

Abstract
Beta-ecdysone (βEcd) is a phytoecdysteroid found in the dry roots and seeds of the asteraceae and achyranthes plants, and is reported to increase osteogenesis in vitro. Since glucocorticoid (GCs) excess is associated with a decrease in bone formation, the purpose of this study was to determine if treatment with βEcd could prevent GC-induced osteoporosis. Two-month-old male Swiss-Webster mice (n=8-10/group) were randomized to either placebo or slow release prednisolone pellets (3.3mg/kg/d) and treated with vehicle control or βEcd (0.5mg/kg/d) for 21 days. GC treatment inhibited age-dependent trabecular gain and cortical bone expansion and this was accompanied by a 30-50% lower bone formation rate (BFR) at both the endosteal and periosteal surfaces. Mice treated with only βEcd significantly increased bone formation on endosteal and periosteal bone surfaces, and increased cortical bone mass were their controls to compare to GC alone. Concurrent treatment of βEcd and GC completely prevented the GC-induced reduction in BFR, trabecular bone volume and partially prevented cortical bone loss. In vitro studies determined that βEcd prevented the GC increase in autophagy of the bone marrow stromal cells as well as in whole bone. In summary, βEcd prevented GC induced changes in bone formation, bone cell viability and bone mass. Additional studies are warranted of βEcd for the treatment of GC induced bone loss.
Keywords: Beta-ecdysone (βEcd), glucocorticoid, bone formation, autophagy
Introduction
Glucocorticoids (GCs) are frequently used in clinical medicine to treat non-infectious inflammatory diseases. However, GC use results in rapid trabecular bone loss and a high incident fracture risk [1, 2]. Lower peak bone mass acquisition, presence of osteopenia and vertebral collapse were often observed in children with primary increase in the endogenous levels of GCs with Cushing's disease [3, 4] or on GC treatments for some chronic diseases such as asthma [5] and other inflammatory diseases [6, 7]. Children treated with chronic GCs normally have growth retardation including the suppression of bone growth [8, 9]. Prevention for and treatment of glucocorticoid-induced osteoporosis (GIOP) in adults include bisphosphonates (BPs) and PTH [10-13]. The former have also been used to treat children with GIOP [14, 15]. However, as bone is highly remodeled during childhood to maintain adequate mineralization of the rapidly growing skeleton, the use of BPs is not ideal as they inhibit bone remodeling and could increase the mineral in the bone matrix, which may not be ideal to use in a growing skeleton [16, 17]. To this end, continued and safety studies for the use of BPs in children have yet to be established [18-20].
Recently, naturally-derived products contain a variety of molecules with potent biological activities. Phytoecdysteroids are plant-derived ecdysteroids that are structural analogs of insect molting hormone ecdysone, which are critical for insects to maintain “eat-to-reproduce” life cycle [21]. Beta-ecdysone (βEcd) is one of the most abundant phytoecdysteroids found in plants, such as in the dry roots and seeds of the asteraceae and achyranthes, as well as in spinach, quinoa and suma root [22, 23]. These plants are often used in the traditional Chinese medicine to help to reduce joint and back pain. It has been shown that βEcd increases protein synthesis and reduces protein degradation in the skeletal muscle cells [24, 25]. As it increases muscle weight in rodents [26-28], βEcd has been referred to as an “anabolic” naturally-derived supplement [29]. Additionally, βEcd is also found to stimulate mesenchymal stem cells' osteogenic differentiation but to inhibit their adipogenic differentiation [30]. βEcd is reported to increase the growth plate width in estrogen deficient rats and to have a marginal beneficial effect on trabecular bone and cartilage preserving following ovariectomy (OVX) [31, 32]. Since GC use in children often results in growth retardation, [6, 7, 33] through GC induced inhibition of osteoblasts through multiple mechanisms [34], we seek to determine if βEcd can rescue the GC-suppression on bone formation. We have hypothesized that βEcd treatment inhibits bone loss and deterioration of mechanical properties associated with GC uses, partially through maintenance of bone formation. Also, we explore osteoblast and osteocyte autophagy following GC or with concurrent βEcd treatment, and evaluate if autophagy is one of the mechanisms explaining the bone anabolic effect we observed for βEcd.
Methods
Animals and Experimental Procedures
Two-month-old male Swiss-Webster mice were maintained on commercial rodent chow (22/5 Rodent Diet; Teklad, Madison, WI) available ad libitum with 0.95% calcium and 0.67% phosphate. Mice were housed in a room that was maintained at 20 °C with a 12-hour light/dark cycle. They were randomized into 4 experimental groups of 8 animals in each group. Slow release pellets (Innovative Research of American, Sarasota, FL) of prednisolone (GC) were implanted respectively: Group 1, the control group, was implanted with a placebo pellet (PL); Group 2 was implanted with PL pellet + βEcd (PL + βEcd 0.5mg/kg, 5×/wk); Group 3 was implanted with a prednisolone 5mg/60 day slow-release pellet, which is equivalent to 3.3 mg/kg/d (GC), and Group 4 was implanted with prednisolone 5mg/60 days slow-release pellet + βEcd (GC + βEcd 0.5mg/kg, 5×/wk). The mice were sacrificed after three weeks of treatments. The βEcd dose was based publications on myogenesis and our in vitro experiments on osteogenesis and osteoclastogenesis using βEcd doses ranging from 10-3 to 10-9M [25, 26].
βEcd was purchased from Sigma-Aldrich (St. Louis, MO). Calcein (30mg/kg) was injected to all mice seven and two days before euthanization. All animals were treated according to the USDA animal care guidelines with the approval of the UC Davis Committee on Animal Research.
Measurements of serum hormonal levels and biochemical markers of Bone Turnover
The mice were fasted overnight before their serums were collected for the measurements of cortisol, leptin and insulin using a luminex multiplexing hormonal panel assay while bone turnover markers, osteocalcin and osteoprotegerin (OPG) levels were measured using a luminex multiplexing bone panel assay (EMD Millipore, Billerica, MA, USA). Serum CTX-1 was measured by ELISA (Immunodiagnostic Systems Inc., Gaithersburg, MD, USA).
Assessment of bone mass and bone microarchitecture
The 5th lumbar vertebral body and the right femur mid-diaphysis from each animal were scanned and measured by MicroCT (VivaCT 40, Scanco Medical, Bassersdorf, Switzerland), with an isotropic resolution of 10.5 μm. Bone samples were scanned at 70 kVp and 145 μA. Three-dimensional trabecular structural parameters were measured directly, as previously described [35]. Ex vivo microCT scans of the central right femur that included a region of total 100 slices. All the slices were used to evaluate total volume (TV), cortical bone volume (BV), and cortical thickness (Ct.Th) [36-39].
Assessment of surface-based bone turnover by bone histomorphometry
The third and fourth lumbar vertebral bodies (LVB) were fixed in 4% paraformaldehyde for 24 hours, and then soaked in 30% sucrose in PBS at 4°C for eight hours and then embedded in optimum cutting temperature compound. Eight μm thick frozen sections were obtained using a Leica microtome coupled with a CyroJane tape transfer system. The slides were mounted using 50% glycerol in PBS. Bone histomorphometry was performed using a semi-automatic image analysis Bioquant system (Bioquant Image Analysis Corporation, Nashville, TN) [35]. Static measurements included total tissue area (T.Ar), bone area (B.Ar) and bone perimeter (B.Pm). Dynamic measurements included single- (sL.Pm) and double-labeled perimeter (dL.Pm), and interlabel width (Ir.L.Wi). These indices were used to calculate 2-D bone volume (B.Ar/T.Ar), trabecular number (Tb.N), trabecular thickness (Tb.Th), and mineralizing surface (MS/BS and mineral apposition rate (MAR). Surface-based bone formation rate (BFR/BS) was calculated by multiplying mineralizing surface (single labeled surface/2 + double labeled surface) by MAR [40]. A separated section was used to stain for tartrate-resistant acid phosphatase (TRAP) to measure osteoclast number at the trabecular bone surface (OC/BS). We used the terminologies following the recommendation of the American Society for Bone and Mineral Research and we have reported similar methodology in other experiments in our laboratory [36, 41].
The femoral shafts were dissected and fixed in 4% paraformaldehyde, dehydrated in graded concentrations of ethanol and xylene, embedded un-decalcified in methyl methacrylate and then cross-sectioned using a SP1600 microtome (Leica, Buffalo Grove, IL, USA) into 40μm sections. Total cross-sectional bone area (T.Ar), cortical area (Ct.Ar), and cortical thickness (Ct.Th) were measured with the Bioquant Image analysis system. Single and double labeled surface, and inter-labeled width were measured separately at the endocortical (Ec.) and periosteal (Ps.) bone surfaces. MAR and BFR/BS were calculated thereafter for both the endocortical and periosteal bone surfaces [36-39].
Biomechanical testing
For the vertebrae, the endplates of the lumbar vertebral body were polished using an 800-grit silicon carbide paper to create two parallel planar surfaces. Before testing, caudal and cranial diameter measurements were taken at the top, middle, and bottom of LVB6 to obtain six measurements which were averaged as the diameter; the height along the long axis was recorded as well and the vertebrae were modeled as a cylinder. Each lumbar vertebra was then loaded to failure under unconfined compression along its long axis using an MTS 831 electro-servo-hydraulic testing system (MTS Systems Corp., Eden Prairie, MN) at a displacement rate of 0.01 mm/s with 1 kN load cell; the tests were performed in 37°C HBSS and sample loads and displacements were continuously recorded throughout each test. Values for the maximum load and maximum stress (bone strength) for compression were then determined, where the stress was calculated using σ=4P/(πdˆ2), with P being the load and d the average diameter.
To analyze the biomechanical properties of the femurs, the femoral samples were subjected to three-point bending tests, with the bone loaded using an MTS 831 electro-servo-hydraulic testing system (MTS Systems Corp., Eden Prairie, MN, USA) such that the posterior surface was in tension and the anterior surface was in compression. The major loading span was 14.5 mm. Each femur was loaded to failure in 37°C HBSS at a displacement rate of 0.01 mm/s while its corresponding load and displacement were measured using a calibrated 1 kN load cell. Two diameter measurements were taken at the fracture location, and averaged to model the femur as a cylinder. Values for the maximum load and ultimate strength of bending tests were then determined, with the stress calculated from σ = PLy/4I, where P is the load, L is the major loading span, y is the distance from the center of mass (d/2), and I is the moment of inertia (πdˆ4/64), with d being the average diameter. A measure of toughness was estimated in terms of the work of fracture, specifically the area under the load vs/ displacement curve normalized by twice the fracture surface area [36, 42].
In vitro osteogenesis and adipogenesis assays
Bone marrow stromal cells (BMSC) were flashed out from long bones obtained from the 2-month-old male mice. For adipogenesis differentiation, the BMSCs were cultured using a STENPRO Adipogenesis Differentiation Kit (GIBCO Invitrogen Cell Culture) for 10 days and stained with Oil Red O for lipoid deposits. RNA was extracted from day 14 cultures for quantitative measurements of RNA levels for genes associated with osteogenesis (Runx2, Bglap1) or adipogenesis (Cebp-α or Ppar-γ). For osteogenic differentiation, BMSCs were cultured for 14 days in osteogenic media and then the colony-forming unit-forming colonies (CFU-F) were stained by crystal violet followed by alizarin red staining for osteogenic colonies (CFU-Ob) (Sigma-Aldrich, St. Loius, MO, USA) [37]. Subsequently, stained cells were eluted from membranes and absorbance was measured at 590 nm (crystal violet) or 410 nm (ALP) [38, 43]. In a separated experiment, BMSC cells were obtained from male dsRed-LC3 reporter mice of Swiss Webster background (made by UC Davis Mutant Mouse Regional Resource Center; property of Drs. Yao and Lane). The BMSC were cultured in osteogenic medium for seven days before they were treated with PBS, Dexamethasone (Dex, 10-6 M), βEcd (10-7M) or combination of Dex + βEcd in serum-starved conditions for eight hours. The cells were either lysed to collect protein or fixed with 4% paraformaldehyde, and examined under a Keyence Imaging System with cell count software (Keyence Corp. of America, Itasca, IL, USA). Autophagic cells were quantified by counting cells exhibiting 10 or more dsRed-LC3 dot/cells.
Real-time RT-PCR
Total RNA was obtained from the distal tibiae. Total RNA was isolated using a modified two-step purification protocol employing homogenization (PRO250 Homogenizer, 10mm × 105mm generator, PRO Scientific IN, Oxford CT) in Trizol (Invitrogen, Carlsbad, CA, USA). The autophagic focus RT-PCR gene pathway arrays and the primer sets were purchased from SABioscience, a Qiangen company, (Frederick, MD, USA). Each pathway gene array has pre-selected 96 genes that are related to autophagy pathways, housekeeping genes, and no primer or cDNA controls. Detailed gene information can be found at http://www.sabiosciences.com/RTPCR.php. We excluded genes with Ct values of > 35 for the pathway analysis [34, 44].
Western Blot
Tibial cortical bones were lysed in RIPA buffer with homogenization. The bone lysates were resolved on SDS-PAGE and electrophoretically transferred to polyvinylidene difluoride membranes. Membranes were incubated with primary antibodies that include β-actin (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Atg 7, anti-Atg-16L and anti-LC3 (Cell Signaling Technology, Danvers, MA, USA) followed by species-specific horseradish peroxidase secondary antibody. Anti-LC3 antibody recognizes both LC3-I, which is cytoplasmic, and LC3-II that binds to the autophagic membranes. Immunoreactive materials were detected by chemiluminescence (Pierce Laboratories, Thermo Fisher Scientific, Rockford, IL, USA), then were imaged and quantitated by BIO-RAD ChemiDoc MP imaging system and analysis software [36].
Statistical analysis
The group means and standard deviations (SDs) were calculated for all outcome variables. The nonparametric Kruskal-Wallis test was used to determine the overall and group-wise differences between the groups. (SPSS Version 14; SPSS Inc., Chicago, IL).
Results
Body weight
Body mass increased by approximately 25% in the PL and PL + βEcd groups and declined in the GC and GC+ βEcd groups by 20% during this three-week study period (p < 0.05 vs. baseline). βEcd treatment did not induce any change in body mass as compared to βEcd naïve groups.
βEcd treatment alone did not alter serum cortisol, ACTH, Leptin or Insulin levels (Table1) or the estrogen, progesterone, T3, T4 levels (data on file). Serum osteocalcin and CTX-1 levels increased significantly in PL + βEcd group as compared to PL group. GC did not significantly change serum hormonal levels but decreased serum osteocalcin by 12% and increased serum CTX-1 concentration by 125% (P < 0.05 vs. PL). GC + βEcd tended to increase osteocalcin level and reduced CTX-1 level (P < 0.05 vs. GC) as compared to GC group (Table1).
Table 1. Serum hormone levels and bone turnover measurements.
| Hormone panel | Bone panel | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Cortisol (ng/ml)) | Leptin (ug/ml) | Insulin (pg/ml) | OC (μg/ml) | OPG (μg/ml) | CTX-1 (μg/ml) | |
| PL | 24.4 ±4.4 | 5.57 ±4.5 | 236 ±27 | 12.8± 1.7 | 1.05 ±0.5 | 19.7 ±4.1 |
| PL+βEcd | 23.4 ± 3.6 | 4.02 ± 3.3 | 233 ± 84 | 16.1 ±2.7* | 1.14 ±0.4 | 31.3 ±6.7* |
| GC | 17.5 ±3.2 | 3.47 ± 3.6 | 176 ±92 | 11.4 ±3.2 | 2.24 ±0.8 | 41.5 ±6.3* |
| GC + βEcd | 17.9 ±4.5 | 2.95 ±1.5 | 189±60 | 12.5 ±3.5 | 1.89 ±0.5 | 20.6 ±1.9# |
, p 0.05 vs. PL;
, P 0.05 vs. GC.
Bone volume and bone turnover changes in the trabecular bones
In PL + βEcd treatment group compared to the PL, trabecular bone volume/tissue volume (BV/TV) was increased by 41% at the 5th lumbar vertebral body (LVB). On the other hand, GC reduced BV/TV by 32% as compared to the PL group (P < 0.05 vs. PL). In GC+ βEcd compared to the GC, BV/TV was increased by 43% (P < 0.05 vs. GC). Trabecular thickness showed similar changes as BV/TV (Figures 1A and B). In the distal femur (DFM), the trends of changes were similar to the LVB but to a lesser degree following GC or with βEcd treatments. In PL + βEcd treatment group compared to the PL, trabecular bone BV/TV and Tb.Th were increased by 73% and 47% (P < 0.05 vs. PL), respectively, at. GCs non-significantly lowered BV/TV and Tb.Th in the DFM and GC+ βEcd had approximately 10% higher BV/TV and Th.N, but these changes were not significant compared to the GC group (Figure 1C).
Figure 1.
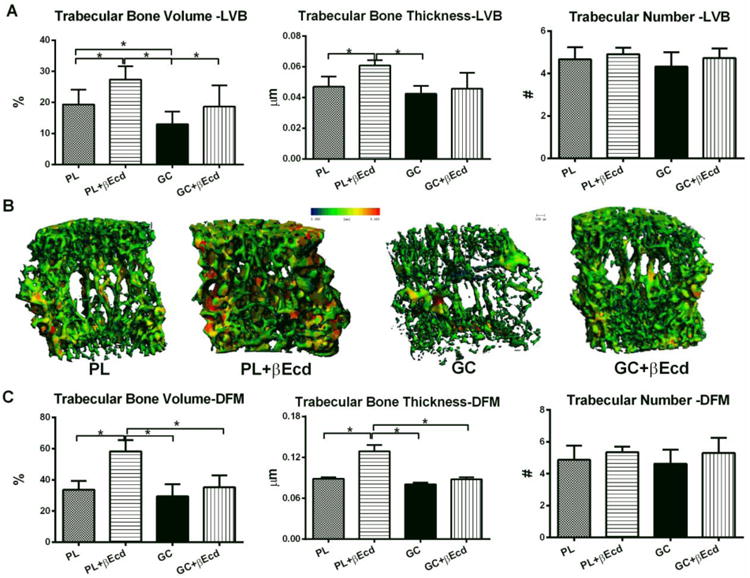
Effects of GC and βEcd treatments on the vertebral and femoral trabecular bone microarchitectures, assessed by microCT. Two-month-old mice were treated with βEcd, GC or concurrent treatment of GC + βEcd for 21 days. (A) Lumbar vertebral trabecular bone (LVB) structure measured by micro-CT. (B) Representative trabecular thickness maps were obtained from the LVB by micro-CT where the trabecular thickness is color coded: with blue-green colors indicate thinner trabeculae whereas yellow-red colors fare used for thicker trabeculae. (C) Distal femoral trabecular bone (DFM) structure as measured by microCT. *: p<0.05 between indicated groups.
More intriguing was that all parameters for trabecular bone formation measured at the 4th LVB, namely bone mineralizing surface (MS/BS), and bone formation rate/BS. In PL + βEcd treatment group compared to the PL, MS/BS and BFR/BS were increased by 43% and 30% (P < 0.05 vs. PL), respectively. In GC treatment group compared to the PL, GC significantly reduced MS/BS by 46%, MAR by 23% and BFR/BS by 60% (P < 0.05 vs. PL) while concurrent treatment of βEcd prevented these inhibitions of GC on bone formation parameters (Figure 2 A and B). In GC treatment group compared to the PL, GC significantly increased the osteoclast number/BS whereas concurrent treatment of βEcd prevented this change (Figure 2A).
Figure 2.
Effects of βEcd on trabecular bone formation. (A) Surface-based bone formation was measured at the un-decalcified LVB frozen sections. (B) Representative LVB sections from the treatment groups. *: p<0.05 between indicated groups.
Bone volume and bone turnover changes in the cortical bone
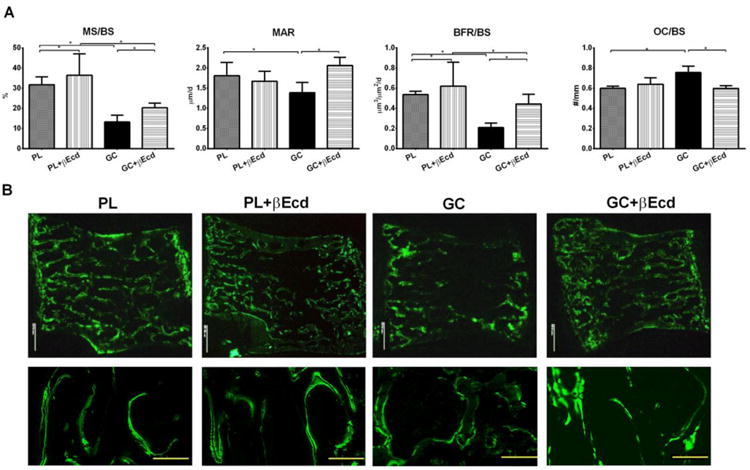
We next measured cortical bone architectural changes at the mid-femoral diaphyses by microCT and bone histomorphometry. When compared to the PL, cortical bone volume (Ct-BV) was increased by 4.5% in PL+ βEcd (P < 0.05 vs. PL), and was reduced by 4% in GC group P < 0.05 vs. PL). βEcd treatment did not prevented the loss in Ct-BV (Figure 3A). When compared to the PL, bone formation at the endocortical bone surface was increased by 213% in PL+ βEcd (P < 0.05 vs. PL), but reduced by 99% in GC group (P < 0.05 vs. PL), and was increased by 93% in the GC+ βEcd group as compared to GC group (P < 0.05 vs. GC). When compared to the PL, bone formation at the periosteal bone surface was increased by 55% in PL+ βEcd but reduced by 120% in GC group; it was 59% higher than GC in GC+ βEcd group (P < 0.05 vs. GC) (Figures 3 A and B).
Figure 3.
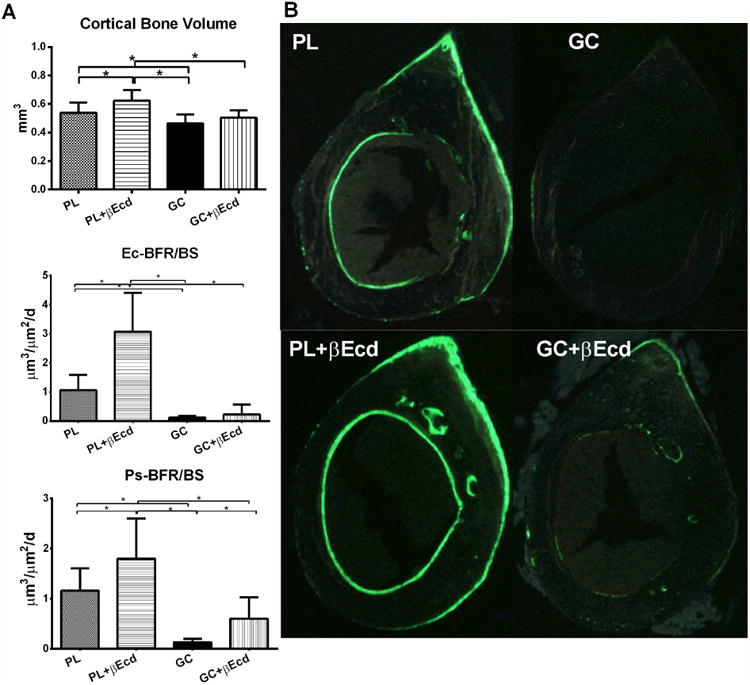
Effect of GC and βEcd treatments on cortical bone structure and surface-based bone turnover, assessed by microCT and bone histomorphometry. (A) Cortical bone volume and bone formation were measured at the mid-shaft of the femur. (B) Representative cross-sectional cortical bone sections from the mid-shaft of the femurs. *: p<0.05 between indicated treatment groups.
Bone strength measurements
The vertebral compression strength was measured by compression test. Compared to PL group, βEcd treatments resulted in a higher maximum load, ultimate stress and toughness by 53%, 21%, and 16% respectively (P < 0.05 vs. PL) (Table 2). On the other hand, GC reduced these parameters by 24%, 19%, and 16% respectively, as compared with the same PL mice (P < 0.05 vs. PL). By contrast, GC+ βEcd mice had higher maximum load, ultimate stress and toughness by 44%, 31%, and 56%, respectively, as compared to GC mice (P < 0.05 vs. GC). Likewise, βEcd induced higher maximum load, ultimate stress and toughness of the femurs by 23%, 42%, and 9% respectively, as compared to PL (P < 0.05 vs. PL). Interestingly, in GC-treated mice, there were no significant differences in the maximum load and ultimate stress but the cortical toughness was reduced by 24% as compared to PL (P < 0.05 vs. PL). GC+βEcd mice had higher maximum load, ultimate stress and toughness by 16%, 18%, and 21%, respectively, as compared to GC mice (P < 0.05 vs. GC), which were similar to the PL levels.
Table 2. Effect of GC and βEcd treatment on bone strength.
| Vertebral compression test | Femoral bending test | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Max Load (N) | Ultimate Stress (MPa) | Toughness (kJ/mˆ2) | Max Load (N) | Ultimate Stress (MPa) | Toughness kJ/mˆ2) | |
| PL | 20.2 ±3.7 | 3.32 ±0.7 | 0.51 ±0.1 | 13.1 ±2.1 | 182± 16 | 2.93 ± 0.9 |
| PL+βEcd | 30.9 ±3.8* | 4.03 ±0.9* | 0.59 ±0.1 | 16.1 ±3.5* | 259±49* | 3.21 ±1.0 |
| GC | 15.4 ±4.5* | 2.70±0.7* | 0.43 ±0.1* | 13.4 ±2.5 | 175±23 | 2.24 ±0.4* |
| GC + βEcd | 22.1 ±5.1* | 3.54±1.2# | 0.67 ±0.1# | 15.6±1.8# | 207 ± 6# | 2.70 ±0.4# |
,p < 0.05 vs. PL;
, P 0.05 vs. GC.
Effect of βEcd on osteogenesis and autophagy
To explore the mechanism of βEcd on bone formation, we first treated bone marrow stromal cells (BMSCs) with βEcd (10-4-10-9M; data from 10-5 and 10-8M was presented) in adipogenic or osteogenic media, we found the expression of pro-adipogenic genes (Cebp-α and Ppar-γ) were lowered by 5-20 fold (P < 0.05 vs. control), accompanied by less lipid formation (Figure 4A). On the other hand, BMSCs osteogenic differentiation was stimulated by βEcd, as shown by an increase in both osteoblastic gene expressions (Runx2 and Bglap1) (P < 0.05 vs. control) and ratio of the CFU-Ob to CFU-F (Figure 4B).
Figure 4.
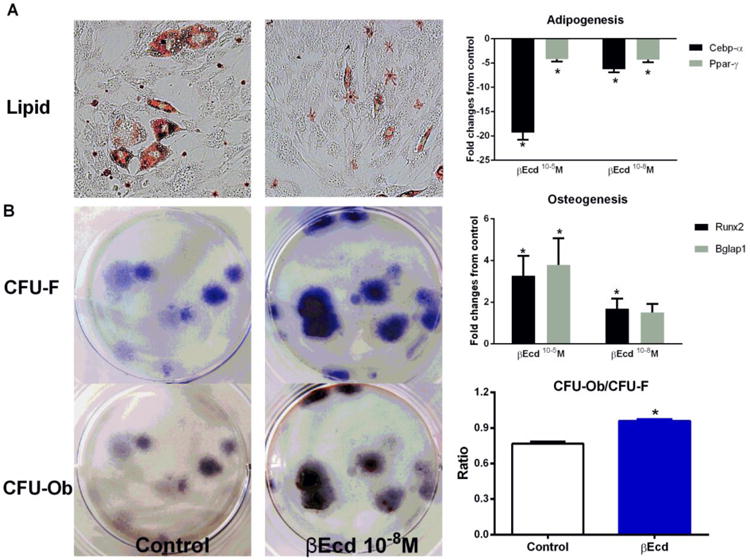
βEcd increases osteogenic differentiation of bone marrow stromal cells. Bone marrow cells were collected from long bones of male mice, two months of age and maintained in α-MEM with 10% FBS and antibiotics for four days. The adherent cells were collected and cultured in adipogenic or osteogenic media for 21 days. (A) Adipogenic differentiation measured by oil red staining and gene expressions related to adipogenesis (Cebp-α and Ppar-γ). (B) Osteogenesis measured the ratio of CFU-Ob/CFU-F and genes associated with osteoblast differentiation (Runx2 and Bglap1). Data are means ± SD. *: p<0.05 vs. control (PBS). All the studies were performed in triplicate.
To further explore how osteoblast and osteocyte viability was affected by treatment with βEcd, we evaluated osteoblast autophagy both in vitro and in vivo. The BMSCs were cultured in ostegenic media for 7 days before they were exposed to Dex or βEcd. We found the ratio of LC3+ primary osteoblast numbers were decreased by more than 50% in Dex –treated osteoblasts and combination treatment of βEcd prevented this decrease (Figure 5A and B).
Figure 5.
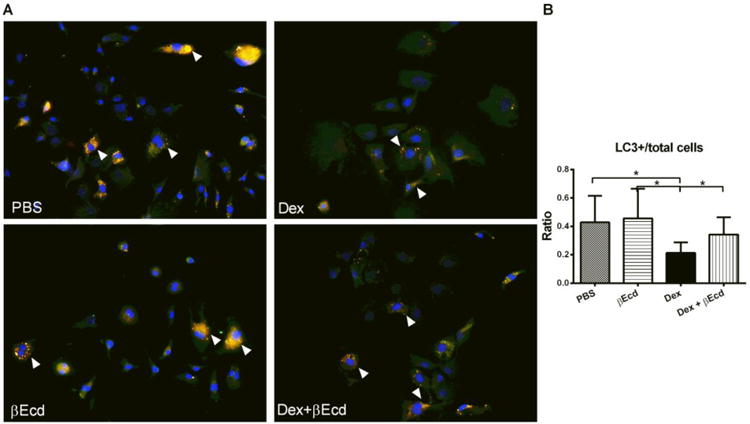
Effects of βEcd on osteoblast autophagy in vitro. BMSC cells were obtained from dsRed-LC3 reporter male mice and cultured in osteogenic medium for 10 days before they were treated with PBS, Dexamethasone (Dex, 10-6 M), βEcd (10-7M) or combination of Dex + βEcd in serum-starved conditions for eight hours. (A and B) Representative images (A) and quantitation of (B) dsRed-LC dots (autophagosomes, white arrows) in BMSC grown in osteogenic medium for 7 days and after 8 hours serum starvation incubation with Dex or βEcd as indicated. More than 200 cells were analyzed per sample. All the studies were performed in triplicate.
To evaluate the contribution of GC or with the combinational treatment of βEcd on autophagy in vivo, we first extracted RNA from the tibial cortical bone and performed RT-PCR array on autophagy assay that contained 80 genes associated with autophagy. We found that GC reduced the autophagic gene expression including key genes associated with autophagy induction, such as Atg7 and Beclin 1[45], by 1-5 folds while co-treatment of βEcd activated these autophagic gene expressions by 5-10 folds (Figure 6A). GC reduced expression of Atg7 and Atg16L by more than 50%, indicating reduced formation of autophagosome. On the other hand, GC decreased LC3-II/I ratio by about 15%, suggesting LC3 conversion might be not significantly affected following GC treatment in vivo. Concurrent of GC and βEcd treatment maintained protein levels of Atg7, Atg16L and the ratio of LC3II/I to PL control level (Figure 6B and C).
Figure 6.
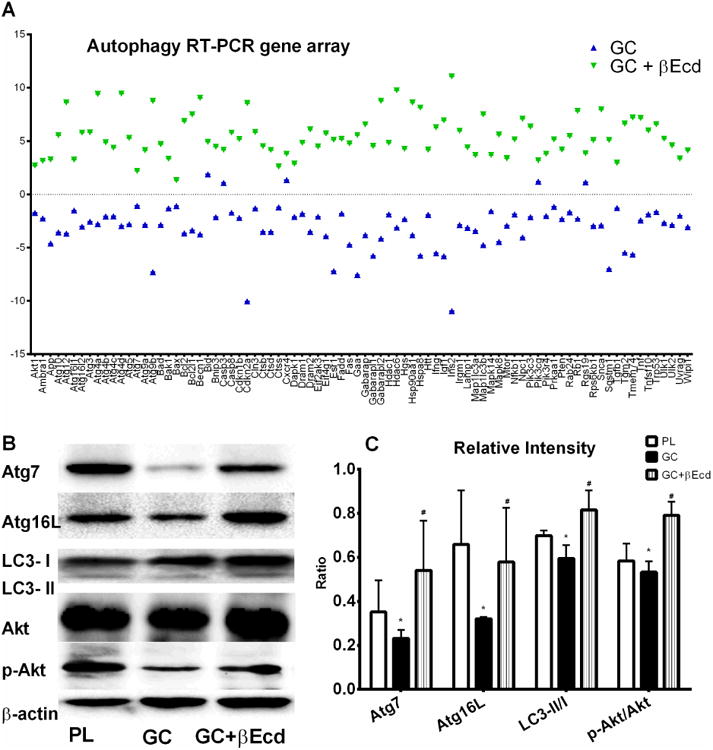
GC decreased while βEcd activated autophagy in bone. (A) RNA was extracted from the tibial shafts of PL, GC or GC+ βEcd treated mice at day 21. The focus RT-PCR gene array for autophagy was performed. RT-PCR data was expressed as fold changes from the PL group. (B) Proteins were extracted from the distal tibiae in animals treated with PL, GC or GC + βEcd. Western blots were performed for Atg7, Atg16L, LC3I/II, total Akt and p-Akt (S473). (C) Relative bend intensity measurements on B (n=3/group). *: p <0.05 vs. PL; #, p<0.05 vs. GC.
Discussion
In this report, we used two-month-old Swiss-Webster male mice and treated them with slow release prednisolone pellets to study the negative effect of GCs on bone growth and to see if treatment with the βEcd and GCs would alter bone growth. We found that three weeks of βEcd treatment alone or with combination of GC treatment altered the gain in body mass. βEcd treatment alone increased bone formation primary by increasing the osteoblast numbers (mineralizing surface) such that surface-based bone formation rate was significantly increased. The substantial improvements in bone formation in βEcd-treated mice translated into substantially higher bone mass with higher vertebral and cortical bone strength. Detrimental effect on both trabecular and cortical bone architecture and bone strength were apparent in mice receiving GCs, which included inhibitions in active bone formation at the trabecular bone and cortical bone expansion. βEcd treatment partially precluded inhibition of bone formation induced by GC, especially at the lumbar vertebrae and at the periosteal surface of the femoral shafts. Furthermore, our results demonstrated that βEcd increased the differentiation of mesenchymal progenitor cells towards osteoblast in vitro, consistent with the observation of increasing the osteoblast surface following βEcd treatment in vivo.
As GCs potently suppress osteoblast activities and adversely affect bone mineralization [41, 46], we hypothesized that therapies that target bone-forming capability would be superior to anti-resorptive agents. In support of this notion, it is reported that anabolic agent, hPTH (1-34), is superior to bisphosphonates in increasing spine and hip BMD and reduces the incidence of new vertebral fractures in glucocorticoid-induced osteoporosis (GIOP) populations for both men and women [11, 47-49]. In clinical practice, both bisphosphonates and PTH increase the spine BMD to a greater extent than the hip BMD [12, 14, 47, 50-53]. Prolonged bisphosphonates treatment may have an adverse effect on cortical bone mineralization and quality such that cortical strength decline independent of bone mass [54]. Prolonged GC treatments further worsen the bone quality [35, 55], and atypical fractures were more common in GIOP patients receiving bisphosphonate treatments ([56]). The effect of hPTH (1-34) on hip fracture reduction has not been shown in clinical studies in GIOP populations. [55, 57]. In our current study, we found GCs reduced vertebral bone strength, an observation that is consistent with bone loss [35, 41]. Interestingly, cortical bone maximum load and maximum stress measurements for cortical bone remained unchanged despite a decrease in bone volume. However, GC significantly reduced toughness, a measure of overall bone quality that measures the resistance to fracture [58]. This finding again suggests the adverse effect of GC on the cortical bone quality may not correlate with whole bone mass or bone mineral density [35, 59]. On the other hand, βEcd completely prevented losses in mechanical (eg. Maximum load) and material strengths (stress and toughness) in both the vertebral trabecular bone and femoral cortical bone despite it did not completely prevent GC-induced bone loss in these two bone sites. These findings suggest βEcd could be a good alternative treatment for GC-induced bone fragility.
βEcd treatment had an inconsistent effect on bone resorption in that it inhibited osteoclasts maturation in vitro (data on file) while increasing serum CTX-1. However, serum was collected only at one single time point (eg, at the end of three months) and thus the serum measurements may not reflect the dynamic changes over the entire experiment on osteoclast activities. Similar to PTH, βEcd treatment may activate bone turnover, increasing both bone resorption and bone formation, with the latter exceeding the former such that a net bone gain was observed [60].
Since GC reduced the body weight of the study mice by more than 20%, it was possible that the reduced body weight might have contributed to the GC-induced bone loss as well as the reduction in treatment efficacy for βEcd as GC dose was increased. We do not think this was the case as we evaluated the changes between weight-bearing bone site (distal femurs) and the non-weight bearing bone site (lumbar vertebral bodies) and found similar trends for GC or GC + βEcd with regards to bone formation and bone mass.
Mechanistically, βEcd is reported to promote muscle cell growth via insulin and PI3K/Akt signaling and is reported to inhibit NF-kB activation in a cancer cell line [21, 25, 27, 61]. βEcd binds to ecdysone receptor with EC50 of 0.3nm, a ligand-activated transcriptional factor found in arthropods [62, 63]. Since βEcd is structurally similar to testosterone, it is considered to have a steroid hormone-like effect. However, βEcd is shown to have no direct binding to the androgen receptor or other cytosolic steroid receptors [25, 64, 65], suggesting that our observation of an anabolic effect of βEcd on male skeleton may be independent of its androgen-like structure. Moreover, we did not observe that βEcd altered serum hormone levels of cortisol, estrogen, progesterone, T3, T4, insulin and leptin, suggesting that βEcd had a direct effect on the bone metabolism. Our finding that βEcd stimulated bone marrow stromal cells differentiating into osteoblasts supports a direct effect on bone cells.
Although far from conclusive, our exploratory experiments on autophagy in vitro and in vivo supported that βEcd might sustain the autophagic level in osteoblastic-like cells and in bone, whereas GC suppressed the induction of autophagy. Endogenous GCs are the main hormones released in response to stress and GC excess has been associated with nutrition deprived and accelerating aging process [66, 67]. We have reported that osteocyte autophagy is crucial to the regulation of bone structure and plays a role in GC induced bone fragility [44, 68]. It was shown that the removal of Atg7, a gene essential for autophagy initiation, resulted in low bone mass [69]. Autophagy is not a simple on – off phenomenon allowing a quick judgment of good/protective or bad/detrimental effect on cell metabolism. Rather, the level of autophagy has been maintained at a fine equilibrium so if or when the stress-inducing agent is removed, the cells might go on to survive. However, a sustained state of cellular stress, such as with high dose or chronic GC treatments, could result in failure of the induction of autophagy. Based on our results we propose that osteoblasts and osteocytes initially respond to GC induced stress by increasing the number of cells that undergo autophagy. The stress from low dose GC is nontoxic, allowing the cells to respond to the initial cellular or nutritional insults, which become no longer effective when the GC dose is high or with prolonged treatment. Defective autophagy may play a crucial role in maintaining bone integrity, especially in GC-induced bone fragility. The maintenance of bone cell viability through autophagy might partially explain βEcd effect on osteogenesis and on bone formation. βEcd may represent an important therapeutic option in the prevention of GC induced bone fragility by sustaining the level of autophagy in bone cells.
This study has several limitations. First, only male mice were studied one time point (21 days), with a single dose regimen of βEcd. Other studies will need to be done to determine if our result is present in female mice and if there is a dose response to the treatment. Second, this study only addressed the effect of βEcd for the prevention of GC induced bone loss. To evaluate if βEcd can increase bone mass in mice treated chronically with GCs will require another study. We used relatedly young mice that we would have to carefully dissect out the effect of GC and βEcd on bone growth inhibition verse a true decrease in bone volume. We did not measure the length of long bones and could not conclude if GC or βEcd affect growth. It has been shown that GCs normally decrease linear bone growth in animals and decrease bone mass, which are different from young patients on GCs who usually gain weight. Glucocorticoid-induced bone changes in children are usually confounded by the underlying disease and the changes in body mass [6, 7, 33]. The mice we used in the study gained about 20% body mass during the study, which might be similar to the skeleton of early adulthood finishing its maturation. Compared to the other studies that we and others have published on GIOP that used skeletal matured mice, we found GC excess in the young growing have more profound inhibition on periosteal expansion with similar degree of trabecular bone loss [35, 41]. Nevertheless, we showed that changes in bone volume correspond to changes in bone strength with GC or following βEcd treatment. These observations were similar between loaded or unloaded skeletal sides suggesting our findings were independent of body mass.
In summary, we have found that short-term administration of βEcd in growing male mice resulted in marked enhancements in both trabecular and cortical bone formation that were associated with significantly increased trabecular and cortical bone volume, both contributing to overall skeletal strength. These findings implicate the potential use of βEcd in augmenting peak bone mass. While GC treatment reduced endosteal bone formation and periosteal bone expansion, βEcd treatment prevented the detrimental effect of GC on bone formation, especially on trabecular bone. These results provide a strong pre-clinical support for testing the ability of βEcd treatment to improve skeletal fragility resulting from GC excess.
Highlights.
Beta-ecdysone (βEcd) completely prevented the GC-induced reduction in trabecular bone formation and bone strength.
βEcd partially prevented the GC-induced reduction in cortical bone formation and bone strength.
βEcd stimulated bone marrow stromal cells differentiation into osteoblast
βEcd maintained the autophagy level in bone, which was otherwise suppressed by GC.
Acknowledgments
This work was funded by National Institutes of Health grant R01 AR061366 (to WY), R01-AR43052 (to NEL), K24 AR048841 (to NEL), the endowed chair for aging at UC Davis (to NEL); Leading Academic Discipline Project of Shanghai Municipal Education Commission (SMEC) # J50301(to W.D), 085 Program, the Scientific and Technology Innovation Project of SMEC #085ZY1204 (to G.J).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goemaere S, Liberman UA, Adachi JD, Hawkins F, Lane N, Saag KG, Schnitzer T, Kaufman JM, Malice MP, Carofano W, Daifotis A. Incidence of nonvertebral fractures in relation to time on treatment and bone density in glucocorticoid-treated patients: a retrospective approach. J Clin Rheumatol. 2003;9:170–5. doi: 10.1097/01.RHU.0000073586.28977.31. [DOI] [PubMed] [Google Scholar]
- 2.Silverman SL, Lane NE. Glucocorticoid-induced osteoporosis. Curr Osteoporos Rep. 2009;7:23–6. doi: 10.1007/s11914-009-0005-4. [DOI] [PubMed] [Google Scholar]
- 3.Chiodini I, Carnevale V, Torlontano M, Fusilli S, Guglielmi G, Pileri M, Modoni S, Di Giorgio A, Liuzzi A, Minisola S, Cammisa M, Trischitta V, Scillitani A. Alterations of bone turnover and bone mass at different skeletal sites due to pure glucocorticoid excess: study in eumenorrheic patients with Cushing's syndrome. J Clin Endocrinol Metab. 1998;83:1863–7. doi: 10.1210/jcem.83.6.4880. [DOI] [PubMed] [Google Scholar]
- 4.Michaud K, Forget H, Cohen H. Chronic glucocorticoid hypersecretion in Cushing's syndrome exacerbates cognitive aging. Brain Cogn. 2009;71:1–8. doi: 10.1016/j.bandc.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Sorva R, Turpeinen M, Juntunen-Backman K, Karonen SL, Sorva A. Effects of inhaled budesonide on serum markers of bone metabolism in children with asthma. J Allergy Clin Immunol. 1992;90:808–15. doi: 10.1016/0091-6749(92)90106-c. [DOI] [PubMed] [Google Scholar]
- 6.Leonard MB. Glucocorticoid-induced osteoporosis in children: impact of the underlying disease. Pediatrics. 2007;119(Suppl 2):S166–74. doi: 10.1542/peds.2006-2023J. [DOI] [PubMed] [Google Scholar]
- 7.Burnham JM, Shults J, Semeao E, Foster B, Zemel BS, Stallings VA, Leonard MB. Whole body BMC in pediatric Crohn disease: independent effects of altered growth, maturation, and body composition. J Bone Miner Res. 2004;19:1961–8. doi: 10.1359/JBMR.040908. [DOI] [PubMed] [Google Scholar]
- 8.Root AW, Bongiovanni AM, Eberlein WR. Studies of the secretion and metabolic effects of human growth hormone in children with glucocorticoid-induced growth retardation. J Pediatr. 1969;75:826–32. doi: 10.1016/s0022-3476(69)80306-9. [DOI] [PubMed] [Google Scholar]
- 9.Allen DB, Julius JR, Breen TJ, Attie KM. Treatment of glucocorticoid-induced growth suppression with growth hormone. National Cooperative Growth Study. J Clin Endocrinol Metab. 1998;83:2824–9. doi: 10.1210/jcem.83.8.5036. [DOI] [PubMed] [Google Scholar]
- 10.Saag KG, Emkey R, Schnitzer TJ, Brown JP, Hawkins F, Goemaere S, Thamsborg G, Liberman UA, Delmas PD, Malice MP, Czachur M, Daifotis AG. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med. 1998;339:292–9. doi: 10.1056/NEJM199807303390502. [DOI] [PubMed] [Google Scholar]
- 11.Saag KG, Shane E, Boonen S, Marin F, Donley DW, Taylor KA, Dalsky GP, Marcus R. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357:2028–39. doi: 10.1056/NEJMoa071408. [DOI] [PubMed] [Google Scholar]
- 12.Reid DM, Devogelaer JP, Saag K, Roux C, Lau CS, Reginster JY, Papanastasiou P, Ferreira A, Hartl F, Fashola T, Mesenbrink P, Sambrook PN. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet. 2009;373:1253–63. doi: 10.1016/S0140-6736(09)60250-6. [DOI] [PubMed] [Google Scholar]
- 13.Lane NE, Sanchez S, Modin GW, Genant HK, Pierini E, Arnaud CD. Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis. Results of a randomized controlled clinical trial. J Clin Invest. 1998;102:1627–33. doi: 10.1172/JCI3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue Y, Shimojo N, Suzuki S, Arima T, Tomiita M, Minagawa M, Kohno Y. Efficacy of intravenous alendronate for the treatment of glucocorticoid-induced osteoporosis in children with autoimmune diseases. Clin Rheumatol. 2008;27:909–12. doi: 10.1007/s10067-008-0864-6. [DOI] [PubMed] [Google Scholar]
- 15.Henderson S, Hoffman N, Prince R. A double-blind placebo-controlled study of the effects of the bisphosphonate risedronate on bone mass in patients with inflammatory bowel disease. Am J Gastroenterol. 2006;101:119–23. doi: 10.1111/j.1572-0241.2006.00372.x. [DOI] [PubMed] [Google Scholar]
- 16.Roldan EJ, Pasqualini T, Plantalech L. Bisphosphonates in children with osteogenesis imperfecta may improve bone mineralization but not bone strength. Report of two patients. J Pediatr Endocrinol Metab. 1999;12:555–9. doi: 10.1515/jpem.1999.12.4.555. [DOI] [PubMed] [Google Scholar]
- 17.Rudge S, Hailwood S, Horne A, Lucas J, Wu F, Cundy T. Effects of once-weekly oral alendronate on bone in children on glucocorticoid treatment. Rheumatology (Oxford) 2005;44:813–8. doi: 10.1093/rheumatology/keh538. [DOI] [PubMed] [Google Scholar]
- 18.Brown JJ, Zacharin MR. Proposals for prevention and management of steroid-induced osteoporosis in children and adolescents. J Paediatr Child Health. 2005;41:553–7. doi: 10.1111/j.1440-1754.2005.00718.x. [DOI] [PubMed] [Google Scholar]
- 19.Papapoulos SE, Cremers SC. Prolonged bisphosphonate release after treatment in children. N Engl J Med. 2007;356:1075–6. doi: 10.1056/NEJMc062792. [DOI] [PubMed] [Google Scholar]
- 20.Hansen KE, Wilson HA, Zapalowski C, Fink HA, Minisola S, Adler RA. Uncertainties in the prevention and treatment of glucocorticoid-induced osteoporosis. J Bone Miner Res. 2011;26:1989–96. doi: 10.1002/jbmr.362. [DOI] [PubMed] [Google Scholar]
- 21.Badisco L, Van Wielendaele P, Vanden Broeck J. Eat to reproduce: a key role for the insulin signaling pathway in adult insects. Front Physiol. 2013;4:202. doi: 10.3389/fphys.2013.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boo KH, Lee D, Jeon GL, Ko SH, Cho SK, Kim JH, Park SP, Hong Q, Lee SH, Lee DS, Riu KZ. Distribution and biosynthesis of 20-hydroxyecdysone in plants of Achyranthes japonica Nakai. Biosci Biotechnol Biochem. 2010;74:2226–31. doi: 10.1271/bbb.100410. [DOI] [PubMed] [Google Scholar]
- 23.Slama K, Koudela K, Tenora J, Mathova A. Insect hormones in vertebrates: anabolic effects of 20-hydroxyecdysone in Japanese quail. Experientia. 1996;52:702–6. doi: 10.1007/BF01925578. [DOI] [PubMed] [Google Scholar]
- 24.Gorelick-Feldman J, MacLean D, Ilic N, Poulev A, Lila MA, Cheng D, Raskin I. Phytoecdysteroids increase protein synthesis in skeletal muscle cells. Journal of Agricultural and Food Chemistry. 2008;56:3532–3537. doi: 10.1021/jf073059z. [DOI] [PubMed] [Google Scholar]
- 25.Esposito D, Komarnytsky S, Shapses S, Raskin I. Anabolic effect of plant brassinosteroid. FASEB J. 2011;25:3708–19. doi: 10.1096/fj.11-181271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng DM, Kutzler LW, Boler DD, Drnevich J, Killefer J, Lila MA. Continuous infusion of 20-hydroxyecdysone increased mass of triceps brachii in C57BL/6 mice. Phytother Res. 2013;27:107–11. doi: 10.1002/ptr.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esposito D, Rathinasabapathy T, Poulev A, Komarnytsky S, Raskin I. Akt-dependent anabolic activity of natural and synthetic brassinosteroids in rat skeletal muscle cells. J Med Chem. 2011;54:4057–66. doi: 10.1021/jm200028h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toth N, Szabo A, Kacsala P, Heger J, Zador E. 20-Hydroxyecdysone increases fiber size in a muscle-specific fashion in rat. Phytomedicine. 2008;15:691–8. doi: 10.1016/j.phymed.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 29.Lafont R, Dinan L. Practical uses for ecdysteroids in mammals including humans: an update. J Insect Sci. 2003;3:7. doi: 10.1093/jis/3.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao L, Cai G, Shi X. Beta-ecdysterone induces osteogenic differentiation in mouse mesenchymal stem cells and relieves osteoporosis. Biol Pharm Bull. 2008;31:2245–9. doi: 10.1248/bpb.31.2245. [DOI] [PubMed] [Google Scholar]
- 31.Kapur P, Wuttke W, Jarry H, Seidlova-Wuttke D. Beneficial effects of beta-Ecdysone on the joint, epiphyseal cartilage tissue and trabecular bone in ovariectomized rats. Phytomedicine. 2010;17:350–5. doi: 10.1016/j.phymed.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Seidlova-Wuttke D, Christel D, Kapur P, Nguyen BT, Jarry H, Wuttke W. Beta-ecdysone has bone protective but no estrogenic effects in ovariectomized rats. Phytomedicine. 2010;17:884–9. doi: 10.1016/j.phymed.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 33.Leonard MB, Feldman HI, Shults J, Zemel BS, Foster BJ, Stallings VA. Long-term, high-dose glucocorticoids and bone mineral content in childhood glucocorticoid-sensitive nephrotic syndrome. N Engl J Med. 2004;351:868–75. doi: 10.1056/NEJMoa040367. [DOI] [PubMed] [Google Scholar]
- 34.Yao W, Cheng Z, Busse C, Pham A, Nakamura MC, Lane NE. Glucocorticoid excess in mice results in early activation of osteoclastogenesis and adipogenesis and prolonged suppression of osteogenesis: a longitudinal study of gene expression in bone tissue from glucocorticoid-treated mice. Arthritis Rheum. 2008;58:1674–86. doi: 10.1002/art.23454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, Habelitz S, Kinney JH, Bonewald LF. Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. J Bone Miner Res. 2006;21:466–76. doi: 10.1359/JBMR.051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao W, Guan M, Jia J, Dai W, Lay YA, Amugongo S, Liu R, Olivos D, Saunders M, Lam KS, Nolta J, Olvera D, Ritchie RO, Lane NE. Reversing bone loss by directing mesenchymal stem cells to bone. Stem Cells. 2013;31:2003–14. doi: 10.1002/stem.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan M, Yao W, Liu R, Lam KS, Nolta J, Jia J, Panganiban B, Meng L, Zhou P, Shahnazari M, Ritchie RO, Lane NE. Directing mesenchymal stem cells to bone to augment bone formation and increase bone mass. Nat Med. 2012;18:456–62. doi: 10.1038/nm.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao W, Dai W, Shahnazari M, Pham A, Chen Z, Chen H, Guan M, Lane NE. Inhibition of the progesterone nuclear receptor during the bone linear growth phase increases peak bone mass in female mice. PLoS ONE. 2010;5:e11410. doi: 10.1371/journal.pone.0011410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao W, Cheng Z, Shahnazari M, Dai W, Johnson ML, Lane NE. Overexpression of Secreted Frizzled-Related Protein 1 Inhibits Bone Formation and Attenuates PTH Bone Anabolic Effects. J Bone Miner Res. 2009 doi: 10.1359/jbmr.090719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28:2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao W, Cheng Z, Pham A, Busse C, Zimmermann EA, Ritchie RO, Lane NE. Glucocorticoid-induced bone loss in mice can be reversed by the actions of parathyroid hormone and risedronate on different pathways for bone formation and mineralization. Arthritis Rheum. 2008;58:3485–3497. doi: 10.1002/art.23954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turner CH, Burr DB. Basic biomechanical measurements of bone: a tutorial. Bone. 1993;14:595–608. doi: 10.1016/8756-3282(93)90081-k. [DOI] [PubMed] [Google Scholar]
- 43.Yao W, Cheng Z, Shahnazari M, Dai W, Johnson ML, Lane NE. Overexpression of secreted frizzled-related protein 1 inhibits bone formation and attenuates parathyroid hormone bone anabolic effects. J Bone Miner Res. 2010;25:190–9. doi: 10.1359/jbmr.090719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jia J, Yao W, Guan M, Dai W, Shahnazari M, Kar R, Bonewald L, Jiang JX, Lane NE. Glucocorticoid dose determines osteocyte cell fate. FASEB J. 2011;25:3366–76. doi: 10.1096/fj.11-182519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Shoji-Kawata S, Sumpter RM, Jr, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110:20364–71. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balooch G, Yao W, Ager JW, Balooch M, Nalla RK, Porter AE, Ritchie RO, Lane NE. The aminobisphosphonate risedronate preserves localized mineral and material properties of bone in the presence of glucocorticoids. Arthritis Rheum. 2007;56:3726–3737. doi: 10.1002/art.22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devogelaer JP, Sambrook P, Reid DM, Goemaere S, Ish-Shalom S, Collette J, Su G, Bucci-Rechtweg C, Papanastasiou P, Reginster JY. Effect on bone turnover markers of once-yearly intravenous infusion of zoledronic acid versus daily oral risedronate in patients treated with glucocorticoids. Rheumatology (Oxford) 2013;52:1058–69. doi: 10.1093/rheumatology/kes410. [DOI] [PubMed] [Google Scholar]
- 48.Gluer CC, Marin F, Ringe JD, Hawkins F, Moricke R, Papaioannu N, Farahmand P, Minisola S, Martinez G, Nolla JM, Niedhart C, Guanabens N, Nuti R, Martin-Mola E, Thomasius F, Kapetanos G, Pena J, Graeff C, Petto H, Sanz B, Reisinger A, Zysset PK. Comparative effects of teriparatide and risedronate in glucocorticoid-induced osteoporosis in men: 18-month results of the EuroGIOPs trial. J Bone Miner Res. 2013;28:1355–68. doi: 10.1002/jbmr.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saag KG, Zanchetta JR, Devogelaer JP, Adler RA, Eastell R, See K, Krege JH, Krohn K, Warner MR. Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: thirty-six-month results of a randomized, double-blind, controlled trial. Arthritis Rheum. 2009;60:3346–55. doi: 10.1002/art.24879. [DOI] [PubMed] [Google Scholar]
- 50.Jacobs JW, de Nijs RN, Lems WF, Geusens PP, Laan RF, Huisman AM, Algra A, Buskens E, Hofbauer LC, Oostveen AC, Bruyn GA, Dijkmans BA, Bijlsma JW. Prevention of glucocorticoid induced osteoporosis with alendronate or alfacalcidol: relations of change in bone mineral density, bone markers, and calcium homeostasis. J Rheumatol. 2007;34:1051–7. [PubMed] [Google Scholar]
- 51.Stoch SA, Saag KG, Greenwald M, Sebba AI, Cohen S, Verbruggen N, Giezek H, West J, Schnitzer TJ. Once-Weekly Oral Alendronate 70 mg in Patients with Glucocorticoid-Induced Bone Loss: A 12-Month Randomized, Placebo-Controlled Clinical Trial. J Rheumatol. 2009 doi: 10.3899/jrheum.081207. [DOI] [PubMed] [Google Scholar]
- 52.Kaji H, Kuroki Y, Murakawa Y, Funakawa I, Funasaka Y, Kanda F, Sugimoto T. Effect of alendronate on bone metabolic indices and bone mineral density in patients treated with high-dose glucocorticoid: a prospective study. Osteoporos Int. 2010;21:1565–71. doi: 10.1007/s00198-009-1110-z. [DOI] [PubMed] [Google Scholar]
- 53.Sambrook PN, Roux C, Devogelaer JP, Saag K, Lau CS, Reginster JY, Bucci-Rechtweg C, Su G, Reid DM. Bisphosphonates and glucocorticoid osteoporosis in men: results of a randomized controlled trial comparing zoledronic acid with risedronate. Bone. 2012;50:289–95. doi: 10.1016/j.bone.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 54.Shahnazari M, Yao W, Dai W, Wang B, Ionova-Martin SS, Ritchie RO, Heeren D, Burghardt AJ, Nicolella DP, Kimiecik MG, Lane NE. Higher doses of bisphosphonates further improve bone mass, architecture, and strength but not the tissue material properties in aged rats. Bone. 2010;46:1267–74. doi: 10.1016/j.bone.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum. 2003;48:3224–9. doi: 10.1002/art.11283. [DOI] [PubMed] [Google Scholar]
- 56.Shane E, Burr D, Ebeling PR, Abrahamsen B, Adler RA, Brown TD, Cheung AM, Cosman F, Curtis JR, Dell R, Dempster D, Einhorn TA, Genant HK, Geusens P, Klaushofer K, Koval K, Lane JM, McKiernan F, McKinney R, Ng A, Nieves J, O'Keefe R, Papapoulos S, Sen HT, van der Meulen MC, Weinstein RS, Whyte M American Society for B, Mineral R. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267–94. doi: 10.1002/jbmr.253. [DOI] [PubMed] [Google Scholar]
- 57.Papapoulos SE, Schimmer RC. Changes in bone remodelling and antifracture efficacy of intermittent bisphosphonate therapy: implications from clinical studies with ibandronate. Ann Rheum Dis. 2007;66:853–8. doi: 10.1136/ard.2006.064931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kruzic JJ, Ritchie RO. Comments on “Measurement of the microstructural fracture toughness of cortical bone using indentation fracture”. J Biomech. 2008;41:1379–80. doi: 10.1016/j.jbiomech.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 59.Saag KG. Bone safety of low-dose glucocorticoids in rheumatic diseases. Ann N Y Acad Sci. 2014;1318:55–64. doi: 10.1111/nyas.12446. [DOI] [PubMed] [Google Scholar]
- 60.Hock JM. Anabolic actions of PTH in the skeletons of animals. J Musculoskelet Neuronal Interact. 2001;2:33–47. [PubMed] [Google Scholar]
- 61.Peschel W, Kump A, Prieto JM. Effects of 20-hydroxyecdysone, Leuzea carthamoides extracts, dexamethasone and their combinations on the NF-kappaB activation in HeLa cells. J Pharm Pharmacol. 2011;63:1483–95. doi: 10.1111/j.2042-7158.2011.01349.x. [DOI] [PubMed] [Google Scholar]
- 62.Riddiford LM, Cherbas P, Truman JW. Ecdysone receptors and their biological actions. Vitam Horm. 2000;60:1–73. doi: 10.1016/s0083-6729(00)60016-x. [DOI] [PubMed] [Google Scholar]
- 63.Lapenna S, Friz J, Barlow A, Palli SR, Dinan L, Hormann RE. Ecdysteroid ligand-receptor selectivity--exploring trends to design orthogonal gene switches. FEBS J. 2008;275:5785–809. doi: 10.1111/j.1742-4658.2008.06687.x. [DOI] [PubMed] [Google Scholar]
- 64.Bathori M. Phytoecdysteroids effects on mammalians, isolation and analysis. Mini Rev Med Chem. 2002;2:285–93. doi: 10.2174/1389557023406269. [DOI] [PubMed] [Google Scholar]
- 65.Toth N, Hunyadi A, Bathori M, Zador E. Phytoecdysteroids and vitamin D analogues--similarities in structure and mode of action. Curr Med Chem. 2010;17:1974–94. doi: 10.2174/092986710791163911. [DOI] [PubMed] [Google Scholar]
- 66.Zheng B, Ohkawa S, Li H, Roberts-Wilson TK, Price SR. FOXO3a mediates signaling crosstalk that coordinates ubiquitin and atrogin-1/MAFbx expression during glucocorticoid-induced skeletal muscle atrophy. FASEB J. 2010;24:2660–9. doi: 10.1096/fj.09-151480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sapolsky RM, Krey LC, McEwen BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci. 1985;5:1222–7. doi: 10.1523/JNEUROSCI.05-05-01222.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, Biswas SK, Lo WK, Jiang JX. Glucocorticoid induced autophagy in osteocytes. J Bone Miner Res. 2010 doi: 10.1002/jbmr.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Onal M, Piemontese M, Xiong J, Wang Y, Han L, Ye S, Komatsu M, Selig M, Weinstein RS, Zhao H, Jilka RL, Almeida M, Manolagas SC, O'Brien CA. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem. 2013;288:17432–40. doi: 10.1074/jbc.M112.444190. [DOI] [PMC free article] [PubMed] [Google Scholar]