

Abstract
Duchenne muscular dystrophy (DMD) is caused by mutations in the dystrophin gene that result in loss of the dystrophin–glycoprotein complex, a laminin receptor that connects the myofiber to its surrounding extracellular matrix. Utrophin, a dystrophin ortholog that is normally localized to the neuromuscular junction, is naturally upregulated in DMD muscle, which partially compensates for the loss of dystrophin. Transgenic overexpression of utrophin causes broad sarcolemma localization of utrophin, restoration of laminin binding and amelioration of disease in the mdx mouse model of DMD. We previously demonstrated that overexpression of sarcospan, a dystrophin- and utrophin-binding protein, ameliorates mdx muscular dystrophy. Sarcospan boosts levels of utrophin to therapeutic levels at the sarcolemma, where attachment to laminin is restored. However, understanding the compensatory mechanism is complicated by concomitant upregulation of α7β1 integrin, which also binds laminin. Similar to the effects of utrophin, transgenic overexpression of α7 integrin prevents DMD disease in mice and is accompanied by increased abundance of utrophin around the extra-synaptic sarcolemma. In order to investigate the mechanisms underlying sarcospan ‘rescue’ of muscular dystrophy, we created double-knockout mice to test the contributions of utrophin or α7 integrin. We show that sarcospan-mediated amelioration of muscular dystrophy in DMD mice is dependent on the presence of both utrophin and α7β1 integrin, even when they are individually expressed at therapeutic levels. Furthermore, we found that association of sarcospan into laminin-binding complexes is dependent on utrophin and α7β1 integrin.
Introduction
Loss of appropriate connections between the muscle cell membrane and laminin in the extracellular matrix (ECM) has emerged as a critical initiating event in many forms of muscular dystrophy and muscle-wasting disorders. ECM attachment to the membrane provides mechanical stability to the sarcolemma during muscle contraction (1) and may also contribute to lateral forces generated by muscle (2). Identification of mechanisms that restore cell surface–ECM connection and promote muscle growth has the potential to address a broad range of muscle-wasting disorders by preventing muscle degeneration and improving regeneration. There are three major laminin-binding adhesion complexes in skeletal muscle, which include the dystrophin–glycoprotein complex (DGC), the utrophin–glycoprotein complex (UGC) and α7β1D integrin. Genetic mutations that affect either the expression or ECM-binding function of these complexes cause many forms of muscular dystrophy (3).
The DGC is comprised of several integral and peripheral membrane protein subcomplexes: the dystroglycans, sarcoglycans and sarcospan (SSPN). The dystroglycans (α- and β-DG) are formed from post-translational processing of a single polypeptide to produce α- and β-subunits (4,5). α-DG is an extensively glycosylated, peripheral membrane proteoglycan that binds directly to ligands in the ECM and is anchored to the cell surface by interaction with its integral membrane partner, β-DG (5–12). The sarcoglycans (α-, β-, γ- and δ-SGs) are transmembrane glycoproteins that bind SSPN to form the SG–SSPN subcomplex, which further contributes to the stable attachment of α-DG to the surface membrane (13–15). Dystrophin is a large, 427-kDa protein that is localized to the intracellular face of the sarcolemma through interaction with β-DG and cytoplasmic F-actin (16–18). Duchenne muscular dystrophy (DMD) is caused by mutations in the dystrophin gene that result in loss of dystrophin protein and, consequently, the entire DGC from the sarcolemma (19–21). Laminin binding to the sarcolemma is severely reduced in the absence of the DGC, rendering the membrane susceptible to contraction-induced injury (1,10,22,23). It is well established from null and transgenic mouse models that the expression of the three laminin-binding complexes is coordinated in skeletal muscle, although the mechanisms that regulate this interplay are unknown and complicated by their concomitant regulation (24).
Although DMD patients and the mdx mouse model for this disease both have mutations in the dystrophin gene, pathological symptoms in dystrophic mice are attenuated by enhanced expression of utrophin (25) and α7β1D integrin (26,27). The UGC and α7β1D integrin are concentrated at neuromuscular and myotendenous junctions (NMJs and MTJs) in normal skeletal muscle, but their abundance at the extra-synaptic sarcolemma is slightly increased in mdx mice in response to dystrophin deficiency (28,29). Utrophin is structurally homologous to dystrophin and forms the UGC by interacting with the DGs and the SG–SSPN subcomplex (30–32). Dystrophin and utrophin bind to F-actin with similar affinities in a non-competitive manner (33). Mice lacking both dystrophin and utrophin develop severe, progressive muscular dystrophy similar to patients with DMD and die prematurely by 20 weeks of age whereas mdx mice have a near-normal lifespan (34,35). Forced overexpression of utrophin in mdx or mdx:utrophin-null mice ameliorates muscular dystrophy, revealing the important role of the UGC in restoring laminin binding and membrane stability during dystrophin deficiency (25,36–40).
α7β1 integrin, predominantly expressed in striated muscles, is a heterodimeric laminin receptor composed of α- and β-subunits that connects to the intracellular actin cytoskeleton via integrin-associated proteins (41–44). Mutations in human ITGA7 have been found in a rare form of congenital muscular dystrophy (45). α7 integrin-deficient mice exhibit mild myopathy, reduced force transmission and viscoelasticity in diaphragm muscle as well as developmental abnormalities of NMJ and MTJ structures (46–52). Forced overexpression of α7 integrin in utrophin-deficient mdx mice alleviates cardiac muscle degeneration and extends lifespan in double-null animals (27,53), and AAV-mediated expression of β1D integrin in mdx muscle ameliorates dystrophic pathology and increased utrophin at the sarcolemma (26). Genetic ablation of α7 integrin in mdx mice leads to an earlier onset of pathology, severely decreased lifespan (4 weeks) and increased severity of muscular dystrophy that is similar to patients with DMD (54,55).
SSPN overexpression in skeletal muscle of dystrophic mdx mice increases the levels of both utrophin and α7β1 integrin around the extra-synaptic sarcolemma (31,56). SSPN transgenes ameliorate mdx disease and restore laminin binding along with sarcolemma integrity, but the mechanism of SSPN-mediated ‘rescue’ remains unclear. We have established that SSPN is a core component of the utrophin– and dystrophin–glycoprotein complexes (14,31,57) and that SSPN-null muscle displays reduction in both of these complexes (52). Interestingly, we observed that α7β1 integrin levels increased in SSPN-null muscle in a compensatory manner (52). The structural similarity of SSPN with the tetraspanin superfamily of proteins led us to hypothesize that SSPN may associate with α7β1 integrin (52). We previously generated α7 integrin and SSPN double-knockout (DKO) mice and determined that DKO mice exhibited exacerbated myopathy, decreased protein levels of the DGC and UGC, aberrant Akt signaling and contraction-induced sarcolemmal damage in the diaphragm muscle (52). While single knockouts displayed reduced specific force activity relative to wild-type controls, the specific force of the DKO diaphragm muscle was further reduced in an additive manner (52). These findings demonstrate that α7 integrin and SSPN function cooperatively to stabilize the levels of the DGC and UGC at the sarcolemma. Coordinated expression of laminin-binding complexes in skeletal muscle is further evidenced by elevated levels of: (1) utrophin in response to α7 integrin deficiency (52), (2) α7 integrin in response to dystrophin deficiency (28), (3) α7 integrin in response to combined loss of utrophin and dystrophin (56) and (4) dystrophin in response to combined loss of utrophin and α7 integrin (58). While these molecular events are well documented, the ability of either utrophin or α7 integrin alone to ‘rescue’ muscular dystrophy is complicated by the findings that overexpression of one compensatory gene causes a concomitant increase in the other. In the current study, we demonstrate that expression of both α7 integrin and utrophin are required to compensate for the loss of dystrophin function and to prevention of muscular dystrophy in mice.
Results
While it is well documented that α7β1 integrin and utrophin are slightly elevated in DMD and mdx muscle and that further boosting their expression at the sarcolemma can fully compensate for loss of dystrophin, it is unclear whether α7β1 integrin and utrophin are both required for SSPN-induced amelioration of muscular dystrophy (24,28,29,34,35,52,54–56,58). Analysis of several lines of SSPN transgenic mdx mice revealed that overexpression of SSPN alone is sufficient to increase abundance of α7β1 integrin and utrophin in a manner that is dependent on the level of SSPN (31,52,56,59). As part of our long-term goal to develop SSPN-based therapies, we sought to investigate the individual contributions of α7β1 integrin and utrophin, induced by SSPN overexpression, to muscle development and disease.
Amelioration of DMD muscular dystrophy requires α7 integrin
To determine whether SSPN ‘rescue’ of muscular dystrophy in mdx mice is dependent on α7 integrin, we introduced the SSPN transgene, under the control of the human skeletal actin promoter, into mdx mice lacking the α7 integrin gene (mdx: α7−/− TG). A final cross between mdx: α7+/− TG and mdx: α7+/− mice was performed to produce mice possessing the SSPN transgene in the absence of dystrophin and α7 integrin. Because mdx:α7−/− mice die prematurely at 4 weeks of age (54,55), our analysis was conducted at 3 weeks of age. We found that 3-fold levels of SSPN overexpression, which has been shown to ameliorate mdx dystrophy (31,56), did not improve the viability, overall body size or kyphosis of 3-week-old mdx:α7−/− mice relative to mdx:α7−/− controls (Fig. 1A and B). In fact, mdx:α7−/− TG mice did not survive past 4 weeks of age, as observed in Kaplan–Meier survival curves (Fig. 1B).
Figure 1.

SSPN amelioration of mdx pathology is dependent on α7 integrin. (A) Photographs of EBD-injected mdx:α7 integrin-null (mdx:α7−/−) and SSPN transgenic mdx:α7 integrin-null (mdx:α7−/− TG) mice at 3 weeks of age. No improvement in the severe kyphosis and muscle wasting is observed with the overexpression of SSPN. (B) The viability of indicated genotypes is shown in the Kaplan–Meier survival curve. All mdx:α7-integrin-null and SSPN transgenic mdx:α7 integrin-null mice die by 28 days. Analysis included 20 animals per genotype. (C) Transverse quadriceps sections from 3-week-old indicated mice were stained for H&E to visualize histopathology (left). Middle panels display sections that were co-stained with laminin (red) and eMHC (green), and right panels are images from sections stained with laminin (green) to outline each myofiber in cryosections from EBD-injected mice (red). Newly regenerating fibers express high levels of eMHC, and membrane-damaged fibers are EBD positive. Bar, 50 μm. (D) Regeneration was quantified from central nucleation counts from H&E-stained transverse quadriceps sections from 3-week-old mdx (n = 8), SSPN transgenic mdx (mdx TG, n = 4), mdx:α7 integrin-null (mdx:α7−/−, n = 8) and SSPN transgenic mdx:α7 integrin-null (mdx:α7−/− TG, n = 4) mice. (E) To investigate myofibers actively undergoing regeneration, eMHC-positive fibers were quantified from laminin co-stained transverse quadriceps sections from 3-week-old mdx (n = 8), SSPN transgenic mdx (mdx TG, n = 4), mdx:α7 integrin-null (mdx:α7−/−, n = 8) and SSPN transgenic mdx:α7 integrin-null (mdx:α7−/− TG, n = 4) mice. (F) Membrane fragility was determined by quantifying EBD-positive fibers in transverse quadriceps cryosections stained with laminin from 3-week-old mdx (n = 7), SSPN transgenic mdx (mdx TG, n = 4), mdx:α7 integrin-null (mdx:α7−/−, n = 6) and SSPN transgenic mdx:α7 integrin-null (mdx:α7−/− TG, n = 4) mice. Data are presented as an average, and error bars represent standard deviation of the mean. Statistics calculated using ANOVAs combined with the Bonferroni correction. P-values are indicated on the plots.
Transgenic and control muscles were evaluated for overall degeneration/regeneration (central nucleation of myofibers), active regeneration of myofibers (marked by expression of embryonic myosin heavy chain or eMHC) and sarcolemma damage (using an Evan's blue dye in vivo tracer assay). Each of these biological parameters was performed and quantitated on transverse quadriceps sections (Fig. 1C–F; Fig. 2A and B). At 3 weeks of age, mdx and SSPN transgenic mdx (mdxTG) myofibers display very few myofibers (<1%) with central nucleation or marked with eMHC and EBD (Fig. 1C–F; Fig. 2A). mdx mice are at a very early, prenecrotic stage of disease and do not yet exhibit characteristic symptoms of muscular dystrophy. As previously documented, genetic removal of α7 integrin in mdx muscle exacerbates all measures of dystrophic pathology relative to mdx controls so that disease symptoms are prevalent even at 3 weeks (Fig. 1C–F) (55). Overexpression of SSPN in mdx:α7−/− mice does not alter the levels of active regeneration or degeneration (Fig. 1C–F). Interestingly, SSPN transgenic mdx:α7−/− mice exhibit increased overall central nucleation compared with mdx:α7−/− controls, whereas the levels of newly regenerating fibers remain unchanged between the two genotypes (Fig. 2B). During muscle regeneration, the nucleus is localized to the center of the myofiber between 30 and 130 days and then migrates to the muscle cell periphery (60,61). We speculate that the elevation of central nucleation in the SSPN transgenic mdx:α7−/− mice may indicate an earlier onset in pathology leading to greater numbers of overall central nucleation at 3 weeks of age relative to mdx:α7−/− controls. Alternatively, SSPN may cause increased spontaneous regeneration in the context of dystrophin and α7 integrin deficiency. The latter possibility is further supported by whole quadriceps staining of mdx:α7−/− double knockouts revealing widespread eMHC-positive fibers that are not localized to the areas of focal damage (compare with mdx:SSPN−/− in Fig. 2A and B). Our data demonstrate that SSPN-mediated amelioration of mdx dystrophic pathology requires α7 integrin.
Figure 2.

Active regeneration is unaffected in SSPN transgenic mdx:α7 integrin-null and mdx:utrophin-null mice. (A–C) Anti-laminin (red) and anti-embryonic myosin heavy chain (eMHC; green) antibodies were used to denote the sarcolemma and newly regenerated fibers in transverse whole quadriceps cryosections from: (A) mdx and mdx:SSPN-null (mdx:SSPN−/−) mice, (B) mdx:α7 integrin-null (mdx:α7−/−) and SSPN transgenic mdx:α7 integrin-null (mdx:α7−/− TG) mice and (C) mdx:utrophin-null (mdx:utr−/−) and SSPN transgenic mdx:utrophin-null (mdx:utr−/− TG) mice. The additional loss of SSPN in mdx mice results in an earlier onset of active regeneration compared with mdx mice at 3 weeks (A). Overexpression of SSPN in mdx:α7 integrin-null mice did not improve active regeneration compared with mdx:α7 integrin-null mice at 3 weeks (B). Overexpression of SSPN in mdx:utrophin-null mice did not improve active regeneration compared with mdx:utrophin-null mice at 6 weeks (C). Bar, 1000 mm.
Amelioration of DMD muscular dystrophy requires utrophin
To determine whether expression of utrophin is required for SSPN amelioration of mdx dystrophic pathology, SSPN transgenic mice were crossed with mdx mice lacking the utrophin gene (mdx:utr−/−) to produce mice overexpressing 3-fold levels of exogenous SSPN in dystrophin- and utrophin-deficient mice (mdx:utr−/− TG). Mice were analyzed at 6-weeks of age because this was the age that SSPN overexpression in mdx muscle was shown to ameliorate disease. Similar to mdx:α7−/− TG, SSPN overexpression did not improve the viability, overall body size or kyphosis of 6-week-old mdx:utr−/− mice (Fig. 3A and B). In fact, mdx:utr−/− TG mice did not survive past 20.5 weeks of age, resulting in Kaplan–Meier survival curves that were similar to mdx:utr−/− controls (Fig. 3B). Furthermore, quantification of biochemical markers of dystrophic pathology (central nucleation, embryonic MHC and Evans blue dye update) was similar between mdx:utr−/− TG and mdx:utr−/− quadriceps muscles at 6 weeks (Fig. 2C and 3C–F). At this age, muscles from mdx:utr−/− and mdx:utr−/− TG quadriceps display statistically significant more total and actively regenerating myofibers compared with mdx controls (Fig. 3C–E).
Figure 3.

SSPN amelioration of mdx pathology is dependent on utrophin. (A) Photographs of EBD-injected mdx, SSPN transgenic mdx (mdx TG), mdx:utrophin-null (mdx:utr−/−) and SSPN transgenic mdx:utrophin-null (mdx:utr−/− TG) mice at 6 weeks of age. No improvement in the severe kyphosis and muscle wasting is observed with the overexpression of SSPN. (B) The Kaplan–Meier survival curve displays the lifespan of mdx:utr−/− and mdx:utr−/− TG mice. All mdx:utr−/− die by 22 weeks, and all mdx:utr−/− TG mice die by 20.5 weeks. Twenty mdx:utr−/− TG and ten mdx:utr−/− animals were included in this study. (C) Transverse quadriceps sections from 6-week-old indicated mice were stained for H&E to visualize histopathology (left). Middle panels display sections that were co-stained with laminin (red) and eMHC (green), and right panels are images from sections stained with laminin (green) to outline each myofiber in cryosections from EBD-injected mice (red). Newly regenerating fibers express high levels of eMHC, and membrane-damaged fibers are EBD positive. Bar, 50 μm. (D) Regeneration was quantified from central nucleation counts from H&E-stained transverse quadriceps sections from 6-week-old mdx (n = 6), SSPN transgenic mdx (mdx TG, n = 8), mdx:utrophin-null (mdx:utr−/−, n = 4) and SSPN transgenic mdx:utrophin-null (mdx:utr−/− TG, n = 6) mice. (E) To investigate myofibers actively undergoing regeneration, eMHC-positive fibers were quantified from laminin co-stained transverse quadriceps sections from 6-week-old mdx (n = 6), SSPN transgenic mdx (mdx TG, n = 8), mdx:utrophin-null (mdx:utr−/−, n = 4) and SSPN transgenic mdx:utrophin-null (mdx:utr−/− TG, n = 4) mice. (F) Membrane fragility was determined by quantifying EBD-positive fibers in transverse quadriceps cryosections stained with laminin from 6-week-old mdx (n = 6), SSPN transgenic mdx (mdx TG, n = 8), mdx:utrophin-null (mdx:utr−/−, n = 4) and SSPN transgenic mdx:utrophin-null (mdx:utr−/− TG, n = 4) mice. Data are presented as an average, and error bars represent standard deviation of the mean. Statistics calculated using ANOVAs combined with the Bonferroni correction. P-values are indicated on the plots.
Stable integration of sarcospan into an adhesion complex is dependent on both utrophin and α7 integrin
We used agarose-bound succinylated wheat germ agglutinin (sWGA) lectin chromatography to purify the adhesion complexes from digitonin-solubilized muscle (Fig. 4A). Protein components of the DGC were initially identified and characterized by their co-purification with dystrophin from digitonin-solubilized skeletal muscle lysates using sWGA lectin affinity chromatography (6). To identify the glycoprotein(s) that directly binds sWGA lectin, we prepared protein lysates from wild-type and mdx muscle under solubilization conditions (RIPA buffer) that are known to eliminate protein–protein interactions within the DGC (Supplementary Material, Fig. S1A). We found that dystrophin, utrophin, the SGs and integrins were present in the sWGA void fraction and α- and β-DG were present in the eluate, demonstrating that they bind directly to sWGA lectin (Supplementary Material, Fig. S1A). Based on these results, we utilized the sWGA lectin assay performed under mild, near native conditions that preserve protein interactions as a method to investigate protein interactions with DG (in other words, retention on sWGA lectin serves as a DG binding assay).
Figure 4.

SSPN association with DG complexes is dependent on utrophin and α7 integrin. (A) Schematic diagram to illustrate sWGA lectin assay. Muscle proteins solubilized in mild digitonin buffer were applied to sWGA agarose, washed and eluted with N-acetylglucosamine (GlcNAc). The sWGA lectin assay in RIPA buffer on wild-type and mdx muscle is shown in Supplementary Material, Figure S1A. (B and C) Skeletal muscle lysates from the indicated murine genotypes were solubilized in digitonin buffer and enriched using sWGA lectin chromatography. Eluates (10 μg) were resolved by SDS–PAGE and immunoblotted with indicated antibodies. Transgenic, exogenous human SSPN (hSSPN) is also shown. Laminin overlays onto immobilized α-DG are also provided (Lam O/L).
To investigate the mechanisms underlying the persistent muscular dystrophy in SSPN transgenic mdx mice lacking α7 integrin, muscles from mdx: α7−/−TG were analyzed using the sWGA lectin assay under gentle solubilization conditions. While SSPN increased the overall abundance of the UGC in mdx:α7−/−TG muscle (Supplementary Material, Fig. S1B), SSPN was unable to affect the detachment of the SG–SSPN subcomplex from DG subcomplex that is characteristic of mdx samples (Fig. 4B). In fact, only a small fraction of SSPN–SG subcomplex is associated with the DGs and the amount of DG bound to the sWGA lectin is decreased in mdx:α7−/−TG samples (Fig. 4B). In the absence of both dystrophin and α7 integrin, SG–SSPN subcomplex is unable to associate in a complex with utrophin and DG.
To investigate the mechanisms underlying the persistent muscular dystrophy in SSPN transgenic mdx mice lacking utrophin, total protein lysates from mdx:utrophin-nullTG skeletal muscle (6 weeks of age) were analyzed by immunoblotting. Consistent with our previous findings (56), loss of utrophin in mdx muscle results in a compensatory increase in the overall levels of α7β1 integrin and near-complete ablation of the UGC (except α-SG; Supplementary Material, Fig. S1C). Overexpression of SSPN increased DG abundance, along with β1D integrin levels (Fig. 4C). Neither ablation nor overexpression of SSPN affected utrophin mRNA levels (Supplementary Material, Fig. S1D). We next analyzed mdx:utrophin-nullTG muscle by purification on sWGA lectin chromatography under gentle, digitonin conditions. The molecular stoichiometry of α7β1 integrin binding to DG was increased in response to the absence of utrophin, as revealed by sWGA lectin assays (Fig. 4C). Although SSPN increases SG subcomplex attachment to DG in double-null samples, SSPN itself does not stably associate with this complex in the absence of utrophin (Fig. 4C). Although levels of α7β1 integrin are further increased and laminin binding is restored to therapeutic levels in mdx:utrophin-nullTG, dystrophic symptoms persist, revealing that utrophin is required to ameliorate disease and is necessary for the compensatory function of SSPN (Fig. 4C).
Collectively, these results reveal that the expression of both utrophin and α7 integrin are necessary for SSPN-based amelioration of DMD muscular dystrophy. Integration of SSPN into laminin-binding adhesion complexes is dependent on the expression of both utrophin and α7 integrin (Fig. 5).
Figure 5.
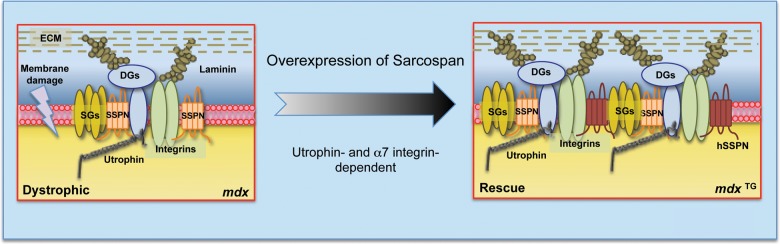
SSPN amelioration of DMD is dependent on two distinct laminin-binding adhesion complexes. Schematic diagrams of the adhesion glycoprotein complexes at the sarcolemma of mdx and SSPN transgenic mdx (mdx TG) mice. SSPN transgenic mdx mice are described relative to mdx muscle, which displays dystrophic pathology (sarcolemmal damage is represented by a ‘lightening bolt’). SSPN overexpression in mdx muscle stabilizes the sarcolemma through the upregulation of both the UGC and α7β1 integrin. SSPN is unable to ameliorate dystrophic pathology in mdx:α7−/− or mdx:utr−/− muscle. This study highlights the necessity of utrophin and α7 integrin for SSPN association with DG subcomplexes, revealing that SSPN-dependent ‘rescue’ of dystrophin function requires both the UGC and α7β1 integrin.
Discussion
Three adhesion glycoprotein complexes, the DGC, UGC and α7β1 integrin, maintain muscle stability during contractions by providing a mechanical linkage across the sarcolemmal membrane between laminin in the ECM and F-actin in the cytoplasm. DMD is caused by mutations in the dystrophin gene, which result in the loss of the entire DGC and subsequently render the sarcolemma vulnerable to contraction-induced damage owing to the loss of the major protein complex responsible for maintaining the connection to the ECM (9,19–22). The upregulation of compensatory adhesion glycoprotein complexes, the UGC and α7β1 integrin, to restore connections to the ECM is a therapeutic approach to treating DMD. Many therapeutic strategies to upregulate utrophin have been developed and proven effective in the mdx mouse model of DMD, including delivery of recombinant human biglycan (62), SMTC 1100 (63) and adeno-associated viral (AAV) delivery of mini-utrophin (36). Recently, AAV delivery of α7 integrin has been shown to ameliorate dystrophic symptoms in the mdx mouse (64). Interestingly, systemic delivery of Engelbreth–Holt–Swarm-derived laminin-111 in the mdx mouse acted as a dual therapy by increasing the protein levels of both the UGC and α7β1 integrin (65). While the compensatory expression of laminin-binding complexes has been documented in several murine models of muscular dystrophy, the molecular mechanisms that account for this regulation are unknown.
This study supports evidence that overexpression of exogenous human SSPN is also a dual targeting therapy. SSPN is a transmembrane protein with biochemical features of both tetraspanins and CD-20 that has been shown to be a core component of the DGC and UGC at the sarcolemma (3,31,52,56,57). Additionally, SSPN has been shown to genetically interact with α7β1 integrin: SSPN- and α7 integrin-deficient mice display more severe myopathy, reduced specific force production and increased percentage of force drop in the diaphragm muscle (52). Genetic removal of α7 integrin from SSPN-deficient mice also results in reduced levels of the DGC and UGC, suggesting that protein–protein interactions between SSPN and α7 integrin are important for DGC and UGC stability at the sarcolemma (52). Overexpression of SSPN in mdx muscle results in the upregulation of both UGC and α7β1 integrin compensatory complexes, activated Akt signaling and increased the GalNAc glycan epitopes per DG protein (31,56). SSPN combines many effects that have individually been shown to reduce dystrophic symptoms in mouse models of DMD (25,27,66–69). The inability of overexpression of SSPN to reduce muscle pathology in the Largemyd mouse model demonstrates that SSPN requires laminin binding to α-DG in the DGC to ameliorate mouse models of muscular dystrophy (56).
In the current study, we tested the requirement of utrophin and α7 integrin for SSPN-mediated amelioration of mdx dystrophic pathology. In creating SSPN transgenic α7 integrin-deficient mdx mice and SSPN transgenic utrophin-deficient mdx mice (two different sets of double-knockout mice), we discovered that SSPN overexpression did not extend the lifespan or decrease central nucleation, actively regenerating myofibers, or degenerating myofibers in these mice. In the current study, it is difficult to discern whether the level of ongoing regeneration is normal or slightly impaired in the various knockout and SSPN transgenic knockout mice, owing to the extent of muscle damage that has occurred in the mice prior to examination. To determine whether regeneration is impaired, further studies on isolated myoblasts to examine their capacity to proliferate and fuse into myotubes are needed. Examination of DG-associated proteins in sWGA eluates revealed that the UGC is reduced in mdx:α7−/−TG muscle compared with controls. In mdx:utr−/− mice overexpressing SSPN, an increase in α7β1 integrins associated with DG proteins was observed. However, the extent of dystrophic pathology was not reduced, and lifespan was not extended, demonstrating that the increase in α7β1 integrin was insufficient in preventing severe dystrophic symptoms. Interestingly, exogenous hSSPN was reduced in sWGA enrichments from both mdx:α7−/−TG and mdx:utr−/−TG samples compared with mdxTG controls, demonstrating that both utrophin and α7 integrin are required to stabilize SSPN association with DG complexes. These studies are useful for furthering the mechanistic understanding of SSPN-mediated amelioration of DMD pathology and allow for more complex design or verification of screens to identify therapeutic compounds for DMD disease that act through SSPN.
While α-DG and integrin bind laminins and other components of the basal lamina, the identification of their distinct functions within skeletal muscle has been elusive. Recent studies have reported that loss of α7 integrin did not affect sarcolemma stability, as evidenced by laser-induced membrane damage assays and lengthening-contraction models of muscle injury (70). Furthermore, electron micrographs of α7 integrin-null muscle appeared normal in contrast to the disruption of basal lamina–sarcolemma connection in hypoglycosylated Large-deficient myd muscle (70). Because DGC levels are not drastically compromised in α7 integrin-null muscle, defects in ECM attachment may be difficult to assess in these experiments. Alternatively, it is feasible that the role of α7β1 integrin may be to regulate signaling pathways that promote muscle cell growth without contributing significantly to ECM-membrane attachment. We and others have reported reduced force transmission in the diaphragm muscle of α7 integrin-deficient muscle demonstrating that while its precise role is unclear, α7β1 integrin is important for muscle function (49,52).
We have previously reported that SSPN-mediated amelioration of mdx dystrophy requires at least 3-fold overexpression (56), so it is not surprising that loss of SSPN association with laminin-binding complexes in α7 integrin-deficient mdx and utrophin-deficient mdx muscle results in the failure to alleviate dystrophic pathology. These data suggest that while α7β1 integrin and the UGC may have distinct roles in muscle, their combined presence at the membrane is important for stability in the context of SSPN overexpression (Fig. 5).
The results presented here illuminate the importance of integrin expression to muscle physiology and suggest that increased expression of utrophin alone may be insufficient to prevent muscle degeneration. Interestingly, it has been reported that overexpression of α7 integrin in utrophin-deficient mdx muscle reduces dystrophic pathology in the heart and significantly extends lifespan; however, no reduction of regeneration was observed in the quadriceps muscle (27,53). Future studies will be focused on determining whether utrophin overexpression ameliorates dystrophic pathology in the absence of dystrophin and α7 integrin.
Materials and Methods
Animal models
Wild-type and mdx mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA), and SSPN-deficient mice were a generous gift from Dr Kevin P. Campbell (University of Iowa Medical School, Howard Hughes Medical Institute, Iowa City, IA, USA) (71). α7 integrin-deficient mice were previously generated by Burkin and colleagues (48) and transferred from University of Nevada (Reno, NV, USA) to University of California (Los Angeles, CA, USA). mdx:utrophin+/− mice were a generous gift from Dr Joshua Sanes and shipped by Dr Jeffrey Chamberlain (35). Genotyping protocols have been previously described (35,48,56,71,72). SSPN transgenic mice, expressing ∼3-fold more SSPN than wild-type, were engineered with the human skeletal actin promoter and VP1 intron upstream of human SSPN, as described previously (56). Transgenic mice were generated by microinjections of transgene DNA into the pronucleus of fertilized single-cell embryos (University of California Irvine, Transgenic Mouse Facility). mdx:SSPN-Tg (mdx TG) males were crossed with mdx:α7 integrin-null females generating mdx/+ or Y:α7 integrin+/−:SSPN-Tg males and females. Siblings were intercrossed to generate mdx, mdx TG, mdx:α7 integrin-null and mdx:α7 integrin-null TG littermates, which were analyzed at 3 weeks. mdx:utrophin+/− mice were crossed with mdx:SSPN-Tg to generate mdx:utrophin+/−:SSPN-Tg mice. The mdx:utrophin+/−:SSPN-Tg males were then crossed with mdx:utrophin+/− females to generate mdx:utrophin−/−:SSPN-Tg mice. mdx, mdx:SSPN-Tg, mdx:utrophin−/− and mdx:utrophin−/−:SSPN-Tg mice were analyzed at 6 weeks. Mice were maintained in the Life Sciences and Terasaki Life Sciences Vivariums, and all procedures were carried out in accordance with guidelines set by the University of California, Los Angeles Institutional Animal Care and Use Committee.
Evans blue dye assay
EBD uptake analysis was performed by an intraperitoneal injection of mice with 50 μl of Evans blue dye (10 mg/ml in 10 mm of sterile phosphate buffer and 150 mm NaCl, pH 7.4) per 10 g of body weight at least 8 h prior to dissection as described previously (73). Quadriceps muscles were processed as described later.
Immunofluorescence assays
Muscles were mounted in 10.2% polyvinyl alcohol/4.3% polyethylene glycol and flash frozen in liquid nitrogen-cooled isopentane. Muscles were stored in the −80°C until further processing. Transverse sections (8 μm) were placed onto positively charged glass slides (Thermo Fisher Scientific) and stored at −80°C. Sections were acclimated to RT for 15 min and blocked with 3% BSA diluted in PBS for 30 min at RT. The avidin/biotin blocking kit (Vector Labs) was used according to manufacturer's instructions. Mouse primary antibodies were prepared with the M.O.M. blocking reagent (Vector Labs) as described by the manufacturer's protocol. Sections were incubated in primary antibody in PBS at 4°C overnight with the following antibodies: laminin (L9393, 1 : 25; Sigma) and eMHC (F1.652, 1 : 25; Developmental Studies Hybridoma Bank). Primary antibodies were detected by biotinylated anti-rabbit (BA-1000, 1 : 500; Vector Labs) and biotinylated anti-mouse (BA-9200, 1 : 500; Vector Labs). Fluorescein-conjugated avidin D (A-2001, 1 : 500; Vector Labs) was used to detect biotinylated secondary antibodies. Both secondary and tertiary probes were diluted in PBS and incubated with sections for 1 h at RT. Sections were mounted in Vectashield (Vector Labs) to prevent photobleaching. Sections were incubated with secondary and tertiary antibodies alone as a control for specificity. Antibody stained sections were visualized using a fluorescent microscope (Axioplan 2; Carl Zeiss, Inc.) equipped with a Plan Neofluar 40× NA 1.3 oil differential interference contrast objective, and images were captured under identical conditions using Axiovision Rel 4.5 software (Carl Zeiss, Inc.).
Histology
Hematoxylin and eosin (H&E) staining was used for visualization of centrally placed nuclei as described previously (74). Transverse quadriceps sections (8 μm) were acclimated to RT for 15 min before beginning the staining procedure. Slides were incubated in hematoxylin for 5 min, washed in water for 2 min, incubated in eosin for 5 min and dehydrated in 70, 80, 90 and 100% ethanol. Sections were then incubated in xylene for 10 min and mounted in Permount. All supplies for this procedure were purchased from Thermo Fisher Scientific. 40× quadriceps images were captured under identical conditions using an Axioplan 2 fluorescent microscope and Axiovision Rel 4.5 software (Carl Zeiss, Inc.). The percentage of centrally nucleated fibers was assessed. The data are represented as an average percentage of the total number of fibers in each whole quadriceps section.
Total protein preparation from skeletal muscle
Total skeletal muscle was snap-frozen in liquid nitrogen and stored at −80°C. For signaling blots, only quadriceps muscles were used. Tissues were ground to a fine powder using a mortar and pestle and then added to ice-cold DGC buffer (50 mm Tris–HCl, pH 7.8, 500 mm NaCl and 0.1% digitonin) or radioimmunoprecipitation assay buffer (RIPA) (89901; Thermo Scientific) containing fresh protease inhibitors (0.6 μg/ml pepstatin A, 0.5 μg/ml aprotinin, 0.5 μg/ml leupeptin, 0.75 mm benzamidine, 0.2 mm PMSF, 5 μm calpain I and 5 μm calpeptin). Homogenates were rotated at 4°C for 1 h. Lysates were clarified by centrifugation at 15,000g for 20 min at 4°C, protein concentration was determined using the DC Protein Assay (Bio-Rad) and lysates were stored at −80°C.
Immunoblot analysis
Equal quantities (10 μg for sWGA and 60 μg for total protein) of protein samples were resolved on 4–20% Pierce precise protein gels (Thermo Scientific) by SDS–PAGE and transferred to nitrocellulose membranes (Millipore). An identical protein gel was stained with Coomassie blue stain to visualize total protein. A 5% blotto solution [non-fat dry milk in TBS with 0.2% tween-20 (Fisher)] was used to block membranes for 30 min at RT and incubated in primary antibodies overnight at 4°C. Incubations were performed with the following primary antibodies: dystrophin (MANDYS1, 1 : 200; Development Studies Hybridoma Bank), utrophin (MANCHO3, 1 : 50; Development Studies Hybridoma Bank), α-DG IIH6 (sc-53987, 1 : 500; Santa Cruz Biotechnology, Inc.), β-DG (MANDAG2, 1 : 250; Development Studies Hybridoma Bank), α-SG (VP-A105, 1 : 100; Vector Labs), β-SG (VP-B206, 1 : 100; Vector Labs), γ-SG (VP-G803, 1 : 100; Vector Labs), laminin (L9393, 1 : 5000; Sigma), β1D Integrin (MAB1900, 1 : 200; Chemicon), α7B Integrin (affinity purified rabbit B2 347, 1 : 500) (55), α5 Integrin (AB1928, 1 : 500; Millipore), hSSPN (affinity purified rabbit 15, 1 : 2000) and SSPN (affinity purified rabbit 3, 1 : 5). Horseradish peroxidase-conjugated anti-rabbit IgG (GE Healthcare), anti-mouse IgG (GE Healthcare) and anti-mouse IgM (Roche) secondary antibodies were used at 1 : 2000 dilutions in 5% blotto and incubated at RT for 3 h. Immunoblots were developed using enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Thermo Scientific).
sWGA lectin enrichment
Protein samples (7 mg) were incubated with 1.2 ml succinylated WGA (sWGA)-conjugated agarose slurry (AL-1023S, Vector labs) and gently rotated overnight at 4°C. sWGA agarose was washed 4× in 10 ml standard DGC solubilization buffer (50 mm Tris–HCl, pH 7.8, 500 mm NaCl and 0.1% digitonin) or radioimmunoprecipitation assay buffer (RIPA) (89901; Thermo Scientific) containing fresh protease inhibitors (0.6 μg/ml pepstatin A, 0.5 μg/ml aprotinin, 0.5 μg/ml leupeptin, 0.75 mm benzamidine, 0.2 mm PMSF, 5 μm calpain I and 5 μm calpeptin) to remove unbound proteins. Bound proteins were eluted with 0.3 M N-acetylglucosamine (sWGA) (Sigma–Aldrich) and concentrated using Centricon Ultracel filtration columns (Millipore) by centrifugation at 4000g for 75 min. Protein concentration was determined with the DC Protein Assay (Bio-Rad). Equal concentrations of eluates (10 μg) were resolved by SDS–PAGE, transferred to nitrocellulose membranes and immunoblotted as described earlier.
Laminin overlay assay
Membranes were prepared as described in sWGA enrichment of protein lysates. Membranes were blocked with 5% BSA in laminin-binding buffer (LBB; 10 mm triethanolamine, 140 mm NaCl, 1 mm MgCl2, 1 mm CaCl2, pH 7.6) followed by incubation of mouse ultrapure Engelbreth–Holm–Swarm laminin (354239, BD Biosciences) in LBB 6 h at 4°C. Membranes were washed and incubated with rabbit anti-laminin (L9393, 1 : 5000; Sigma) overnight at 4°C followed by horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (GE Healthcare) at RT for 3 h. Blots were developed by enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Thermo Scientific).
Kaplan–Meier survival curve
The dates of birth and death of the mice were recorded in order to calculate the weeks of survival. These data were then inputted into an Excel spreadsheet to create a step graph with the number of weeks along the X-axis and percent survival of the mice along the Y-axis.
Whole-body images
Mice were first safely euthanized before they were skinned, according to the guidelines of the University of California, Los Angeles Institutional Animal Care and Use Committee. PBS was used to dampen and remove any excess hair on the body. Once dissected, whole-body pictures were captured. Bodies may appear blue owing to Evans blue dye.
Quantification of dystrophic pathology
ImageJ was used to quantify individual fibers in a whole quadriceps muscle sections. In H&E-stained sections, centrally nucleated and total fibers were quantified in order to determine the level of regeneration. In muscle sections treated with EBD to visualize degeneration, EBD-positive and total fibers outlined with anti-laminin were quantified. eMHC-positive and total fibers outlined with anti-laminin were quantified to determine the level of active regeneration. Dividing the number of H&E-, EBD- or eMHC-positive fibers by the total number of fibers yielded the percentage of dystrophic pathology seen in one whole muscle section. Averaging the percentages of whole muscle sections within one genotype gives the mean level of dystrophic pathology. Excel was used to create the bar graphs visualizing regeneration and degeneration.
Statistics
All statistics were analyzed using the one-way analysis of variance (ANOVA) with a Bonferroni correction or Tukey's multiple comparison tests to determine differences between groups. Statistical analysis between two samples was performed using the Student's T-test. n- and P-values are indicated on the plots. Statistical significance was accepted for P < 0.05.
Supplementary Material
Funding
This work was supported by the grants from the Genetic Mechanisms Pre-doctoral Training Fellowship USPHS National Research Service Award GM07104, the Edith Hyde Fellowship, the Eureka Pre-doctoral Training Fellowship and the Ruth L. Kirschstein National Service Award T32AR059033 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases to J.L.M.; Undergraduate Research Fellows Program to J.O.; Undergraduate Research Fellows Program to E.C.; Tegger Foundation and Swedish Research Council (524-2009-619) to J.H.; NIH/NIAMS (R01 AR053697) and MDA (238981) to D.J.B and NIH/NIAMS (R01 AR048179) to R.C.W.
Supplementary Material
Acknowledgements
The authors thank B. Burton, S. Clark, A. Grossi, M.P. Huang, A. Kwok and A.C. Ocampo for technical expertise, Dr T. Kitada for assistance with statistical analysis and Drs. M. Alexander, E. Gibbs, T. Kitada and M. Parvatiyar for critically reading the manuscript. We thank Dr Paul T. Martin (The Research Institute at Children's Nationwide Hospital, The Ohio State University) and Dr Federica Montanaro (The Research Institute at Children's Nationwide Hospital, The Ohio State University) for thoughtful discussion.
Conflict of Interest statement. None declared.
References
- 1.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramaswamy K.S., Palmer M.L., van der Meulen J.H., Renoux A., Kostrominova T.Y., Michele D.E., Faulkner J.A. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J. Physiol. 2011;589:1195–1208. doi: 10.1113/jphysiol.2010.201921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall J.L., Watson R.H. Sarcospan: a small protein with large potential for Duchenne muscular dystrophy. Skelet. Muscle. 2013;3:1. doi: 10.1186/2044-5040-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holt K.H., Crosbie R.H., Venzke D.P., Campbell K.P. Biosynthesis of dystroglycan: processing of a precursor propeptide. FEBS Lett. 2000;468:79–83. doi: 10.1016/s0014-5793(00)01195-9. [DOI] [PubMed] [Google Scholar]
- 5.Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., Slaughter C.A., Sernett S.W., Campbell K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 6.Campbell K.P., Kahl S.D. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- 7.Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 8.Ervasti J.M., Campbell K.P. Dystrophin and the membrane skeleton. Curr. Opin. Cell Biol. 1993;5:82–87. doi: 10.1016/s0955-0674(05)80012-2. [DOI] [PubMed] [Google Scholar]
- 9.Ervasti J.M., Campbell K.P. Dystrophin-associated glycoproteins: their possible roles in the pathogenesis of Duchenne muscular dystrophy. Mol. Cell Biol. Hum. Dis. Ser. 1993;3:139–166. doi: 10.1007/978-94-011-1528-5_6. [DOI] [PubMed] [Google Scholar]
- 10.Ervasti J.M., Campbell K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ervasti J.M., Kahl S.D., Campbell K.P. Purification of dystrophin from skeletal muscle. J. Biol. Chem. 1991;266:9161–9165. [PubMed] [Google Scholar]
- 12.Yoshida M., Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- 13.Holt K.H., Campbell K.P. Assembly of the sarcoglycan complex. Insights for muscular dystrophy. J. Biol. Chem. 1998;273:34667–34670. doi: 10.1074/jbc.273.52.34667. [DOI] [PubMed] [Google Scholar]
- 14.Crosbie R.H., Lebakken C.S., Holt K.H., Venzke D.P., Straub V., Lee J.C., Grady R.M., Chamberlain J.S., Sanes J.R., Campbell K.P. Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. J. Cell Biol. 1999;145:153–165. doi: 10.1083/jcb.145.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crosbie R.H., Lim L.E., Moore S.A., Hirano M., Hays A.P., Maybaum S.W., Collin H., Dovico S.A., Stolle C.A., Fardeau M., et al. Molecular and genetic characterization of sarcospan: insights into sarcoglycan-sarcospan interactions. Hum. Mol. Genet. 2000;9:2019–2027. doi: 10.1093/hmg/9.13.2019. [DOI] [PubMed] [Google Scholar]
- 16.Koenig M., Monaco A.P., Kunkel L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–226. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 17.Koenig M., Kunkel L.M. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J. Biol. Chem. 1990;265:4560–4566. [PubMed] [Google Scholar]
- 18.Rybakova I.N., Amann K.J., Ervasti J.M. A new model for the interaction of dystrophin with F-actin. J. Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman E.P., Brown R.H., Jr., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 20.Hoffman E.P., Knudson C.M., Campbell K.P., Kunkel L.M. Subcellular fractionation of dystrophin to the triads of skeletal muscle. Nature. 1987;330:754–758. doi: 10.1038/330754a0. [DOI] [PubMed] [Google Scholar]
- 21.Bonilla E., Samitt C.E., Miranda A.F., Hays A.P., Salviati G., DiMauro S., Kunkel L.M., Hoffman E.P., Rowland L.P. Duchenne muscular dystrophy: deficiency of dystrophin at the muscle cell surface. Cell. 1988;54:447–452. doi: 10.1016/0092-8674(88)90065-7. [DOI] [PubMed] [Google Scholar]
- 22.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 23.Weller B., Karpati G., Carpenter S. Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J. Neurol. Sci. 1990;100:9–13. doi: 10.1016/0022-510x(90)90005-8. [DOI] [PubMed] [Google Scholar]
- 24.Marshall J.L., Kwok Y., McMorran B.J., Baum L.G., Crosbie-Watson R.H. The potential of sarcospan in adhesion complex replacement therapeutics for the treatment of muscular dystrophy. FEBS J. 2013;280:4210–4229. doi: 10.1111/febs.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 26.Liu J., Milner D.J., Boppart M.D., Ross R.S., Kaufman S.J. β1D chain increases α7β1 integrin and laminin and protects against sarcolemmal damage in mdx mice. Hum. Mol. Genet. 2011;21:1592–1603. doi: 10.1093/hmg/ddr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burkin D.J., Wallace G.Q., Nicol K.J., Kaufman D.J., Kaufman S.J. Enhanced expression of the α7β1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodges B.L., Hayashi Y.K., Nonaka I., Wang W., Arahata K., Kaufman S.J. Altered expression of the alpha7beta1 integrin in human and murine muscular dystrophies. J. Cell Sci. 1997;110:2873–2881. doi: 10.1242/jcs.110.22.2873. [DOI] [PubMed] [Google Scholar]
- 29.Pons F., Robert A., Marini J.F., Leger J.J. Does utrophin expression in muscles of mdx mice during postnatal development functionally compensate for dystrophin deficiency? J. Neurol. Sci. 1994;122:162–170. doi: 10.1016/0022-510x(94)90295-x. [DOI] [PubMed] [Google Scholar]
- 30.Pearce M., Blake D.J., Tinsley J.M., Byth B.C., Campbell L., Monaco A.P., Davies K.E. The utrophin and dystrophin genes share similarities in genomic structure. Hum. Mol. Genet. 1993;2:1765–1772. doi: 10.1093/hmg/2.11.1765. [DOI] [PubMed] [Google Scholar]
- 31.Peter A.K., Marshall J.L., Crosbie R.H. Sarcospan reduces dystrophic pathology: stabilization of the utrophin-glycoprotein complex. J. Cell Biol. 2008;183:419–427. doi: 10.1083/jcb.200808027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tinsley J.M., Blake D.J., Roche A., Fairbrother U., Riss J., Byth B.C., Knight A.E., Kendrick-Jones J., Suthers G.K., Love D.R., et al. Primary structure of dystrophin-related protein. Nature. 1992;360:591–593. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- 33.Rybakova I.N., Humston J.L., Sonnemann K.J., Ervasti J.M. Dystrophin and utrophin bind actin through distinct modes of contact. J. Biol. Chem. 2006;281:9996–10001. doi: 10.1074/jbc.M513121200. [DOI] [PubMed] [Google Scholar]
- 34.Deconinck A.E., Rafael J.A., Skinner J.A., Brown S.C., Potter A.C., Metzinger L., Watt D.J., Dickson J.G., Tinsley J.M., Davies K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 35.Grady R.M., Teng H., Nichol M.C., Cunningham J.C., Wilkinson R.S., Sanes J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 36.Odom G.L., Gregorevic P., Allen J.M., Finn E., Chamberlain J.S. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol. Ther. 2008;16:1539–1545. doi: 10.1038/mt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sonnemann K.J., Heun-Johnson H., Turner A.J., Baltgalvis K.A., Lowe D.A., Ervasti J.M. Functional substitution by TAT-utrophin in dystrophin-deficient mice. PLoS Med. 2009;6:e1000083. doi: 10.1371/journal.pmed.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tinsley J.M., Potter A.C., Phelps S.R., Fisher R., Trickett J.I., Davies K.E. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 39.Wakefield P.M., Tinsley J.M., Wood M.J., Gilbert R., Karpati G., Davies K.E. Prevention of the dystrophic phenotype in dystrophin/utrophin-deficient muscle following adenovirus-mediated transfer of a utrophin minigene. Gene Ther. 2000;7:201–204. doi: 10.1038/sj.gt.3301066. [DOI] [PubMed] [Google Scholar]
- 40.Squire S., Raymackers J.M., Vandebrouck C., Potter A., Tinsley J., Fisher R., Gillis J.M., Davies K.E. Prevention of pathology in mdx mice by expression of utrophin: analysis using an inducible transgenic expression system. Hum. Mol. Genet. 2002;11:3333–3344. doi: 10.1093/hmg/11.26.3333. [DOI] [PubMed] [Google Scholar]
- 41.Hynes R.O. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 42.Song W.K., Wang W., Foster R.F., Bielser D.A., Kaufman S.J. H36-alpha 7 is a novel integrin alpha chain that is developmentally regulated during skeletal myogenesis. J. Cell Biol. 1992;117:643–657. doi: 10.1083/jcb.117.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von der Mark H., Durr J., Sonnenberg A., von der Mark K., Deutzmann R., Goodman S.L. Skeletal myoblasts utilize a novel beta 1-series integrin and not alpha 6 beta 1 for binding to the E8 and T8 fragments of laminin. J. Biol. Chem. 1991;266:23593–23601. [PubMed] [Google Scholar]
- 44.Carmignac V., Durbeej M. Cell-matrix interactions in muscle disease. J. Pathol. 2011;226:200–218. doi: 10.1002/path.3020. [DOI] [PubMed] [Google Scholar]
- 45.Hayashi Y.K., Chou F.L., Engvall E., Ogawa M., Matsuda C., Hirabayashi S., Yokochi K., Ziober B.L., Kramer R.H., Kaufman S.J., et al. Mutations in the integrin α7 gene cause congenital myopathy. Nat. Genet. 1998;19:94–97. doi: 10.1038/ng0598-94. [DOI] [PubMed] [Google Scholar]
- 46.Burkin D.J., Gu M., Hodges B.L., Campanelli J.T., Kaufman S.J. A functional role for specific spliced variants of the α7β1 integrin in acetylcholine receptor clustering. J. Cell Biol. 1998;143:1067–1075. doi: 10.1083/jcb.143.4.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burkin D.J., Kim J.E., Gu M., Kaufman S.J. Laminin and alpha7beta1 integrin regulate agrin-induced clustering of acetylcholine receptors. J. Cell Sci. 2000;113:2877–2886. doi: 10.1242/jcs.113.16.2877. [DOI] [PubMed] [Google Scholar]
- 48.Flintoff-Dye N.L., Welser J., Rooney J., Scowen P., Tamowski S., Hatton W., Burkin D.J. Role for the α7β1 integrin in vascular development and integrity. Dev. Dyn. 2005;234:11–21. doi: 10.1002/dvdy.20462. [DOI] [PubMed] [Google Scholar]
- 49.Lopez M.A., Mayer U., Hwang W., Taylor T., Hashmi M.A., Jannapureddy S.R., Boriek A.M. Force transmission, compliance, and viscoelasticity are altered in the alpha7-integrin-null mouse diaphragm. Am. J. Physiol. Cell Physiol. 2005;288:C282–C289. doi: 10.1152/ajpcell.00362.2003. [DOI] [PubMed] [Google Scholar]
- 50.Mayer U., Saher G., Fassler R., Bornemann A., Echtermeyer F., von der Mark H., Miosge N., Poschl E., von der Mark K. Absence of integrin α7 causes a novel form of muscular dystrophy. Nat. Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- 51.Nawrotzki R., Willem M., Miosge N., Brinkmeier H., Mayer U. Defective integrin switch and matrix composition at alpha 7-deficient myotendinous junctions precede the onset of muscular dystrophy in mice. Hum. Mol. Genet. 2003;12:483–495. doi: 10.1093/hmg/ddg047. [DOI] [PubMed] [Google Scholar]
- 52.Marshall J.L., Chou E., Oh J., Kwok A., Burkin D.J., Crosbie-Watson R.H. Dystrophin and utrophin expression require sarcospan: loss of alpha7 integrin exacerbates a newly discovered muscle phenotype in sarcospan-null mice. Hum. Mol. Genet. 2012;21:4378–4393. doi: 10.1093/hmg/dds271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burkin D.J., Wallace G.Q., Milner D.J., Chaney E.J., Mulligan J.A., Kaufman S.J. Transgenic expression of α7β1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am. J. Pathol. 2005;166:253–263. doi: 10.1016/s0002-9440(10)62249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo C., Willem M., Werner A., Raivich G., Emerson M., Neyses L., Mayer U. Absence of α7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet. 2006;15:989–998. doi: 10.1093/hmg/ddl018. [DOI] [PubMed] [Google Scholar]
- 55.Rooney J.E., Welser J.V., Dechert M.A., Flintoff-Dye N.L., Kaufman S.J., Burkin D.J. Severe muscular dystrophy in mice that lack dystrophin and α7 integrin. J. Cell Sci. 2006;119:2185–2195. doi: 10.1242/jcs.02952. [DOI] [PubMed] [Google Scholar]
- 56.Marshall J.L., Holmberg J., Chou E., Ocampo A.C., Oh J., Lee J., Peter A.K., Martin P.T., Crosbie-Watson R.H. Sarcospan-dependent Akt activation is required for utrophin expression and muscle regeneration. J. Cell Biol. 2012;197:1009–1027. doi: 10.1083/jcb.201110032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crosbie R.H., Heighway J., Venzke D.P., Lee J.C., Campbell K.P. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J. Biol. Chem. 1997;272:31221–31224. doi: 10.1074/jbc.272.50.31221. [DOI] [PubMed] [Google Scholar]
- 58.Welser J.V., Rooney J.E., Cohen N.C., Gurpur P.B., Singer C.A., Evans R.A., Haines B.A., Burkin D.J. Myotendinous junction defects and reduced force transmission in mice that lack α7 integrin and utrophin. Am. J. Pathol. 2009;175:1545–1554. doi: 10.2353/ajpath.2009.090052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peter A.K., Miller G., Crosbie R.H. Disrupted mechanical stability of the dystrophin-glycoprotein complex causes severe muscular dystrophy in sarcospan transgenic mice. J. Cell Sci. 2007;120:996–1008. doi: 10.1242/jcs.03360. [DOI] [PubMed] [Google Scholar]
- 60.Li Y., Lee Y., Thompson W.J. Changes in aging mouse neuromuscular junctions are explained by degeneration and regeneration of muscle fiber segments at the synapse. J. Neurosci. 2011;31:14910–14919. doi: 10.1523/JNEUROSCI.3590-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y., Thompson W.J. Nerve terminal growth remodels neuromuscular synapses in mice following regeneration of the postsynaptic muscle fiber. J. Neurosci. 2011;31:13191–13203. doi: 10.1523/JNEUROSCI.2953-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amenta A.R., Yilmaz A., Bogdanovich S., McKechnie B.A., Abedi M., Khurana T.S., Fallon J.R. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc. Natl Acad. Sci. USA. 2010;108:762–767. doi: 10.1073/pnas.1013067108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tinsley J.M., Fairclough R.J., Storer R., Wilkes F.J., Potter A.C., Squire S.E., Powell D.S., Cozzoli A., Capogrosso R.F., Lambert A., et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One. 2011;6:e19189. doi: 10.1371/journal.pone.0019189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heller K.N., Montgomery C.L., Janssen P.M., Clark K.R., Mendell J.R., Rodino-Klapac L.R. AAV-mediated overexpression of human α7 integrin leads to histological and functional improvement in dystrophic mice. Mol. Ther. 2013;21:520–525. doi: 10.1038/mt.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rooney J.E., Gurpur P.B., Burkin D.J. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc. Natl Acad. Sci. USA. 2009;106:7991–7996. doi: 10.1073/pnas.0811599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blaauw B., Mammucari C., Toniolo L., Agatea L., Abraham R., Sandri M., Reggiani C., Schiaffino S. Akt activation prevents the force drop induced by eccentric contractions in dystrophin-deficient skeletal muscle. Hum. Mol. Genet. 2008;17:3686–3696. doi: 10.1093/hmg/ddn264. [DOI] [PubMed] [Google Scholar]
- 67.Kim M.H., Kay D.I., Rudra R.T., Chen B.M., Hsu N., Izumiya Y., Martinez L., Spencer M.J., Walsh K., Grinnell A.D., et al. Myogenic Akt signaling attenuates muscular degeneration, promotes myofiber regeneration and improves muscle function in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2011;20:1324–1338. doi: 10.1093/hmg/ddr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peter A.K., Ko C.Y., Kim M.H., Hsu N., Ouchi N., Rhie S., Izumiya Y., Zeng L., Walsh K., Crosbie R.H. Myogenic Akt signaling upregulates the utrophin-glycoprotein complex and promotes sarcolemma stability in muscular dystrophy. Hum. Mol. Genet. 2009;18:318–327. doi: 10.1093/hmg/ddn358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nguyen H.H., Jayasinha V., Xia B., Hoyte K., Martin P.T. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc. Natl Acad. Sci. USA. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han R., Kanagawa M., Yoshida-Moriguchi T., Rader E.P., Ng R.A., Michele D.E., Muirhead D.E., Kunz S., Moore S.A., Iannaccone S.T., et al. Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of α-dystroglycan. Proc. Natl Acad. Sci. USA. 2009;106:12573–12579. doi: 10.1073/pnas.0906545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lebakken C.S., Venzke D.P., Hrstka R.F., Consolino C.M., Faulkner J.A., Williamson R.A., Campbell K.P. Sarcospan-deficient mice maintain normal muscle function. Mol. Cell Biol. 2000;20:1669–1677. doi: 10.1128/mcb.20.5.1669-1677.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shin J.H., Hakim C.H., Zhang K., Duan D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve. 2011;43:283–286. doi: 10.1002/mus.21873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Straub V., Rafael J.A., Chamberlain J.S., Campbell K.P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peter A.K., Crosbie R.H. Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp. Cell Res. 2006;312:2580–2591. doi: 10.1016/j.yexcr.2006.04.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.