

Abstract
With the emergence of drug-resistant strains and the cumulative toxicities associated with current therapies, demand remains for new inhibitors of HIV-1 replication. The inhibition of HIV-1 entry is an attractive, yet underexploited therapeutic approach with implications for salvage and preexposure prophylactic regimens, as well as topical microbicides. Using the combination of a field-derived bioactive conformation template to perform virtual screening and iterative bioisosteric replacements, coupled with in silico predictions of absorption, distribution, metabolism, and excretion, we have identified new leads for HIV-1 entry inhibitors.
Keywords: Field-based virtual screening, Three-dimensional virtual screening, HIV-1 Env, Antiviral, Bioisostere, Entry inhibitor, Field-based scaffold hopping
With the emergence of drug-resistant strains and the cumulative toxicities associated with current therapies, demand remains for new inhibitors of HIV-1 replication. The inhibition of HIV-1 entry is an attractive, yet underexploited therapeutic approach with implications for salvage and preexposure prophylactic regimens, as well as topical microbicides. The entry of HIV-1 into permissible cells is a complex series of events orchestrated by the viral envelope glycoprotein complex, the only exposed viral components on the virion surface. Being the only viral products accessible to the host cell immune system, the Env glycoproteins gp120 and gp41 have evolved several strategies to mask functionally important regions from the neutralizing antibody response. These strategies include the presence of surface-exposed variable loops on gp120, a high degree of glycosylation, the lability and defectiveness of many envelope glycoprotein spikes (possible immunologic decoys), and conformational flexibility.1-4
The Env complex is organized on the virion surface as trimeric spikes composed of three gp120 molecules noncovalently linked to three gp41 molecules. The heavily glycosylated surface gp1205 contains a core composed of conserved regions (C1 to C5) and hypervariable regions that are mostly disulfide-constrained, surface-exposed loop structures (V1 to V5) that retain a large degree of flexibility.4, 6-9 The transmembrane glycoprotein gp41 contains the fusion peptide that is inserted into the membrane of the target cells,10 as well as two heptad repeat (HR) domains (aminoterminal or HR1 and carboxyterminal or HR2) that are implicated in the formation of a six-helix-bundle fusion intermediate via a conformational change following receptor interaction. HIV-1 infection usually occurs only after two sequential and specific binding steps: first, the Env gp120 protein binding to the CD4 antigen present in CD4+ T cells, monocyte/macrophages, and other immune and nonimmune cells; and second, gp120 binding to a member of the chemokine receptor subfamily, within the large G protein–coupled family of receptors, mainly CCR5 and/or CXCR4.
Advances in knowledge of the molecular mechanisms of HIV-1 entry have allowed the discovery and development of molecules that target discrete steps in the entry process and have shown success in the clinic. Successful examples include maraviroc (Selzentry; Pfizer, New York, NY), which binds to CCR5 and blocks the interaction of the Env complex with the coreceptor, and enfuvirtide (Fuzeon; Hoffman-La Roche, Nutley, NJ), which binds to gp41 and stops the fusion of the viral and cell membrane. However, as of yet, no gp120-targeted therapies have been approved for use in the clinic.
In the HIV-1 entry field, two main Env-targeted inhibitor chemotypes predominate: the NBD-556 analogues11 and the BMS-378806 analogues (Figure 1).12 NBD-556 and its analogues bind to the conserved CD4-binding site in gp120 and block the interaction of the Env complex with cellular CD4.13-15 The binding site for BMS-378806 and its analogues is poorly understood, and based on resistance mutation data it may actually be a composite site composed of regions of gp120 and g41.16 The mechanism of action of BMS-378806 and its derivatives is also under debate, with some studies claiming a CD4 binding inhibition mechanism and others describing an allosteric mechanism that prevents the propagation of the receptor binding signals from gp120 to gp41.17, 18 Given the huge therapeutic potential of inhibiting HIV-1 entry, the development of new chemotypes that target viral entry with broad activities is highly desirable. In this study we describe the use of high-content field-based pharmacophore screening as a first step in identifying new chemotypes for this inhibitor class.
Figure 1.

Structures of entry inhibitors developed by Bristol-Myers Squibb and the New York Blood Bank. The chemical structures were drawn with ChemAxon software (Budapest, Hungary).
To date, the piperazine-based entry inhibitors as first described by Bristol-Myers Squibb12 are the most broadly acting and potent HIV-1 entry inhibitors. This indicates that the binding site for these compounds, although currently not well described, is well conserved and available for targeting on the virion and that compounds such as BMS-663068 may have great therapeutic value. In fact, the entry inhibitor BMS-663068 recently performed favorably in a phase IIb clinical trial (presented at the Conference on Retroviruses and Opportunistic Infections, March 3–March 6, 2014, Boston MA).
Although the agents are potent and have a broad therapeutic spectrum, the piperazine class of entry inhibitors developed by Bristol-Myers Squibb has been plagued by the problems of low solubility and poor intrinsic dissolution properties. To circumvent these issues, a prodrug approach was adopted. BMS-663068 is a phosphonooxymethyl prodrug of BMS-62652914,16-18 designed to have increased solubility in the gut. The prodrug is thought to be cleaved by alkaline phosphatase, located on the luminal surface of the small intestine brush border membranes, releasing BMS-626529, which is then rapidly absorbed.19 Despite the success of this approach, an entry inhibitor candidate with more intrinsic druglike properties would be preferable.
As a first step towards discovering entry inhibitor candidates with intrinsic druglike properties, we conducted a series of field-based three-dimensional similarity virtual screening experiments using Blaze (Cresset, Litlington, UK) with a high-content field-based pharmacophore template derived from BMS-62652917, 19-21 (Forge/FieldTemplater, Cresset) in order to identify novel scaffolds that could function as entry inhibitors. To perform a Blaze virtual screen, an active ligand in its three-dimensional bioactive conformation should be used as a search query. In the absence of structural information, a binding mode hypothesis may be calculated.
As no structural information is currently available for BMS-626529 in its target-bound state, we used FieldTemplater (Forge) to determine a hypothesis for the three-dimensional conformation adopted in binding to the target using field and shape information to create a template using compounds BMS-626529, BMS-488043, and BMS-378806.16, 22 The FieldTemplater-derived hypothesis for the bioactive conformation was then annotated with its calculated field points, resulting in a three-dimensional field point pattern. The field point pattern provides a condensed representation of the compound’s shape, electrostatics, and hydrophobicity. It is well established that when two diverse structures have conformations with similar field point patterns, they are experienced by the receptor in a similar fashion.23-33 The field-based alignment for the three selected templating molecules is shown in Figure 2.
Figure 2.

(a) Field-based template A containing a single conformation of compounds BMS-378806 (pink), BMS-488043 (lime green), and BMS-626529 (teal green) aligned based on their three-dimensional field point patterns. Negatively charged field points are shown in blue; positively charged field points are red; van der Waals/shape field points are displayed in yellow; centers of hydrophobicity are shown in orange. (b) Conformation 69 of BMS-626529 with its associated field points. This conformation was common to the top three templates identified and was used as the query structure for the Blaze (Cresset, Litlington, UK) field-based virtual screen
The field point pattern for the hypothesized three-dimensional bioactive conformation of BMS-626529 was subsequently used to query a database of approximately 6 million commercially available compounds using Blaze. The steps involved in the Blaze virtual screening procedure are shown schematically in Figure 3. The Blaze procedure resulted in the rank ordering of the top 1000 Blaze library compounds whose three-dimensional arrangements of field points had similarities to that of the BMS-626529 template structure. Fifty compounds were chosen and purchased for biological testing using the single-round infection assay.34 The structures of the selected compounds are shown as supplementary data. Those compounds found to be active and specific, as judged by their activity against HIV-1YU-2 Env pseudotyped and amphotropic murine leukemia virus (AMLV) Env pseudotyped HIV-1 virus, are shown in Table 1. Five compounds with specific anti-HIV-1 activity were discovered using this approach with half-maximal inhibitory concentrations (IC50) in the range of 13 to 150 μM. Compound 11 was found to be the most potent, inhibiting HIV-1YU-2 with an IC50 value of 13.1 ± 1.7 μM.
Figure 3.

Steps involved in a Blaze (Cresset, Litlington, UK) field-based virtual screen experiment. (1) An active molecule is selected and converted to a relevant bioactive conformation. (2) Field points are added to this search ligand in the specified conformation to produce the Blaze pharmacophore seed. (3) The Blaze search query consists of the field point pattern of the pharmacophore seed, which is used to search the Blaze database by alignment of every structure based on field point patterns (of its up to 100 conformations). (4) Top scoring compounds are retrieved as three-dimensional alignments to the search query, along with their score (molecular similarity based on 50% shape, 50% fields).
Table 1.
Structure and potency of compounds identified within this study with specific activity against HIV-1 YU-2. Chemical structures were drawn with ChemAxon software (Budapest, Hungary). NA = not active over concentration range tested.
| Compound | IC50 YU-2 (μM) | IC50 AMLV (μM) |
|---|---|---|
| 11 | ||
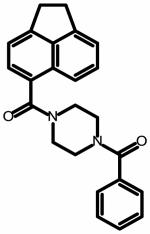
|
13.1 ± 1.7 | NA |
|
| ||
| 12 | ||
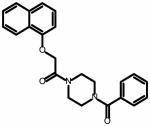
|
53.5 ± 3.0 | NA |
|
| ||
| 28 | ||
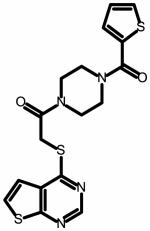
|
33.7 ± 4.5 | NA |
|
| ||
| 32 | ||
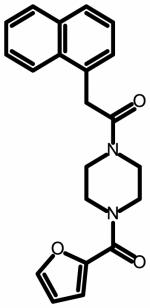
|
79.4 ± 11 | NA |
|
| ||
| 34 | ||
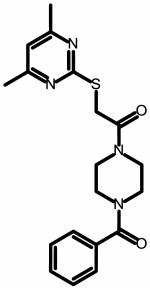
|
153 ± 44 | NA |
The active and specific compounds identified from this study, despite showing a degree of chemical diversity, all share a piperazine core comparable to the Bristol-Myers Squibb compounds. Therefore, we investigated whether the piperazine core could be replaced with another moiety that would function in the same capacity but be chemically distinct. Pfizer reported a study in which replacement of an N-methylpiperazine in a lead compound, JNJ-7777120, with 2-methyloctahydropyrrolo[3,4-c]pyrrole led to a compound that retained binding affinity and selectivity but also had improved resistance to metabolism.35 We therefore decided to determine whether this dipyrrolidine group could successfully replace the piperazine in compound 11. In silico bioisostere replacement (scaffold-hopping) experiments using Spark (Cresset, UK) conducted to assess the potential for this group to substitute the piperazine in 11 indicated that it may be a viable, if not imperfect replacement. This conclusion was based upon the value of the calculated bioisostere factor (BIF%), a rescaled score, which indicates how much better or worse the replacement is compared with simply capping the attachment point(s) with hydrogen(s). Positive BIF% values indicate good bioisosteres; negative values correspond to replacements where the geometry of the original molecule is reproduced; however, the fragment is not a good mimic of the replaced part.
A BIF% of 57 was obtained for the dipyrrolidine group, and therefore this replacement for piperazine was investigated. Substitution of the piperazine core in 11 with 2-methyloctahydropyrrolo[3,4-c]pyrrole yielded compound SC04 ([5-(1,2-dihydro acenaphthylene-5-carbonyl)-1,3,3a,4,6,6a-hexahydropyrrolo[3,4-c]pyrrol-2-yl]-phenyl-methanone]), a novel compound that retains the properties of 11, albeit with lower potency (IC50 HIV-1YU-2 = 70 ± 6 μM; IC50 HIV-1JR-CSF = 100 ± 30 μM).
Having identified a new scaffold, we focused on redesigning compound SC04 to improve its potency and to remove any potential toxophores. The head region of SC04, like compound 11, is an acenaphthene group, which may be carcinogenic owing to its potential to intercalate into DNA sequences. Moreover, several studies by Bristol-Myers Squib have demonstrated that this head region greatly influences the potency of compounds in the piperazine class. Subsequently, in order to improve the potency of SC04, we downloaded from PubChem 453 compounds that displayed similarity to BMS-488043. These compounds were exhaustively fragmented (Spark DBGen, Cresset, UK), and we used the resulting fragments in iterative Spark experiments, seeking fragments that could function as bioisosteric substitutions for the acenaphthene moiety but with greater potential for hydrogen bonding interactions. This resulted in the identification of two head groups, 7-chloro-4-methoxy-1H-pyrrolo[2,3-c]pyridine and 4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridine, with BIF% values (compared with acenaphthene) of 57 and 64, respectively. Two compounds bearing these head groups were synthesized (SC07 and SC08) and assessed in the single-round infection assay. Owing to the relative decreased sensitivity of HIV-1JR-CSF to SC04, as compared with HIV-1YU-2, this isolate was selected for use in potency optimization. Both compounds were specific to HIV-1 (no inhibition of AMLV-pseudotyped HIV-1) and inhibited HIV-1JR-CSF pseudotyped HIV-1 virus with IC50 values of 0.98 ± 0.06 μM and 0.09 ± 0.01 μM, for SC07 and SC08, respectively (Table 2).
Table 2.
Structure and potency of second-generation compounds based on the dipyrrolidine core scaffold. Chemical structures were drawn with ChemAxon software (Budapest, Hungary). AMLV = amphotropic murine leukemia virus; NA = not active over concentration range tested; ND = not determined. Chemical structures were drawn with ChemAxon software (Budapest, Hungary).
| IC50 (MM) | |||
|---|---|---|---|
|
| |||
| Compound | JR-CSF | HxBc2 | AMLV |
| SC04 | |||
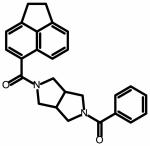
|
100 ± 30 | ND | NA |
|
| |||
| SC07 | |||
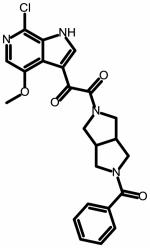
|
0.98 ± 0.06 | 16.8 ± 6.2 | NA |
|
| |||
| SC08 | |||
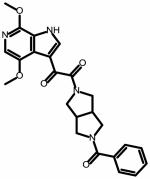
|
0.09 ±0.01 | 0.19 ±0.08 | NA |
|
| |||
| SC11 | |||
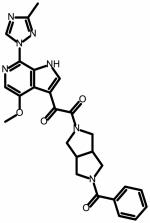
|
0.0008 ± 0.0004 | 0.001 ± 0.0001 | NA |
|
| |||
| SC26 | |||
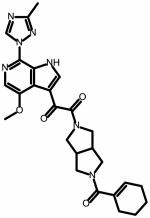
|
0.002 ± 0.0001 | 0.0006 ± 0.00008 |
NA |
The head group of SC08 is the same as in BMS-488043.36, 37 Replacement of the methoxy in the 7 position on the azaindole ring with a methyltriazole greatly improved the potency of the compound, resulting in the creation of BMS-626529.38, 39 Therefore, we explored this substitution in SC08 to determine whether it would result in a similar enhancement of potency (SC11). SC11 was synthesized and tested for specificity and activity against HIV-1JR-CSF using the single-round infection assay. SC11 displayed greatly enhanced potency as compared with SC08, retaining HIV-1 specificity and inhibiting HIV-1JR-CSF with an IC50 value of 0.0008 ± 0.0004 μM (Table 2).
Synthesis and assessment of SC11 revealed that the dipyrrolidine core can support compounds of nanomolar potency that prevent HIV-1 entry. The next step was to analyze the predicted ADME properties (absorption, distribution, metabolism, and excretion) of this compound and compare them with those of the BMS piperazine-based entry inhibitors (Figure 4). To accomplish this comparison, we used a combination of computationally guided bioisosteric replacement using Spark (focusing on the terminal phenyl group of SC11) and in silico prediction of druglike metrics of the results as implemented in the oral non–central nervous system (CNS) drug profile in StarDrop 5.5 (Optibrium, Ltd., Cambridge, UK).40
Figure 4.

(a) The individual models that comprise the oral non-CNS (central nervous system) drug profile and their respective importance to the profile. HIA = human intestinal absorption; hERG (human ether-à-go-go-related gene); IC50 = half-maximal inhibitory concentration; BBB = blood–brain barrier. (b) Plot showing the StarDrop (Optibrium, Ltd., Cambridge, UK)–derived logS versus the score from a multimetric oral non-CNS profile for the BMS and SC compounds.
Optibrium’s oral non-CNS drug profile is composed of the following metrics: logS (intrinsic aqueous solubility); classification for human intestinal absorption; logP (octanol/water); hERG (human ether-à-go-go-related gene) pIC50 (mammalian cells); cytochrome P450 CYP2D6 classification; cytochrome P450 CYP2C9 pKi values; classification of P-glycoprotein transport; classification of blood–brain barrier (BBB) penetration; and predicted BBB penetration value. The models and their respective importance to the profile are shown in Figure 4a.
Details for the specific models are provided in the StarDrop Reference Guide from Optibrium and are online at the StarDrop FAQs (http://www.optibrium.com/community/faq/adme-qsar-models). A probabilistic scoring algorithm41 is then used to combine the model predictions in the oral non-CNS drug profile into an overall score.
This combined Spark/StarDrop analysis suggested that a simple replacement of phenyl with cyclohexene may be a good transformation to perform in order to improve ADME properties while retaining the potency associated with the dipyrrolidine scaffold. StarDrop analysis of the cyclohexene variant of SC11 (designated SC26) versus BMS-663068 using the oral non-CNS drug profile provided scores for SC26 and BMS-663068 of 0.3815 ± 0.2323 and 0.0799 ± 0.1014, respectively (Figure 4b). For reference, scores range from 0 to 1, with 0 suggesting extremely non–druglike, and 1 suggesting the perfect drug.
Given two oral non-CNS drug profile scores with errors corresponding to 1 standard deviation:
X1 +/− σ1
X2 +/− σ2;
and assuming that the error is normally distributed, the difference in the scores will take a normal distribution with mean X1-X2 and standard deviation given by . Therefore, the probability that X1 > X2 is the probability that X1 - X2 > 0 was calculated in Excel (Microsoft; Redmond, WA) using the NORMDIST function. This analysis predicted an 88% likelihood that SC26 has better druglike properties than BMS-663068. Thus, we synthesized compound SC26 and subjected it to specificity and potency analysis using the single-round infection assay. Gratifyingly, this compound exhibited IC50 values of 2.0 ± 0.1 nM and 0.6 ± 0.01 nM against HIV-1JR-CSF and HIV-1HxBc2, respectively (Table 2).
Because it demonstrated good potency against two isolates in the single-round infection assay as well as specificity to HIV-1, we next sought to quantify the potency of SC26 against the fully infectious virus. Moreover, this assay was performed using healthy, primary cells to gain further insight into potential toxicities of the compound towards natural target cells. Additionally, as a key issue in the development of novel HIV drugs is their ability to inhibit the replication of genetically diverse isolates (especially isolates from the most globally prevalent subtypes A, B, C, and D), we chose to assess the potency of SC26 to inhibit the replication of isolates from subtypes A, B, C, and D in primary human peripheral blood mononuclear cells (PBMCs).42-44 Concomitantly, we assessed the toxicity of SC26 to the PBMCs using an MTS assay. The results of these analyses are summarized in Table 3. As can be observed from the IC50 values, the potency of SC26 against different isolates ranges quite dramatically, most likely reflecting differences in the binding site between subtypes. Moreover, SC26 had no effect on the replication of an HIV-2 isolate, mirroring the specificity analyses using the AMLV pseudoptyped recombinant HIV-1 virus, and further demonstrating specificity to HIV-1.
Table 3.
Therapeutic spectrum of SC26 against highly prevalent HIV-1 subtypes and HIV-2. IC50 = half-maximal inhibitory concentration; NA = not applicable; TC50 = half-maximal toxic concentration.
| Virus isolate | Subtype | IC50 (MM) | C50 (MM) | Antiviral index (TC50/IC50) |
|---|---|---|---|---|
|
|
|
|||
| 92UG037 | A | 25.2 ± 6.9 | >100 | >3.97 |
|
|
|
|||
| 91US004 | B | 0.195 ±0.012 | >513 | |
|
|
|
|||
| 94US_33931 N | B | 0.0006 ± 0.0005 | > 166,666 | |
|
|
|
|||
| 98US_MSC5016 | C | 3.9 ± 0.8 | >25.6 | |
|
|
|
|||
| 99UG_A07412M1 | D | 0.49 ± 0.06 | >204 | |
|
|
|
|||
| HIV-2CDC310319 | NA | > 100 | NA | |
In summary, we have identified a novel scaffold for the HIV-1 Env-directed entry inhibitors using field-based computational methods and multiparameter optimization. The most potent in the new class of dipyrrolodine-scaffolded entry inhibitors displayed potency comparable to the BMS piperazine-based entry inhibitors but was characterized by an improved predicted ADME profile. Like the BMS piperazine-based entry inhibitors, the new dipyrrolidine scaffold displayed varying degrees of potency against HIV-1 isolates from different subtypes, probably reflecting isolate-specific differences in the binding site on Env. Further investigation and determination of the exact binding site of these class of entry inhibitors will undoubtedly be invaluable in the redesign and optimization of such compounds, while illuminating a new site of vulnerability in the Env complex of HIV-1. Taken as a whole, these results extend the chemotypes for this class of HIV-1 inhibitor and validate the use of multiparameter optimization using high-content three-dimensional field-based models, bioisosteric replacement, and consideration of druglike metrics in drug design.
Supplementary Material
Acknowledgements
This work was supported in part by NIH/NIAID grant 1R21AI104354-01A1 (Cocklin, PI) and W. W. Smith Charitable Trust grant #A1301 (Cocklin, PI). We thank Diana Winters (Academic Publishing Services, Drexel University College of Medicine) for proofreading the manuscript.
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- AMLV
amphotropic murine leukemia virus
- BBB
blood–brain barrier
- BIF
bioisostere factor
- CNS
central nervous system
- DCM
dichloromethane
- DiEA
diisopropylethylamine
- DIPEA
N,N-eiisopropylethylamine
- DMF
dimethylformamide
- EtOAc
ethyl acetate
- HATU
hexafluorophosphate
- hERG
human ether-à-go-go-related gene
- HIA
human intestinal absorption
- HPLC
high-performance liquid chromatography
- HR
heptad repeat
- IC50
half-maximal inhibitory concentration
- PBMCs
peripheral blood mononuclear cells
- Pd/C
palladium on carbon
- TC50
half-maximal toxic concentration
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, Katinger H, Parren PW, Robinson J, Van Ryk D, Wang L, Burton DR, Freire E, Wyatt R, Sodroski J, Hendrickson WA, Arthos J. Nature. 2002;420:678. doi: 10.1038/nature01188. [DOI] [PubMed] [Google Scholar]
- 2.Modrow S, Hahn BH, Shaw GM, Gallo RC, Wong-Staal F, Wolf H. Journal of virology. 1987;61:570. doi: 10.1128/jvi.61.2.570-578.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. Nature. 2003;422:307. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 4.Wyatt R, Sodroski J. Science. 1998;280:1884. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 5.Leonard CK, Spellman MW, Riddle L, Harris RJ, Thomas JN, Gregory TJ. The Journal of biological chemistry. 1990;265:10373. [PubMed] [Google Scholar]
- 6.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Nature. 1998;393:648. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rizzuto CD, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, Sodroski J. Science. 1998;280:1949. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 8.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. Nature. 1998;393:705. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 9.Moore JP, Sattentau QJ, Wyatt R, Sodroski J. Journal of virology. 1994;68:469. doi: 10.1128/jvi.68.1.469-484.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosch ML, Earl PL, Fargnoli K, Picciafuoco S, Giombini F, Wong-Staal F, Franchini G. Science. 1989;244:694. doi: 10.1126/science.2541505. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Virology. 2005;339:213. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 12.Wang T, Zhang Z, Wallace OB, Deshpande M, Fang H, Yang Z, Zadjura LM, Tweedie DL, Huang S, Zhao F, Ranadive S, Robinson BS, Gong YF, Ricarrdi K, Spicer TP, Deminie C, Rose R, Wang HG, Blair WS, Shi PY, Lin PF, Colonno RJ, Meanwell NA. Journal of medicinal chemistry. 2003;46:4236. doi: 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
- 13.Kwon YD, LaLonde JM, Yang Y, Elban MA, Sugawara A, Courter JR, Jones DM, Smith AB, 3rd, Debnath AK, Kwong PD. PloS one. 2014;9:e85940. doi: 10.1371/journal.pone.0085940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Courter JR, Madani N, Sodroski J, Schon A, Freire E, Kwong PD, Hendrickson WA, Chaiken IM, Lalonde JM, Smith AB., 3rd Accounts of chemical research. 2014 doi: 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5663. doi: 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:11013. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Zhou N, Sun Y, Ray N, Lataillade M, Hanna GJ, Krystal M. Antimicrobial agents and chemotherapy. doi: 10.1128/AAC.00513-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, 3rd, Sodroski JG. Proc Natl Acad Sci U S A. 2004;101:5036. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nowicka-Sans B, Gong YF, McAuliffe B, Dicker I, Ho HT, Zhou N, Eggers B, Lin PF, Ray N, Wind-Rotolo M, Zhu L, Majumdar A, Stock D, Lataillade M, Hanna GJ, Matiskella JD, Ueda Y, Wang T, Kadow JF, Meanwell NA, Krystal M. Antimicrobial agents and chemotherapy. 2012 doi: 10.1128/AAC.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charpentier C, Larrouy L, Visseaux B, Landman R, Levittas M, Storto A, Damond F, Yazdanpanah Y, Yeni P, Brun-Vezinet F, Descamps D. The Journal of antimicrobial chemotherapy. 2012 doi: 10.1093/jac/dks067. [DOI] [PubMed] [Google Scholar]
- 21.Singh IP, Chauthe SK. Expert opinion on therapeutic patents. 2011;21:399. doi: 10.1517/13543776.2011.550876. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z, Zadjura LM, Marino AM, D'Arienzo CJ, Malinowski J, Gesenberg C, Lin PF, Colonno RJ, Wang T, Kadow JF, Meanwell NA, Hansel SB. Journal of pharmaceutical sciences. 2010;99:2135. doi: 10.1002/jps.21948. [DOI] [PubMed] [Google Scholar]
- 23.Cheeseright T, Mackey M, Rose S, Vinter A. Journal of chemical information and modeling. 2006;46:665. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- 24.Cheeseright T, Mackey M, Rose S, Vinter A. Expert opinion on drug discovery. 2007;2:131. doi: 10.1517/17460441.2.1.131. Phd. Phd. Phd. [DOI] [PubMed] [Google Scholar]
- 25.Cheeseright TJ, Holm M, Lehmann F, Luik S, Gottert M, Melville JL, Laufer S. Journal of medicinal chemistry. 2009;52:4200. doi: 10.1021/jm801399r. [DOI] [PubMed] [Google Scholar]
- 26.Cheeseright TJ, Mackey MD, Melville JL, Vinter JG. Journal of chemical information and modeling. 2008;48:2108. doi: 10.1021/ci800110p. [DOI] [PubMed] [Google Scholar]
- 27.Cheeseright TJ, Mackey MD, Scoffin RA. Current computer-aided drug design. 2011;7:190. doi: 10.2174/157340911796504314. [DOI] [PubMed] [Google Scholar]
- 28.Collins JC, Armstrong A, Chapman KL, Cordingley HC, Jaxa-Chamiec AA, Judd KE, Mann DJ, Scott KA, Tralau-Stewart CJ, Low CMR. Medchemcomm. 2013;4:1148. [Google Scholar]
- 29.Kirpotina LN, Khlebnikov AI, Schepetkin IA, Ye RD, Rabiet MJ, Jutila MA, Quinn MT. Molecular pharmacology. 2010;77:159. doi: 10.1124/mol.109.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Low CM, Buck IM, Cooke T, Cushnir JR, Kalindjian SB, Kotecha A, Pether MJ, Shankley NP, Vinter JG, Wright L. Journal of medicinal chemistry. 2005;48:6790. doi: 10.1021/jm049069y. [DOI] [PubMed] [Google Scholar]
- 31.Low CM, Vinter JG. Journal of medicinal chemistry. 2008;51:565. doi: 10.1021/jm070880t. [DOI] [PubMed] [Google Scholar]
- 32.Mackey MD, Melville JL. Journal of chemical information and modeling. 2009;49:1154. doi: 10.1021/ci8003978. [DOI] [PubMed] [Google Scholar]
- 33.Webster SP, Binnie M, McConnell KM, Sooy K, Ward P, Greaney MF, Vinter A, Pallin TD, Dyke HJ, Gill MI, Warner I, Seckl JR, Walker BR. Bioorganic & medicinal chemistry letters. 2010;20:3265. doi: 10.1016/j.bmcl.2010.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biorn AC, Cocklin S, Madani N, Si Z, Ivanovic T, Samanen J, Van Ryk DI, Pantophlet R, Burton DR, Freire E, Sodroski J, Chaiken IM. Biochemistry. 2004;43:1928. doi: 10.1021/bi035088i. [DOI] [PubMed] [Google Scholar]
- 35.Mowbray CE, Bell AS, Clarke NP, Collins M, Jones RM, Lane CA, Liu WL, Newman SD, Paradowski M, Schenck EJ, Selby MD, Swain NA, Williams DH. Bioorganic & medicinal chemistry letters. 2011;21:6596. doi: 10.1016/j.bmcl.2011.07.125. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z, Zadjura LM, Marino AM, D'Arienz CJ, Malinowski J, Gesenberg C, Lin PF, Colonno RJ, Wang T, Kadow JF, Meanwell NA, Hansel SB. Journal of pharmaceutical sciences. 2010;99:2135. doi: 10.1002/jps.21948. [DOI] [PubMed] [Google Scholar]
- 37.Wang T, Yin ZW, Zhang ZX, Bender JA, Yang Z, Johnson G, Yang Z, Zadjura LM, D'Arieno CJ, Parker DD, Gesenberg C, Yamanaka GA, Gong YF, Ho HT, Fang H, Zhou NN, McAuliffe BV, Eggers BJ, Fan L, Nowicka-Sans B, Dicker IB, Gao Q, Colonno RJ, Lin PF, Meanwell NA, Kadow JF. Journal of medicinal chemistry. 2009;52:7778. doi: 10.1021/jm900843g. [DOI] [PubMed] [Google Scholar]
- 38.Li ZF, Zhou NN, Sun YN, Ray N, Lataillade M, Hanna GJ, Krystal M. Antimicrobial agents and chemotherapy. 2013;57:4172. doi: 10.1128/AAC.00513-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nowicka-Sans B, Gong YF, McAuliffe B, Dicker I, Ho HT, Zhou NN, Eggers B, Lin PF, Ray N, Wind-Rotolo M, Zhu L, Majumdar A, Stock D, Lataillade M, Hanna GJ, Matiskella JD, Ueda Y, Wang T, Kadow JF, Meanwell NA, Krystal M. Antimicrobial agents and chemotherapy. 2012;56:3498. doi: 10.1128/AAC.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Segall M, Champness E, Obrezanova O, Leeding C. Chem Biodivers. 2009;6:2144. doi: 10.1002/cbdv.200900148. [DOI] [PubMed] [Google Scholar]
- 41.Segall MD. Current pharmaceutical design. 2012;18:1292. doi: 10.2174/138161212799436430. [DOI] [PubMed] [Google Scholar]
- 42.Zentner I, Sierra LJ, Fraser AK, Maciunas L, Mankowski MK, Vinnik A, Fedichev P, Ptak RG, Martin-Garcia J, Cocklin S. ChemMedChem. 2013;8:426. doi: 10.1002/cmdc.201200577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zentner I, Sierra LJ, Maciunas L, Vinnik A, Fedichev P, Mankowski MK, Ptak RG, Martin-Garcia J, Cocklin S. Bioorganic & medicinal chemistry letters. 2013;23:1132. doi: 10.1016/j.bmcl.2012.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kortagere S, Madani N, Mankowski MK, Schon A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang X, Jones DM, Stavale E, Krebs FC, Martin-Garcia J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB., 3rd Journal of virology. 2012;86:8472. doi: 10.1128/JVI.05006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.