosuppressive myeloid-derived suppressor cells (MSDCs) and blocking T cell-driven anti-tumour immunity221,222. This altered cytokine expression and/or the MSDCs themselves might be exploitable for immunotherapeutic attack. Similarly, one focus of the NCI Ras Project involves mapping the surface of mutant K-Ras to identify sites of tumour-selective immune recognition.
Finally, silencing of RAS by interfering RNA is another possible future direction. While still not ready for prime time, progress continues to be made in addressing key limitations of RNAi-based therapy, particularly to facilitate efficient delivery to and entry into tumour cells, and to obtain the effective and prolonged gene suppression that will be required to achieve a clinical response223.
In summary, while the task of developing effective therapeutic strategies for Ras-dependent cancers remains a daunting initiative, with new knowledge and strategies, there is cautious optimism that what has been an impossible mission may become Mission Possible.
Supplementary Material
Acknowledgments
The work of C.J.D. and A.D.C. is supported by US National Institutes of Health (NIH) grants CA042978, CA179193 and CA175747, and by grants from the Lustgarten Foundation for Pancreatic Cancer Research and the Pancreatic Cancer Action Network-American Association for Cancer Research. The work of S.W.F. is supported by NIH grants DP1OD006933/DP1CA174419 (NIH Director’s Pioneer Award; S.W.F.), P50A095103-12 (NCI SPORE in GI Cancer; R.J. Coffey), and RC2CA148375 (NIH ARRA Stimulus Grant; L.J. Marnett), and by the Lustgarten Foundation for Pancreatic Cancer Research. The work of A.C.K. is supported by NIH grant R01CA157490, American Cancer Society Research Scholar Grant RSG-13-298-01-TBG, and by the Lustgarten Foundation. A.C.K. is a consultant for Forma Therapeutics. The work of J.L. is supported by the US National Cancer Institute Intramural Program.
Footnotes
LINKS TO WEB SITES
Cancer Cell Line Encyclopedia (CCLE): http://www.broadinstitute.org/ccle
canSAR knowledgebase: http://cansar.icr.ac.uk
ClinicalTrials.gov: http://clinicaltrials.gov/
COSMIC: http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/
International Cancer Genome Consortium: http://icgc.org
The Ras Project: http://deainfo.nci.nih.gov/advisory/ncab/165_0613/McCormick.pdf
The Cancer Genome Atlas Data Portal: https://tcga-data.nci.nih.gov/tcga/
DATABASES
cBioPortal for Cancer Genomics: http://www.cbioportal.org/public-portal/
ClinicalTrials.gov: http://clinicaltrials.gov/
COSMIC: http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/
ICGC Data Portal: https://dcc.icgc.org/
The Cancer Genome Atlas: https://tcga-data.nci.nih.gov/tcga/
TumorPortal - Broad Institute: http://cancergenome.broadinstitute.org
Bibliography
- 1.Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson H. US National Cancer Institute’s new Ras project targets an old foe. Nat Med. 2013;19:949–50. doi: 10.1038/nm0813-949. [DOI] [PubMed] [Google Scholar]
- 4.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11:775–91. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 6.Ji H, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–10. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 7.Haigis KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–8. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2:243–7. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 9.Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell. 2005;8:381–92. doi: 10.1016/j.ccr.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Singh A, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin L, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–72. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 12.Collins MA, et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One. 2012;7:e49707. doi: 10.1371/journal.pone.0049707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisher GH, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–62. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwong LN, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–10. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ying H, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–77. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 17.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–81. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 18.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiss Y, Goldstein JL, Seabra MC, Casey PJ, Brown MS. Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell. 1990;62:81–8. doi: 10.1016/0092-8674(90)90242-7. [DOI] [PubMed] [Google Scholar]
- 20.James GL, Goldstein JL, Brown MS. Polylysine and CVIM sequences of K-RasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J Biol Chem. 1995;270:6221–6. doi: 10.1074/jbc.270.11.6221. [DOI] [PubMed] [Google Scholar]
- 21.Whyte DB, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–64. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 22.Rowell CA, Kowalczyk JJ, Lewis MD, Garcia AM. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J Biol Chem. 1997;272:14093–7. doi: 10.1074/jbc.272.22.14093. [DOI] [PubMed] [Google Scholar]
- 23.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–57. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grant BJ, et al. Novel allosteric sites on Ras for lead generation. PLoS One. 2011;6:e25711. doi: 10.1371/journal.pone.0025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buhrman G, et al. Analysis of binding site hot spots on the surface of Ras GTPase. J Mol Biol. 2011;413:773–89. doi: 10.1016/j.jmb.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Fang G, Rudolph J. Ras inhibition via direct Ras binding--is there a path forward? Bioorg Med Chem Lett. 2012;22:5766–76. doi: 10.1016/j.bmcl.2012.07.082. [DOI] [PubMed] [Google Scholar]
- 27.Taveras AG, et al. Ras oncoprotein inhibitors: the discovery of potent, ras nucleotide exchange inhibitors and the structural determination of a drug-protein complex. Bioorg Med Chem. 1997;5:125–33. doi: 10.1016/s0968-0896(96)00202-7. [DOI] [PubMed] [Google Scholar]
- 28.Peri F, et al. Design, synthesis and biological evaluation of sugar-derived Ras inhibitors. Chembiochem. 2005;6:1839–48. doi: 10.1002/cbic.200400420. [DOI] [PubMed] [Google Scholar]
- 29.Herrmann C, et al. Sulindac sulfide inhibits Ras signaling. Oncogene. 1998;17:1769–76. doi: 10.1038/sj.onc.1202085. [DOI] [PubMed] [Google Scholar]
- 30.Waldmann H, et al. Sulindac-derived Ras pathway inhibitors target the Ras-Raf interaction and downstream effectors in the Ras pathway. Angew Chem Int Ed Engl. 2004;43:454–8. doi: 10.1002/anie.200353089. [DOI] [PubMed] [Google Scholar]
- 31.Karaguni IM, et al. The new sulindac derivative IND 12 reverses Ras-induced cell transformation. Cancer Res. 2002;62:1718–23. [PubMed] [Google Scholar]
- 32.Karaguni IM, et al. New indene-derivatives with anti-proliferative properties. Bioorg Med Chem Lett. 2002;12:709–13. doi: 10.1016/s0960-894x(01)00839-3. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez-Perez V, et al. Genetic and functional characterization of putative Ras/Raf interaction inhibitors in C. elegans and mammalian cells. J Mol Signal. 2010;5:2. doi: 10.1186/1750-2187-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato-Stankiewicz J, et al. Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert Ras-dependent transformation phenotypes in human cancer cells. Proc Natl Acad Sci USA. 2002;99:14398–403. doi: 10.1073/pnas.222222699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosnizeck IC, et al. Stabilizing a weak binding state for effectors in the human ras protein by cyclen complexes. Angew Chem Int Ed Engl. 2010;49:3830–3. doi: 10.1002/anie.200907002. [DOI] [PubMed] [Google Scholar]
- 36.Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol. 2011;7:585–7. doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang YS, et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci USA. 2013;110:E3445–54. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maurer T, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci USA. 2012;109:5299–304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Q, et al. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012;51:6140–3. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shima F, et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci USA. 2013;110:8182–7. doi: 10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen KX, et al. A novel class of highly potent irreversible hepatitis C virus NS5B polymerase inhibitors. J Med Chem. 2012;55:2089–101. doi: 10.1021/jm201322r. [DOI] [PubMed] [Google Scholar]
- 43.Ward RA, et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR) J Med Chem. 2013;56:7025–48. doi: 10.1021/jm400822z. [DOI] [PubMed] [Google Scholar]
- 44.Lim SM, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burns MC, et al. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc Natl Acad Sci USA. 2014;111:3401–6. doi: 10.1073/pnas.1315798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Makarewicz JM, Knauf JA, Johnson LK, Fagin JA. Transformation by Hras is consistently associated with mutant allele copy gains and is reversed by farnesyl transferase inhibition. Oncogene. 2013 doi: 10.1038/onc.2013.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu M, et al. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc Natl Acad Sci USA. 2010;107:6471–6. doi: 10.1073/pnas.0908396107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marom M, et al. Selective inhibition of Ras-dependent cell growth by farnesylthiosalisylic acid. J Biol Chem. 1995;270:22263–70. doi: 10.1074/jbc.270.38.22263. [DOI] [PubMed] [Google Scholar]
- 49.Gana-Weisz M, et al. The Ras antagonist S-farnesylthiosalicylic acid induces inhibition of MAPK activation. Biochem Biophys Res Commun. 1997;239:900–4. doi: 10.1006/bbrc.1997.7582. [DOI] [PubMed] [Google Scholar]
- 50.Haklai R, et al. Dislodgment and accelerated degradation of Ras. Biochemistry. 1998;37:1306–14. doi: 10.1021/bi972032d. [DOI] [PubMed] [Google Scholar]
- 51.Makovski V, Haklai R, Kloog Y. Farnesylthiosalicylic acid (salirasib) inhibits Rheb in TSC2-null ELT3 cells: a potential treatment for lymphangioleiomyomatosis. Int J Cancer. 2012;130:1420–9. doi: 10.1002/ijc.26139. [DOI] [PubMed] [Google Scholar]
- 52.McMahon LP, Yue W, Santen RJ, Lawrence JC., Jr Farnesylthiosalicylic acid inhibits mammalian target of rapamycin (mTOR) activity both in cells and in vitro by promoting dissociation of the mTOR-raptor complex. Mol Endocrinol. 2005;19:175–83. doi: 10.1210/me.2004-0305. [DOI] [PubMed] [Google Scholar]
- 53.Hanker AB, et al. Differential requirement of CAAX-mediated posttranslational processing for Rheb localization and signaling. Oncogene. 2010;29:380–91. doi: 10.1038/onc.2009.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weisz B, et al. A new functional Ras antagonist inhibits human pancreatic tumor growth in nude mice. Oncogene. 1999;18:2579–88. doi: 10.1038/sj.onc.1202602. [DOI] [PubMed] [Google Scholar]
- 55.Haklai R, Elad-Sfadia G, Egozi Y, Kloog Y. Orally administered FTS (salirasib) inhibits human pancreatic tumor growth in nude mice. Cancer Chemother Pharmacol. 2008;61:89–96. doi: 10.1007/s00280-007-0451-6. [DOI] [PubMed] [Google Scholar]
- 56.Laheru D, et al. Integrated preclinical and clinical development of S-trans, trans-Farnesylthiosalicylic Acid (FTS, Salirasib) in pancreatic cancer. Invest New Drugs. 2012;30:2391–9. doi: 10.1007/s10637-012-9818-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wahlstrom AM, et al. Rce1 deficiency accelerates the development of K-RAS-induced myeloproliferative disease. Blood. 2007;109:763–8. doi: 10.1182/blood-2006-05-024752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wahlstrom AM, et al. Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood. 2008;112:1357–65. doi: 10.1182/blood-2007-06-094060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Court H, et al. Isoprenylcysteine carboxylmethyltransferase deficiency exacerbates KRAS-driven pancreatic neoplasia via Notch suppression. J Clin Invest. 2013 doi: 10.1172/JCI65764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Majmudar JD, et al. Amide-modified prenylcysteine based Icmt inhibitors: Structure-activity relationships, kinetic analysis and cellular characterization. Bioorg Med Chem. 2012;20:283–95. doi: 10.1016/j.bmc.2011.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manandhar SP, Hildebrandt ER, Schmidt WK. Small-molecule inhibitors of the Rce1p CaaX protease. J Biomol Screen. 2007;12:983–93. doi: 10.1177/1087057107307226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Winter-Vann AM, et al. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc Natl Acad Sci USA. 2005;102:4336–41. doi: 10.1073/pnas.0408107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chiu VK, et al. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002;4:343–50. doi: 10.1038/ncb783. [DOI] [PubMed] [Google Scholar]
- 64.Cuiffo B, Ren R. Palmitoylation of oncogenic NRAS is essential for leukemogenesis. Blood. 2010;115:3598–605. doi: 10.1182/blood-2009-03-213876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swarthout JT, et al. DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem. 2005;280:31141–8. doi: 10.1074/jbc.M504113200. [DOI] [PubMed] [Google Scholar]
- 66.Goodwin JS, et al. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J Cell Biol. 2005;170:261–72. doi: 10.1083/jcb.200502063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rocks O, et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell. 2010;141:458–71. doi: 10.1016/j.cell.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 68.Rocks O, et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–52. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 69.Dekker FJ, et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol. 2010;6:449–56. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 70.Resh MD. Targeting protein lipidation in disease. Trends Mol Med. 2012;18:206–14. doi: 10.1016/j.molmed.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bivona TG, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–93. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 72.Sung PJ, et al. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc Natl Acad Sci USA. 2013;110:20593–8. doi: 10.1073/pnas.1306431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kollar P, Rajchard J, Balounova Z, Pazourek J. Marine natural products: Bryostatins in preclinical and clinical studies. Pharm Biol. 2013 doi: 10.3109/13880209.2013.804100. [DOI] [PubMed] [Google Scholar]
- 74.Barcelo C, et al. Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-13-1750. [DOI] [PubMed] [Google Scholar]
- 75.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–9. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lampson BL, et al. Targeting eNOS in pancreatic cancer. Cancer Res. 2012;72:4472–82. doi: 10.1158/0008-5472.CAN-12-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang H, Constantine R, Frederick JM, Baehr W. The prenyl-binding protein PrBP/delta: a chaperone participating in intracellular trafficking. Vision Res. 2012;75:19–25. doi: 10.1016/j.visres.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chandra A, et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2012;14:148–58. doi: 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
- 79.Zimmermann G, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–42. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 80.Philips MR. Ras hitchhikes on PDE6delta. Nat Cell Biol. 2012;14:128–9. doi: 10.1038/ncb2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. Differential modification of Ras proteins by ubiquitination. Mol Cell. 2006;21:679–87. doi: 10.1016/j.molcel.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 82.Sasaki AT, et al. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2011;4:ra13. doi: 10.1126/scisignal.2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sumita K, et al. Degradation of Activated K-Ras Orthologue via K-Ras-specific Lysine Residues Is Required for Cytokinesis. J Biol Chem. 2014;289:3950–9. doi: 10.1074/jbc.M113.531178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang MH, et al. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol Cancer Res. 2013;11:1072–7. doi: 10.1158/1541-7786.MCR-13-0040-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blasco RB, et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–63. doi: 10.1016/j.ccr.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Collisson EA, et al. A central role for RAF-->MEK-->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2:685–93. doi: 10.1158/2159-8290.CD-11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–43. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 89.Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer. 2001;8:219–25. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- 90.Wilhelm SM, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 91.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–9. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 92.Hatzivassiliou G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 93.Heidorn SJ, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hall-Jackson CA, et al. Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol. 1999;6:559–68. doi: 10.1016/s1074-5521(99)80088-x. [DOI] [PubMed] [Google Scholar]
- 96.Oberholzer PA, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30:316–21. doi: 10.1200/JCO.2011.36.7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Su F, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–15. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell. 2013;49:751–8. doi: 10.1016/j.molcel.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gilmartin AG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 100.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hatzivassiliou G, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–6. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 102.Ishii N, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 2013;73:4050–60. doi: 10.1158/0008-5472.CAN-12-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Duncan JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–21. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Little AS, et al. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal. 2011;4:ra17. doi: 10.1126/scisignal.2001752. [DOI] [PubMed] [Google Scholar]
- 105.Bollag G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morris EJ, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–50. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 107.Gupta S, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–68. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 108.Castellano E, et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell. 2013;24:617–30. doi: 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ebi H, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121:4311–21. doi: 10.1172/JCI57909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71:1395–409. doi: 10.1007/s00280-013-2121-1. [DOI] [PubMed] [Google Scholar]
- 111.Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8:133–40. doi: 10.1038/nrc2296. [DOI] [PubMed] [Google Scholar]
- 112.Neel NF, et al. The RalGEF-Ral Effector Signaling Network: The Road Less Traveled for Anti-Ras Drug Discovery. Genes Cancer. 2011;2:275–87. doi: 10.1177/1947601911407329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gonzalez-Garcia A, et al. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell. 2005;7:219–26. doi: 10.1016/j.ccr.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 114.Vigil D, et al. Aberrant overexpression of the Rgl2 Ral small GTPase-specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral-dependent and Ral-independent mechanisms. J Biol Chem. 2010;285:34729–40. doi: 10.1074/jbc.M110.116756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lim KH, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385–94. doi: 10.1016/j.cub.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 116.Peschard P, et al. Genetic deletion of RALA and RALB small GTPases reveals redundant functions in development and tumorigenesis. Curr Biol. 2012;22:2063–8. doi: 10.1016/j.cub.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 117.Wu JC, et al. Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J Biol Chem. 2005;280:9013–22. doi: 10.1074/jbc.M411068200. [DOI] [PubMed] [Google Scholar]
- 118.Lim KH, et al. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2010;30:508–23. doi: 10.1128/MCB.00916-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang H, et al. Phosphorylation of RalB is important for bladder cancer cell growth and metastasis. Cancer Res. 2010;70:8760–9. doi: 10.1158/0008-5472.CAN-10-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Martin TD, Mitin N, Cox AD, Yeh JJ, Der CJ. Phosphorylation by protein kinase Calpha regulates RalB small GTPase protein activation, subcellular localization, and effector utilization. J Biol Chem. 2012;287:14827–36. doi: 10.1074/jbc.M112.344986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chien Y, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–70. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 122.Martin TD, et al. Ral and Rheb GTPase Activating Proteins Integrate mTOR and GTPase Signaling in Aging, Autophagy, and Tumor Cell Invasion. Mol Cell. 2014;53:209–20. doi: 10.1016/j.molcel.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Malliri A, et al. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002;417:867–71. doi: 10.1038/nature00848. [DOI] [PubMed] [Google Scholar]
- 124.Lambert JM, et al. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol. 2002;4:621–5. doi: 10.1038/ncb833. [DOI] [PubMed] [Google Scholar]
- 125.Welch HC, et al. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–21. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- 126.Hodis E, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Krauthammer M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–14. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kissil JL, et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007;67:8089–94. doi: 10.1158/0008-5472.CAN-07-2300. [DOI] [PubMed] [Google Scholar]
- 129.Heid I, et al. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterol. 2011;141:719–30. 730 e1–7. doi: 10.1053/j.gastro.2011.04.043. [DOI] [PubMed] [Google Scholar]
- 130.Cardama GA, et al. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med Chem. 2013 doi: 10.2174/18715206113136660334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101:7618–23. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chow HY, et al. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–75. doi: 10.1158/0008-5472.CAN-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fritsch R, et al. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell. 2013;153:1050–63. doi: 10.1016/j.cell.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chan JJ, Katan M. PLCvarepsilon and the RASSF family in tumour suppression and other functions. Adv Biol Regul. 2013;53:258–79. doi: 10.1016/j.jbior.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 135.Hartman JLt, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science. 2001;291:1001–4. doi: 10.1126/science.1056072. [DOI] [PubMed] [Google Scholar]
- 136.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 137.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 138.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 139.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu BL, Ji, editors. Synthetic lethal genetic screens in Ras mutant cancers. Academic Press; 2013. [DOI] [PubMed] [Google Scholar]
- 141.Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22:8999–9006. doi: 10.1038/sj.onc.1207111. [DOI] [PubMed] [Google Scholar]
- 142.Kumar MS, et al. Cell. 2012:642–655. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 143.Luo J, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Steckel M, et al. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012;22:1227–45. doi: 10.1038/cr.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Carretero J, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–59. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kim HS, et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell. 2013;155:552–66. doi: 10.1016/j.cell.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- 149.Kapoor A, et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell. 2014 doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shao DD, et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell. 2014 doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cheung HW, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci USA. 2011;108:12372–7. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Marcotte R, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2:172–89. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Anderson SN, Towne DL, Burns DJ, Warrior U. A high-throughput soft agar assay for identification of anticancer compound. J Biomol Screen. 2007;12:938–45. doi: 10.1177/1087057107306130. [DOI] [PubMed] [Google Scholar]
- 155.Meacham CE, Ho EE, Dubrovsky E, Gertler FB, Hemann MT. In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat Genet. 2009;41:1133–7. doi: 10.1038/ng.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zuber J, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628–40. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–50. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Murugaesu N, et al. An in vivo functional screen identifies ST6GalNAc2 sialyltransferase as a breast cancer metastasis suppressor. Cancer Discov. 2014;4:304–17. doi: 10.1158/2159-8290.CD-13-0287. [DOI] [PubMed] [Google Scholar]
- 159.Schramek D, et al. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science. 2014;343:309–13. doi: 10.1126/science.1248627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zender L, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell. 2008;135:852–64. doi: 10.1016/j.cell.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kumar MS, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149:642–55. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 162.Luo T, et al. STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proc Natl Acad Sci USA. 2012;109:2860–5. doi: 10.1073/pnas.1120589109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Muvaffak A, et al. Evaluating TBK1 as a Therapeutic Target in Cancers with Activated IRF3. Mol Cancer Res. 2014 doi: 10.1158/1541-7786.MCR-13-0642. [DOI] [PubMed] [Google Scholar]
- 164.Zhu Z, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4:452–65. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Corcoran RB, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–4. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148:1132–44. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 172.Stine ZE, Dang CV. Stress eating and tuning out: Cancer cells re-wire metabolism to counter stress. Crit Rev Biochem Mol Biol. 2013;48:609–19. doi: 10.3109/10409238.2013.844093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340–4. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 174.Racker E, Resnick RJ, Feldman R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc Natl Acad Sci USA. 1985;82:3535–8. doi: 10.1073/pnas.82.11.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wise DR, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782–7. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.White E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013;27:2065–71. doi: 10.1101/gad.228122.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mah LY, Ryan KM. Autophagy and cancer. Cold Spring Harb Perspect Biol. 2012;4:a008821. doi: 10.1101/cshperspect.a008821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Guo JY, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–61. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lock R, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–78. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yang S, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rao S, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 187.Rosenfeldt MT, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 188.Yang A, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Amaravadi RK, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–66. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget. 2011;2:1302–6. doi: 10.18632/oncotarget.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bar-Sagi D, Feramisco JR. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233:1061–8. doi: 10.1126/science.3090687. [DOI] [PubMed] [Google Scholar]
- 194.Lim JP, Gleeson PA. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol. 2011;89:836–43. doi: 10.1038/icb.2011.20. [DOI] [PubMed] [Google Scholar]
- 195.Commisso C, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–7. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kamphorst JJ, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci USA. 2013;110:8882–7. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yun J, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–9. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.McCleland ML, et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin Cancer Res. 2013;19:773–84. doi: 10.1158/1078-0432.CCR-12-2638. [DOI] [PubMed] [Google Scholar]
- 199.Patra KC, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213–28. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gaglio D, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Weinberg F, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–93. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Shukla K, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012;55:10551–63. doi: 10.1021/jm301191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Thornburg JM, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–5. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.To MD, et al. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat Genet. 2008;40:1240–4. doi: 10.1038/ng.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lampson BL, et al. Rare codons regulate KRas oncogenesis. Curr Biol. 2013;23:70–5. doi: 10.1016/j.cub.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Abubaker J, et al. Prognostic significance of alterations in KRAS isoforms KRAS-4A/4B and KRAS mutations in colorectal carcinoma. J Pathol. 2009;219:435–45. doi: 10.1002/path.2625. [DOI] [PubMed] [Google Scholar]
- 209.De Roock W, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–20. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 210.Tejpar S, et al. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012;30:3570–7. doi: 10.1200/JCO.2012.42.2592. [DOI] [PubMed] [Google Scholar]
- 211.Ihle NT, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–39. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhang Z, et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 213.Bremner R, Balmain A. Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell. 1990;61:407–17. doi: 10.1016/0092-8674(90)90523-h. [DOI] [PubMed] [Google Scholar]
- 214.Li H, et al. Growth inhibitory effect of wild-type Kras2 gene on a colonic adenocarcinoma cell line. World J Gastroenterol. 2007;13:934–8. doi: 10.3748/wjg.v13.i6.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Qiu W, et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2:862–73. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lin WC, et al. H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc Natl Acad Sci USA. 2014;111:2996–3001. doi: 10.1073/pnas.1321155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Xu J, et al. Dominant role of oncogene dosage and absence of tumor suppressor activity in Nras-driven hematopoietic transformation. Cancer Discov. 2013;3:993–1001. doi: 10.1158/2159-8290.CD-13-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Grabocka E, et al. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25:243–56. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013;3:112–23. doi: 10.1158/2159-8290.CD-12-0231. [DOI] [PubMed] [Google Scholar]
- 220.Vonderheide RH, Nathanson KL. Immunotherapy at large: the road to personalized cancer vaccines. Nat Med. 2013;19:1098–100. doi: 10.1038/nm.3317. [DOI] [PubMed] [Google Scholar]
- 221.Bayne LJ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–47. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11:59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Thompson HJ, et al. Sulfone metabolite of sulindac inhibits mammary carcinogenesis. Cancer Res. 1997;57:267–71. [PubMed] [Google Scholar]
- 225.Sarthy AV, et al. Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol Cancer Ther. 2007;6:269–76. doi: 10.1158/1535-7163.MCT-06-0560. [DOI] [PubMed] [Google Scholar]
- 226.Morgan-Lappe SE, et al. Identification of Ras-related nuclear protein, targeting protein for xenopus kinesin-like protein 2, and stearoyl-CoA desaturase 1 as promising cancer targets from an RNAi-based screen. Cancer Res. 2007;67:4390–4398. doi: 10.1158/0008-5472.CAN-06-4132. [DOI] [PubMed] [Google Scholar]
- 227.Scholl C, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 228.Vicent S, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J Clin Invest. 2010;120:3940–3952. doi: 10.1172/JCI44165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wang Y, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010;29:4658–4670. doi: 10.1038/onc.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Singh A, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148:639–650. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Cullis J, et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell. 2014;25:181–95. doi: 10.1016/j.ccr.2014.01.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.