

Abstract
Urofacial syndrome (UFS) is an autosomal recessive disease with severe dysfunctional urination including urinary incontinence (UI). Biallelic mutations of HPSE2 are discovered from UFS patients, suggesting that HPSE2 is a candidate disease gene. Here, we show that deletion of Hpse2 is sufficient to cause the UFS-like phenotype in mice. Hpse2 knockout mutants display a distended bladder (megacystis) phenotype and abnormal voiding behavior similar to that found in patients. While Hpse2 is largely dispensable for detrusor smooth muscle and urothelial cell fate determination, the mutants have significantly lower rates of cell proliferation than wild-type littermate controls. All Hpse2 mutants have a growth retardation phenotype and die within a month after birth. Comprehensive blood chemistry and urinalysis indicate that Hpse2 mutants have renal dysfunction and malnutrition. We provide evidence that transforming growth factor beta (Tgfβ) signaling is attenuated at birth. However, Tgfβ activity is significantly enhanced at later stages when the urological phenotype is severe, and the mutant bladders have accumulated excessive amount of fibrotic tissue. Together, these findings strongly suggest that Hpse2 is a causative gene of human UFS and further uncover unexpected roles of Hpse2 in bladder physiology, tissue remodeling and Tgfβ signaling.
Introduction
Dysfunctional urination including urinary incontinence (UI) is a common debilitating disease from pediatric patients to senior citizens. The underlying molecular and cellular basis, however, remains elusive and poorly understood. Urofacial syndrome (UFS, aka Ochoa syndrome) is a rare autosomal recessive birth defect with severe UI phenotype (1). A better understanding of the etiology of UFS may therefore yield clues about the pathophysiology of UI.
Clinical symptoms of UFS are largely confined to the urinary tract with an exception of the unusual facial expression, i.e. patients appear to grimace when they smile (1,2). As such, UFS is an ideal disease model to investigate molecular biology and physiology of the urinary tract. Major urological features include UI, frequency, urgency and an enlarged urinary bladder (megacystis). Radiological investigations demonstrate urinary retention and occasionally vesicoureteric reflux and hydronephrosis without any detectable anatomical obstruction. Cystometry indicates high intravesical pressures with detrusor-sphincter dyssynergia. Many patients also experience repeated episodes of urinary tract infections (UTI) and renal dysfunction.
Genetic characterization of multiple UFS families has identified Heparanase 2 (HPSE2) as the first candidate disease gene (3,4). Both alleles of HPSE2 are mutated in the affected patients, consistent with the autosomal recessive nature of the disease. At least 12 mutations are reported so far from patients of diverse ethnicities (3–6). These mutations spread across all 12 exons of HPSE2 gene locus that spans over 700 kb of the human genome. Most of them are non-sense and deletion mutations with an exception of one missense mutation. Clinical features are almost indistinguishable among patients harboring these mutations, predicting that all of these are functional null mutations. HPSE2 protein sequence is similar to heparanase 1 (HPSE1), a heparan sulfate (HS)-degrading enzyme (7). Despite sequence similarity, HPSE2 has no detectable enzymatic activity; and it functions as an endogenous inhibitor of HPSE1 (7).
Leucine-rich repeats and immunoglobulin-like domains-2 (LRIG2) is the second candidate UFS gene (8). LRIG2 is a member of the conserved LRIG-family of transmembrane proteins that modulate a variety of signal pathways (9). HPSE2 and LRIG2 proteins appear to colocalize with a neuronal marker, β3-tubulin, in human bladder (2,8), implying their potential involvement in neuronal modulation. Surprisingly, Lrig2 mutant mice with an insertion mutation have normal life spans with no detectable phenotype (9). A second Lrig2 mutant allele exhibits a mild and transient growth retardation phenotype as well as a slight increase in spontaneous mortality rates (10), but the proposed function of Lrig2 in urination is yet to be determined.
An estimated 64% of families affected by classical UFS have mutations in HPSE2 and 21% have mutations in LRIG2 (8). Most of these mutations cause frame shift, truncation or non-sense mediated RNA decay, suggesting that UFS patients may suffer from the loss of HPSE2 or LRIG2. A major remaining question is whether mutation of either gene is sufficient to cause the disease. Here we report for the first time that deletion of Hpse2 but not Lrig2 causes UFS-like urological phenotypes in mice. Our findings strongly suggest that HPSE2 is a causative gene of UFS. Molecular examination reveals an unexpected role of Hpse2 in the Tgfβ signal pathway as well as pathological remodeling of the bladder. Together, these findings begin to shed light on the molecular basis of bladder pathophysiology.
Results
Hpse2 is selectively enriched in the bladder
The prominent urologic phenotype observed in UFS patients prompted us to examine whether Hpse2 expression is enriched in the bladder. Using a real time quantitative polymerase chain reaction (PCR) (RT-qPCR) assay (Fig. 1), we measured mouse Hpse2 mRNA level and its tissue distribution pattern. At postnatal day 30 (P30), high levels of Hpse2 transcripts were detected in bladder, large and small intestines (Fig. 1A). Expression in other tissues, including kidney, skeletal muscle, rectum, heart and brain, was significantly lower or nearly undetectable. As a comparison, significantly higher levels of Hpse1 transcripts were also detected in bladder, rectum and large intestine (Fig. 1B). Examination of the temporal gene expression pattern revealed that levels of both Hpse1 and Hpse2 transcripts in the bladder were relatively low at birth but reached their maximal levels at P30 and persisted in aged bladder (Fig. 1C). Further analysis indicated that Hpse2 was enriched in bladder detrusor (Fig. 1D), similar to the human HPSE2 distribution pattern (3). Conversely, Hpse1 expression was higher in urothelial tissue than in detrusor tissue. This restricted expression pattern of Hpse2 is consistent with the notion that Hpse2 has a tissue-specific role in the lower urinary tract.
Figure 1.

Tissue-specific expression patterns of Hpse1 and Hpse2. RT-qPCR analysis of Hpse2 (A) and Hpse1 (B) in tissues from P30 mice. Relative levels of expression are normalized against a Gapdh internal control. Bl, bladder, Kd, kidney; Skm1, hind leg skeletal muscle; Skm2, facial skeletal muscle; Ret, rectum; SI, small intestine; LI, large intestine; H, heart; Br, brain cortex. (C) Increased amounts of Hpse1 and Hpse2 transcripts during postnatal development of mouse bladder. (D) Hpse2 transcripts are detected predominately in bladder detrusor smooth muscle tissue (gray bar) while Hpse1 is preferentially detected in bladder mucosal tissue (open bar) as shown in the right panel. Tissue identities were confirmed based on expression of specific molecular markers (left panel), including Upk3a (an urothelium marker, U) and Mlck (a smooth muscle cell marker, SM).
Generation of Hpse2 knockout mice
Human HPSE2 and LRIG2 are candidate UFS genes (3,4,8). Cystometrogram (CMG) of Lrig2 knockout mice (9) was performed and failed to detect any obvious defect, suggesting that LRIG2 is unlikely to be a major causative gene (data not shown). To determine whether mutation of Hpse2 is sufficient to cause UFS-like phenotype in mice, we obtained a gene trap allele for Hpse2 from Texas A&M Institute for Genomic Medicine (TIGM, Hpse2Gt(OST411605)Lex) and confirmed germline transmission of the mutant allele (Fig. 2A and B). Sequence analysis demonstrated that the gene trap vector (VICTR48) was inserted into intron 6 of Hpse2, about 32.0 Kb from the splice donor site of exon 6 and 2.4 Kb upstream of the splice acceptor site of exon 7. Insertion of the gene trap vector resulted in truncation of the carboxyl-terminus of the Hpse2 protein. Instead of producing wild-type transcript, the gene trap allele is expected to generate two separate transcripts—the marker fusion transcript (MFT) that consists of a truncated 5′ region of Hpse2 and the neomycin (Neo) selection marker, and the OmniBank sequence tag (OST) fusion transcript (OFT). Molecular weights of the predicted MFT fusion protein and endogenous Hpse2 are comparable.
Figure 2.

Generation of Hpse2 knockout mice. (A) Schematic diagram of an insertional mutagenesis strategy to knockout Hpse2 gene. The gene trap vector, VICTR48, was inserted into intron 6 of Hpse2. The gene trap allele was expected to generate two separate transcripts, the MFT and the OmniBank sequence tag (OST) fusion transcript (OFT). Black arrows, genotyping oligos; gray arrowheads, RT-qPCR oligos. (B) Representative results of PCR genotyping using oligos indicated in A (black arrows). (C) RT-qPCR analysis of Hpse2 transcripts from P14 wild-type (wt) and mutant (mut) bladders using 5′ and 3′ primer sets indicated in A (gray arrowheads). *P < 0.05, Student's t-test. (D) Immunoblotting of tissue samples using Hpse2-specific antibody. Gapdh expression was used as an internal control.
To confirm that Hpse2 is successfully inactivated, we first examined expression levels of all predicted transcripts. Transcripts containing 5′-Hpse2 sequence were detected from mutant bladder tissue but were significantly reduced compared with wild-type controls (∼90% decrease, Fig. 2C). These transcripts most likely corresponded to MFT but not full length Hpse2 because 3′-Hpse2 transcripts were nearly undetectable using the Hpse2 3′-specific qPCR oligos (Fig. 2C). A faint signal was detected from the mutant bladder tissue using an Hpse2-specific antibody (Fig. 2D), which recognizes an N-terminal epitope of Hpse2 (7). Because the OFT RNA, i.e. the 3′-end of Hpse2 transcripts, was undetectable (Fig. 2C), this residual immunoreactivity was most likely the MFT fusion protein with unknown function. A similar truncation was identified from a patient with UFS (3), suggesting that the gene trap allele mimic a disease-causing mutation in human. Together, these findings suggest that the gene trap allele is a functional null mutation of Hpse2 (Hpse2−/−).
Growth defect and postnatal lethality of Hpse2 mutant mice
Heterozygous Hpse2 mutants had no detectable phenotype. Homozygous mutants, on the other hand, were noticeably smaller than littermate controls and died within 1 month of birth (Fig. 3A–C). During the first two weeks of postnatal life, Hpse2 mutants gained body weight but began to lose weight thereafter (Fig. 3B).
Figure 3.

Hpse2 mutants die within a month after birth. (A) A representative image of Hpse2 wild-type (wt) and mutant (mut) mice at P18. (B) A growth retardation phenotype of Hpse2 mutants. (C) The Kaplan–Meier plot of postnatal survival. P < 0.05, log-rank test. (D) Distended bladder (megacystis) of Hpse2 mutant at P18 (∼90%, n = 28). (E) Hpse2 mutants have significantly decreased albumin and glucose (GLU) levels but increased alkaline phosphatase (ALP) levels in blood samples. (F) Proteinuria was detected in Hpse2 mutants. ALB, albumin; CRE, creatinine; A/C, ALB and CRE ratio. *P < 0.05, Student's t-test.
Gross inspection of Hpse2 mutants revealed that nearly 90% of Hpse2 homozygous mutants had distended bladders (megacystis) (Fig. 3D, n = 28). This is consistent with the clinical presentation of UFS. Comprehensive blood chemistry revealed a significant decrease in blood albumin and glucose levels and an increase in alkaline phosphatase level in the mutants (Fig. 3E and Table 1). Urinalysis indicated that Hpse2 mutants had a significantly elevated albumin content (14.1 ± 2.7 mg/l in mutant versus 6.5 ± 0.8 mg/l in littermate controls) and a decreased urine creatinine level (Fig. 3F). The urine albumin/creatinine ratio, an indicator of glomerular permeability and renal dysfunction, was significantly increased from 13.9 ± 1.9 to 34.8 ± 5.9 mg/g. Mutant kidneys were significantly smaller but otherwise grossly normal (data not shown). We did not detect any retrograde labeling of ureter or kidney, a sign of vesicoureteral reflux, from bladder infused with methylene blue. Analysis of serial histological sections indicated that the mutant kidneys had comparable total glomeruli counts despite smaller size. Periodic acid–Schiff staining did not reveal any obvious glomerulus defect (data not shown). Because of high incidence of UTI of UFS patients, we also cultured urine and bladder homogenate on MacConkey agar plate and did not detect any significant difference of bacterial colony formation between control and Hpse2 mutants. Expression of an inflammatory marker gene Cox2 was not altered either. Taken together, the exact cause of proteinuria phenotype remains to be defined but renal dysfunction likely contributed to the lethality phenotype of Hpse2 mutants. In addition, malnutrition and digestive tract defect were additional possible causative factors because of the gross reduction of intestinal contents in the mutants (data not shown).
Table 1.
Blood chemistry
| ALB (g/dl)* | ALP (U/l)* | ALT (U/l) | AMY (U/l) | BUN (mg/dl) | Ca2+ (mg/dl) | |
|---|---|---|---|---|---|---|
| WT | 3.5 ± 0.1 | 290.5 ± 23.9 | 57.5 ± 10.0 | 797.7 ± 25.7 | 18.3 ± 2.0 | 12.3 ± 0.3 |
| MUT | 3.0 ± 0.2 | 453.0 ± 30.9 | 39.0 ± 2.8 | 881.4 ± 84.9 | 15.4 ± 1.2 | 12.1 ± 0.2 |
| GLOB (g/dl) | GLU (mg/dl)* | Na+ (mmol/l) | PHOS (mg/dl) | TBIL (mg/dl) | TP (g/dl) | |
| WT | 1.5 ± 0.1 | 323.3 ± 30.3 | 150.5 ± 1.0 | 13.1 ± 1.1 | 0.3 ± 0.0 | 5.0 ± 0.1 |
| MUT | 1.8 ± 0.2 | 224.2 ± 27.6 | 147.4 ± 1.9 | 11.5 ± 0.4 | 0.3 ± 0.0 | 4.9 ± 0.1 |
ALB, albumin; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AMY, amylase; BUN, blood urea nitrogen; Ca2+, calcium; GLOB, globulin; GLU, glucose; Na+, sodium; Phos, phosphorus; TBIL, total bilirubin; TP, total protein.
Comprehensive blood chemistry of wild-type (WT) and Hpse2 mutants (MUT) at P15–18.
*P < 0.05, Student's t-test.
Dysfunctional urination of Hpse2 mutants
Whole mount evaluation of urethra after India ink injection did not detect any obvious narrowing of the mutant urethra (data not shown), suggesting that the megacystis phenotype (Fig. 3D) is less likely due to physical or anatomical obstruction. Instead, Hpse2 mutants may have functional obstructions including possible neurological defect. To access the urinary functions, we first used the voided stain on paper (VSOP) assay to examine spontaneous micturition pattern of unrestrained animals (11,12). Urine spots of Hpse2 mutants were significantly smaller and more frequent than that of age-matched littermate controls (Fig. 4A–C), suggesting that Hpse2 mutants have UI phenotype. To further evaluate this observation using an independent assay, we analyzed bladder contractile force during micturition cycles based on the CMG (11,13). The 2-week-old wild-type control mice (P14–P18) showed a periodicity (2.73 ± 0.89 min/cycle) of an alternating low-pressure non-voiding phase (basal pressure, 13.7 ± 2.7 cm H2O) and the high-pressure voiding phase (voiding pressure, 21.7 ± 1.70 cm H2O) (Fig. 4D, n = 5). Hpse2 mutant, however, leaked constantly and had much higher resting intravesical pressure (basal pressure, 27.8 ± 1.32 cm H2O) and maximum intravesical pressure (39.8 ± 1.92 cm H2O) (Fig. 4D, n = 8). Occasionally, mutants would cease to leak and displayed a concurrent steady increase in intravesical pressure until the reappearance of leaking/voiding. In all Hpse2 mutants examined, the CMG pattern was highly irregular and the sharp spikes of acute increase of intravesical pressure were blunted. Hpse2 mutants thus exhibit the UFS-like urination defect.
Figure 4.

Voiding dysfunction of Hpse2 mutants. (A–C) Representative images (A) of the VSOP, which were quantified in (B) and (C). Quantifications of VSOP. (D) CMG of P14 wt (n = 3) and Hpse2 mutants (n = 5). y-axis, water pressure (cm); x-axis, time in minutes. Color codes on the x-axis indicate periods of voiding (light gray), non-voiding (black) and leaking (dark gray).
Hpse2 is required for cell proliferation but not urethelial cell fate determination
The growth retardation phenotype of Hpse2 mutants suggested a possible cell proliferation defect. We therefore used Ki67 as well as the mitotic marker phospho-histone H3 (p-HH3) to interrogate cell proliferation rates of the bladder tissues (Fig. 5, data not shown). To distinguish different tissue layers, we co-immunostained bladder with cell proliferation markers and cell-type specific markers including Uroplakin 3a (Upk3a) and Smooth Muscle Actin (Sma), which label the urothelial and detrusor smooth muscle cells, respectively. Quantitative evaluation of P18 and newborn animals indicated that proliferation rates of cells within all three bladder tissue layers, namely urothelium, lamina propria and detrusor smooth muscle, were significantly reduced in the mutants (Fig. 5E and data not shown), demonstrating that Hpse2 is important for normal cell proliferation.
Figure 5.
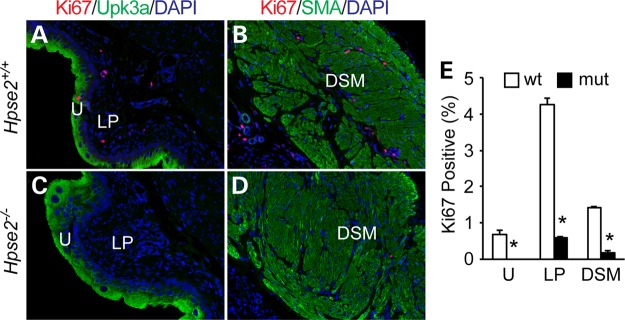
Hpse2 mutants have a reduced rate of cell proliferation. (A–D) Wild-type (A and B) and Hpse2 mutant, (C and D) p18 bladders were stained with a proliferation marker Ki67 (red), urothelial marker Upk3a (A and C, green) and smooth muscle marker SMA (B and D, green). Blue, DAPI counter staining. U, Urothelium; LP, lamina propria; DSM, detrusor smooth muscle. (E) Quantitative analysis of results shown in A–D. Student's t-test, *P < 0.05.
Distribution patterns of the Upk3a and Sma-positive cells were comparable between Hpse2 mutants and controls (Fig. 5A–D), suggesting that the mucosal epithelium and detrusor smooth muscle tissue types were specified in the absence of Hpse2. To further confirm this observation, we examined the urothelium using several independent cell-type specific molecular markers (Fig. 6). Comparable numbers of Ck5-positive basal cells were detected in Hpse2 mutant bladders (Fig. 6). Intermediate cells, which are positive for the p63 transcription regulator but negative for Ck5, were also detected at the similar frequency. Superficial umbrella cells were stained with Ck20 and there was no significant difference between control and Hpse2 mutants. Likewise, comparable expression patterns of the tight junction protein Z0-1 of umbrella cells were also observed (Fig. 6G and H). The overall size and shape of umbrella cells were similar between Hpse2 mutants and controls. These findings suggest that Hpse2 is required for normal cell proliferation but not tissue type determination.
Figure 6.

Tissue differentiation of Hpse2 mutant bladder. (A–F) Double staining of wild-type control (A–C) and Hpse2 mutants (D–F) with the basal cell marker Ck5 (red), umbrella cell markers Ck20 (green) and Upk3a (green), and a transcription regulator p63 (green), which is expressed in both basal and intermediate cells (arrows point to intermediate cells). Boxed regions in A–F are shown in a–f. (G) and (H). Whole mount immunostaining of tight junction protein Z0-1 (red) of wild-type (G) and Hpse2 mutant (H) bladders. Superficial umbrella cells, large cells that are in focus; basal and intermediate cells, small cells in the background that are out of focus plane. DAPI counter staining (blue) is shown in all panels except a and d.
Aberrant Tgfβ signaling and pathological remodeling of Hpse2 mutant bladders
Hpse2 mutant bladders were palpably stiffer than controls. We, therefore, examined tissue collagen content using the Sirius Red staining assay (Fig. 7). Wild-type bladder tissue had weak or no staining at birth, suggesting minimal collagen deposition in the newborn immature bladder (Fig. 7A and B). However, strong red signal was detected at P18, particularly within the sub-urothelial region (Fig. 7C and D). Minimal amount of signal was observed within the interstitial region of the detrusor smooth muscle layer. While the staining pattern of Hpse2 mutants was similar to controls at birth, higher levels of collagen deposition were detected from mutants at P18. In particular, aberrantly strong staining was observed within the interstitium of the detrusor layer (Fig. 7H), indicating pathological remodeling of the mutant bladder tissue.
Figure 7.
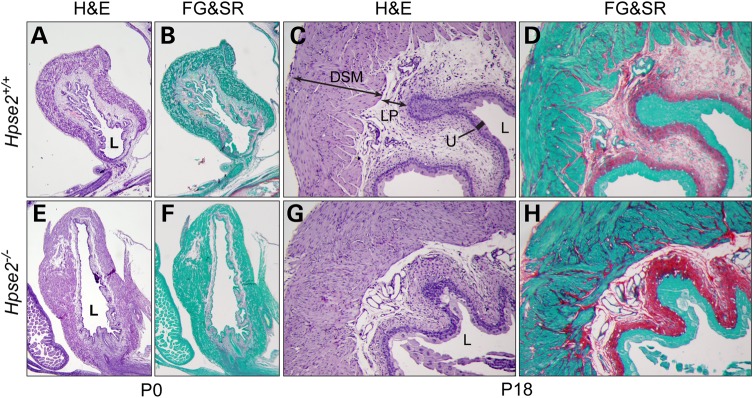
Hpse2 mutant bladders have excessive fibrotic tissue. Adjacent bladder sections of wild-type (A–D) and Hpse2 mutants (E–H) were stained with hematoxylin/eosin (H&E) and Sirius red/fast green stain (FG&SR) as indicated. An increased amount of collagen deposition (red) was observed in P18 mutant bladders (compare H and D) but not newborn bladders (compare F and B). DSM, detrusor smooth muscle; LP, lamina propria; U, urothelium; L, lumen.
HS binds and potentiates Tgfβ signaling (14), which plays a major role in pathological remodeling after bladder outlet obstruction (15), and in parturition-induced stress UI (16). We therefore investigated whether Tgfβ signaling was affected in the absence of Hpse2 (Fig. 8). Immunoblot indicated that phosphorylation level of Smad2 (p-Smad2), a downstream effector of Tgfβ signaling, was reduced in Hpse2 mutant bladder tissue at P6 (Fig. 8A). However, a significant increase in p-Smad2 levels was detected at P18, a stage when mutant bladders had excessive amounts of fibrotic tissue. Consistently, expression of a Tgfβ downstream target gene, connective tissue growth factor (Ctgf), was also significantly reduced at P6 but increased at P18 bladder (Fig. 8B). Thus, aberrant Tgfβ signaling may contribute to the pathological tissue remodeling of Hpse2 mutants.
Figure 8.
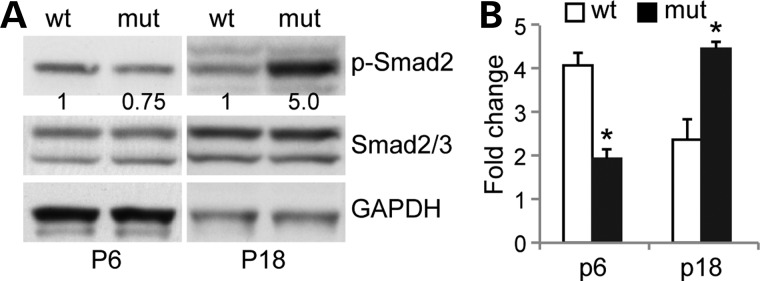
Abnormal Tgfβ signaling of Hpse2 mutant bladder. (A) Immunoblotting of tissue samples from P6 and P18 bladders using phospho-specific Smad2 (p-Smad2), Smad2/3 and Gapdh antibodies. Numbers underneath the top panels indicated relative signal density of p-Smad2 after normalization using both Gapdh and Smad2/3. (B) RT-qPCR analysis of Ctgf transcripts in wild-type (wt) and Hpse2 mutants (mut) at postnatal day 6 (P6) and P18. *P < 0.05, Student's t-test.
Discussion
A possible genetic basis of UI has been suspected but few genes have been identified or implicated (1,2). We show here that Hpse2 is a critical regulator and causative gene of dysfunctional urination and report the first mouse disease model of human UFS. These findings provide a starting point to dissect the pathophysiology of voiding dysfunction.
HPSE2 and LRIG2 are candidate disease genes identified from multiple UFS patients of diverse ethnic backgrounds (3,4,8). Our characterizations of mouse mutants harboring mutations of either one of these genes suggest that loss-of-function of Hpse2 but not Lrig2 is sufficient to cause the UFS-like phenotypes in mouse. In contrast to Lrig2 mutants, which do not have detectable urological phenotypes, Hpse2 mutants have enlarged bladders. This megacystis phenotype occurs in both sexes, suggesting that the defect is independent of the dimorphic features of the urethra. Consistent with CMG findings of patients with UFS (2), Hpse2 mutants have an abnormally high resting and voiding intravesical pressures. Despite of this, we did not detect any obvious sign of vesicoureteral reflux and hydronephrosis in Hpse2 mutants, which are believed to cause renal damage of human UFS patients. Comprehensive blood chemistry and urinalysis indicate that Hpse2 mutants indeed have dysfunctional renal system. Hpse2 is a de facto inhibitor of Hpse1 enzymatic activity (7). Reminiscent to Hpse2 knockout mice, Hpse1 transgenic mice also exhibit renal phenotypes including proteinuria (17). Therefore, a potential increase of Hpse1 enzymatic activity in Hpse2 mutants may contribute to the renal phenotypes associated with UFS patients.
We show here for the first time that Hpse2 mutant bladders undergo pathological fibrotic remodeling. Bladder tissue fibrosis is associated with a number of the lower urinary tract symptoms and is a major cause of the late stage bladder dysfunction. For instance, pediatric patients with non-compliant bladders, regardless of the neurogenic or non-neurogenic etiology, have increased connective tissue infiltration within the detrusor smooth muscle bundles and abnormal collagen deposition (18), similar to that found in Hpse2 mutants. In the rabbit models of the atherosclerosis-induced chronic bladder ischemia and hypercholesterolemia, animals develop severe bladder fibrosis and non-compliance (19). Tissue fibrosis and increased Tgfβ signaling are also observed in rat models of parturition-induced stress UI (16,20). Our findings suggest that Hpse2 may modulate tissue remodeling via the Tgfβ signal pathway because p-Smad2 levels and expression of a Tghβ downstream target gene Ctgf are significantly enhanced in Hpse2 mutants. Surprisingly, activity of the Tgfβ signal pathway is attenuated at birth. Hpse2 is highly expressed in both developing and mature bladders but it is unclear what causes such dichotomous responses in Tgfβ signaling in the absence of Hpse2. It is possible that Hpse2 may have stage-specific activities. On the other hand, it is also possible that over time high intravesical pressure of Hpse2 mutants damages bladder tissue and causes pathological fibrotic remodeling. Indeed, we have observed excessive fibrotic tissue formation of Hpse2 mutant bladders at late but not early stages. We suspect that aberrant Tgfß signaling together with the persistent high intravesical pressure cause a rapid deterioration of urinary tract functions in Hpse2 mutants, thereby contributing to the pathogenesis of UFS.
Potential neurological impairment of UFS has been postulated because of the unusual facial expression pattern. However, the underlying mechanism remains to be determined. In addition, it remains to be determined whether Hpse2-dependent program is important for maturation and contractile phenotype of smooth muscle cells. Future studies using the Hpse2 mutant mouse model may shed light on the neurogenic and myogenic basis of micturition as well as facial grimace upon smiling.
Materials and Methods
Mouse lines
Hpse2−/− mice were generated using an insertional mutagenesis strategy. The gene trap vector, VICTR48 from Texas A&M Institute for Genomic Medicine (TIGM, Hpse2Gt(OST411605)Lex), was inserted into intron 6 of Hpse2. Insertion site and germline transmission of the mutant allele were confirmed by direct sequencing and PCR analyses. Sequences of PCR genotyping oligos are 5′-TGT GGC CAG TAT TTT TCT TTA TTT G-3′, 5′-GAG CTG TTT CAC AGA GTG CC-3′ and 5′-CCA ATA AAC CCT CTT GC AGT TGC-3′. Mice were maintained on a C57BL/6 genetic background. All animal studies were performed according to protocols reviewed and approved by the Institutional Animal Care and Use Committee at the Children's Hospital Boston.
Weight and survival analysis
Newborn mutants and littermate controls were weighed every 2 days for a period of 30 days. Data are presented as mean ± SEM. These data were plotted using Microsoft Excel. In addition, survival of mutant mice was monitored daily and the results were analyzed using Kaplan–Meier plots and the log-rank test. Paired two-tailed Student's t-test was used to analyze body weight. P < 0.05 was considered to be statistically significant.
Gene expression analysis
Specific tissues were microdissected in diethyl pyrocarbonate treated phosphate buffered saline (PBS) under a microscope (Olympus SZX16). RNA was extracted using the Trizol extraction kit following the suggested protocol (Invitrogen). Total RNA was treated with the TURBO DNA-free™ kit (Invitrogen) and reverse-transcribed into cDNA using the SuperScript® III kit (Invitrogen). RT-qPCR analyses were performed using the SYBR Green method (Roche) on an ABI-7500 detector (Applied BioSystems). Relative gene expression levels were normalized to an internal control Gapdh. Gene-specific oligos were as follows: Hpse1 transcript forward, 5′-GGC AAT GAG CCC AAC AGT TT-3′; reverse, 5′-TTG GTA GCG ATG CGT CCA TT-3′; Hpse2 5′ transcript forward, 5′-GGG CAA AAG GAC GGA TTT C-3′; reverse, 5′-TGT CCA AGG CAA CAT CAC TC-3′; Hpse2 3′ transcript forward, 5′-CCA CAA CTA TGT CCG TGG CT-3′; reverse, 5′-GAG CTG CAC TGA CTT GGA CT-3′; Ctgf transcript forward, 5′-CTC CAC CCG AGT TAC CAA TG-3′; reverse, 5′-TGG CGA TTT TAG GTG TCC G-3′. Data are presented as mean ± SEM. P < 0.05 was considered to be statistically significant using Student's t-test.
For protein immunoblotting, tissues were homogenized in radioimmunoprecipitation assay buffer (50 mM Tris–HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS). Furthermore, 100 g total protein lysate were subjected to western immunoblotting. Primary antibodies used include anti-Hpse2 (1:1000 from Dr Israel Vlodavsky) (7), p-Smad2 (1:1000, Cell signaling), Smad2/3 (1:1000, Cell signaling) and Gapdh (1:2000, Santa Cruz).
Histology and immunofluorescence staining analyses
Bladders were dissected, embedded into either OCT or paraffin, and sectioned at 4–10 µM as described previously (21–23). Paraffin sections were stained with hematoxylin and eosin (H&E, Thermo Scientific) and/or Masson Trichrome blue (Histology Core in Beth Israel Deaconess Medical Center) and/or Sirius red and fast green (FG&SR, Sigma) using standard procedures. Cryostat sections and paraffin sections were subjected to immunofluorescence staining. Anti-Ki67 (Abcam), Upk3a (Grifted from Dr TT Sun), Sma (Sigma), Ck5 (Abcam), Ck20 (Thermo Scientific) and p63 (Santa Cruz) primary antibodies were incubated at 1:50, 1:200, 1:500, 1:500, 1:50 and 1:200, respectively, in blocking solution at 4°C overnight. Sections were then stained with donkey anti-mouse or anti-rabbit secondary antibodies (1:200 in blocking solution) at room temperature for 1 h. Images were captured using a Zeiss fluorescence microscope. Results of three sections from each bladder were counted. The cell proliferation rate was calculated by dividing number of proliferating cells with total cell number of each bladder tissue, namely, urothelium, lamina propria and smooth muscle, and analyzed by Student's t-test. P < 0.05 was considered to be significant.
Urine and blood test
Urine samples were collected from wild-type control and Hpse2 mutant mice at P15–18. After centrifugation, 40 µl urine was loaded into urine capillary tubes and analyzed using DCA™ System (Siemens) to measure albumin and creatinine levels by DCA Vantage Analyzer (Siemens). Blood samples were collected using heparin-coated tubes from wild-type and Hpse2 mutant mice at P15–18. Blood tests were performed immediately (within 1.5 h) using VetScan® comprehensive Diagnostic Profile (Abaxis Veterinary Diagnostics). Briefly, 150 µl blood was loaded into blood capillary disk to measure 12 chemical parameters by VetScan® VS2 Analyzer (Abaxis). The resulting data were analyzed using Student's t-test. P < 0.05 is considered to be significant.
VSOP analysis
VSOP was performed as described previously (11,12). Briefly, Whatman grade 1 filter paper (catalog no. 1001-917) was placed at the bottom of cages. Mice were placed individually in each cage for 3 h and the urine drops were collected onto the filter paper as voided stain. At the end the assay period, filter paper was retrieved and urine spots left behind were imaged under ultraviolet light. All pictures were converted to binary images so that size and number of urine spots can be analyzed using ImageJ. Total area and number of void spots from littermate control and Hpse2 mutants were compared and subjected to Student's t-test. P < 0.05 is considered to be significant.
CMG analysis
CMG was performed as previously described with minimal modifications (11). Briefly, cystometry was performed under general anesthesia with urethane (1.8 g/kg, Sigma). Bladders were surgically exposed, and a 26-gauge needle was introduced into the dome and connected via a three-way adapter to a fluid-filled pressure line at one end and an injection pump (Harvard Apparatus) at the other. The pressure line was connected to a physiologic pressure transducer (MLT844 AD Instruments), and the bladder was filled with PBS at a constant rate (20.5 μl/min). The output signal was amplified by BridgeAmp ML221 (AD Instruments) and recorded using the LabChart system V6 (AD instruments).
Funding
This work was supported by grants from NIH/NIDCR (1R01DE019823, X.L.) and NIH/NIDDK (1R01DK091645-01A1, X.L.).
Acknowledgements
We would like to thank Drs Zarine Balsara and Jingying Wang for their critical input on the manuscript; Drs Valerie Schumacher, YeunGoo Chung and Ms Debra Frank for their technical assistance. We thank Dr Lisa Goodrich for providing us with Lrig2−/− mice. C.M.G. is an America Urology Association Foundation Research Scholar.
Conflict of Interest statement. The authors declare no conflict of interest.
References
- 1.Ochoa B. Can a congenital dysfunctional bladder be diagnosed from a smile? The Ochoa syndrome updated. Pediatr Nephrol. 2004;19:6–12. doi: 10.1007/s00467-003-1291-1. [DOI] [PubMed] [Google Scholar]
- 2.Woolf A.S., Stuart H.M., Roberts N.A., McKenzie E.A., Hilton E.N., Newman W.G. Urofacial syndrome: a genetic and congenital disease of aberrant urinary bladder innervation. Pediatr Nephrol. 2014;29:513–518. doi: 10.1007/s00467-013-2552-2. [DOI] [PubMed] [Google Scholar]
- 3.Daly S.B., Urquhart J.E., Hilton E., McKenzie E.A., Kammerer R.A., Lewis M., Kerr B., Stuart H., Donnai D., Long D.A., et al. Mutations in HPSE2 cause urofacial syndrome. Am J Hum Genet. 2010;86:963–969. doi: 10.1016/j.ajhg.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pang J., Zhang S., Yang P., Hawkins-Lee B., Zhong J., Zhang Y., Ochoa B., Agundez J.A., Voelckel M.A., Gu W., et al. Loss-of-function mutations in HPSE2 cause the autosomal recessive urofacial syndrome. Am J Hum Genet. 2010;86:957–962. doi: 10.1016/j.ajhg.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al Badr W., Al Bader S., Otto E., Hildebrandt F., Ackley T., Peng W., Xu J., Li J., Owens K.M., Bloom D., et al. Exome capture and massively parallel sequencing identifies a novel HPSE2 mutation in a Saudi Arabian child with Ochoa (urofacial) syndrome. J Pediatr Urol. 2011;7:569–573. doi: 10.1016/j.jpurol.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahmood S., Beetz C., Tahir M., Imran M., Mumtaz R., Bassmann I., Jahic A., Malik M., Nurnberg G., Hassan S., et al. First HPSE2 missense mutation in urofacial syndrome. Clin Genet. 2012;81:88–92. doi: 10.1111/j.1399-0004.2011.01649.x. [DOI] [PubMed] [Google Scholar]
- 7.Levy-Adam F., Feld S., Cohen-Kaplan V., Shteingauz A., Gross M., Arvatz G., Naroditsky I., Ilan N., Doweck I., Vlodavsky I. Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. J Biol Chem. 2010;285:28010–28019. doi: 10.1074/jbc.M110.116384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stuart H.M., Roberts N.A., Burgu B., Daly S.B., Urquhart J.E., Bhaskar S., Dickerson J.E., Mermerkaya M., Silay M.S., Lewis M.A., et al. LRIG2 mutations cause urofacial syndrome. Am J Hum Genet. 2013;92:259–264. doi: 10.1016/j.ajhg.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Del Rio T., Nishitani A.M., Yu W.M., Goodrich L.V. In vivo analysis of Lrig genes reveals redundant and independent functions in the inner ear. PLoS Genet. 2013;9:e1003824. doi: 10.1371/journal.pgen.1003824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rondahl V., Holmlund C., Karlsson T., Wang B., Faraz M., Henriksson R., Hedman H. Lrig2-deficient mice are protected against PDGFB-induced glioma. PLoS One. 2013;8:e73635. doi: 10.1371/journal.pone.0073635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu W., Ackert-Bicknell C., Larigakis J.D., MacIver B., Steers W.D., Churchill G.A., Hill W.G., Zeidel M.L. Spontaneous voiding by mice reveals strain-specific lower urinary tract function to be a quantitative genetic trait. Am J Physiol Renal Physiol. 2014;306:F1296–F1307. doi: 10.1152/ajprenal.00074.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugino Y., Kanematsu A., Hayashi Y., Haga H., Yoshimura N., Yoshimura K., Ogawa O. Voided stain on paper method for analysis of mouse urination. Neurourol Urodyn. 2008;27:548–552. doi: 10.1002/nau.20552. [DOI] [PubMed] [Google Scholar]
- 13.Andersson K.E., Soler R., Fullhase C. Rodent models for urodynamic investigation. Neurourol Urodyn. 2011;30:636–646. doi: 10.1002/nau.21108. [DOI] [PubMed] [Google Scholar]
- 14.Lyon M., Rushton G., Gallagher J.T. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J Biol Chem. 1997;272:18000–18006. doi: 10.1074/jbc.272.29.18000. [DOI] [PubMed] [Google Scholar]
- 15.Metcalfe P.D., Wang J., Jiao H., Huang Y., Hori K., Moore R.B., Tredget E.E. Bladder outlet obstruction: progression from inflammation to fibrosis. BJU Int. 2010;106:1686–1694. doi: 10.1111/j.1464-410X.2010.09445.x. [DOI] [PubMed] [Google Scholar]
- 16.Li G.Y., Cui W.S., Zhou F., Gao Z.Z., Xin H., Liu T., Li W.R., Gong Y.Q., Bai G.Y., Guo Y.L., et al. Pathology of urethral fibromuscular system related to parturition-induced stress urinary incontinence and TGF-beta1/Smad pathway. Mol Cel Biochem. 2012;364:329–335. doi: 10.1007/s11010-012-1234-x. [DOI] [PubMed] [Google Scholar]
- 17.Zcharia E., Metzger S., Chajek-Shaul T., Aingorn H., Elkin M., Friedmann Y., Weinstein T., Li J.P., Lindahl U., Vlodavsky I. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. FASEB J. 2004;18:252–263. doi: 10.1096/fj.03-0572com. [DOI] [PubMed] [Google Scholar]
- 18.Deveaud C.M., Macarak E.J., Kucich U., Ewalt D.H., Abrams W.R., Howard P.S. Molecular analysis of collagens in bladder fibrosis. J Urol. 1998;160:1518–1527. [PubMed] [Google Scholar]
- 19.Azadzoi K.M., Tarcan T., Siroky M.B., Krane R.J. Atherosclerosis-induced chronic ischemia causes bladder fibrosis and non-compliance in the rabbit. J Urol. 1999;161:1626–1635. [PubMed] [Google Scholar]
- 20.Lin G., Shindel A.W., Banie L., Deng D., Wang G., Hayashi N., Lin C.S., Lue T.F. Molecular mechanisms related to parturition-induced stress urinary incontinence. Eur Urol. 2009;55:1213–1222. doi: 10.1016/j.eururo.2008.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang C., Gargollo P., Guo C., Tang T., Mingin G., Sun Y., Li X. Six1 and Eya1 are critical regulators of peri-cloacal mesenchymal progenitors during genitourinary tract development. Dev Biol. 2011;360:186–194. doi: 10.1016/j.ydbio.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo C., Sun Y., Guo C., MacDonald B.T., Borer J.G., Li X. Dkk1 in the peri-cloaca mesenchyme regulates formation of anorectal and genitourinary tracts. Dev Biol. 2014;385:41–51. doi: 10.1016/j.ydbio.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo C., Sun Y., Zhou B., Adam R.M., Li X., Pu W.T., Morrow B.E., Moon A., Li X. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J Clin Invest. 2011;121:1585–1595. doi: 10.1172/JCI44630. [DOI] [PMC free article] [PubMed] [Google Scholar]