

Abstract
MicroRNAs (miRs) play important roles in regulation of a variety of cell functions, including immune responses. We have previously demonstrated that miR-17-92 expression in T-cells enhances Th1 phenotype and provides a long-term protection against glioblastoma when co-expressed as a transgene in T-cells along with a chimeric antigen receptor. To further elucidate the function of miR-17-92 in tumor antigen-specific CD8+ T-cells, we generated transgenic (Tg) mice in which CD8+ T-cells overexpress transgene-derived miR-17-92 under the lck promoter as well as T-cell receptor specific for human gp10025-33 (Pmel-1) (miR-17-92/Pmel-Tg). CD8+ T-cells from miR-17-92/Pmel-Tg mice demonstrated enhanced interferon (IFN)-γ production and cytotoxicity in response to the cognate antigen compared with those from control Pmel-Tg mice without the transgene for miR-17-92. In addition, miR-17-92/Pmel-Tg mouse-derived CD8+CD44+ T-cells demonstrated increased frequencies of cells with memory phenotypes and IFN-γ production. We also found that miR-17-92/Pmel-Tg-derived CD8+ T-cells expressed decreased levels of transforming growth factor (TGF)-β type II receptor (TGFBR2) on their surface, thereby resisting against suppressive effects of TGF-β1. Our findings suggest that engineering of tumor antigen-specific CD8+ T-cells to express miR-17-92 may improve the potency of cancer immunotherapy.
Keywords: miRNA-17-92, antigen-specific CD8+ T-cells, IFN-γ, cytotoxicity, TGFBR2
1. Introduction
Adoptive cell transfer (ACT) of tumor-specific T-cells has shown promise as a potentially curative treatment for cancer [1]. Based on the cytokine-producing profiles and functions, T-cell immune responses are classified into at least four distinct subsets: type-1, type-2, type-17 and regulatory T-cells [2–4]. Although type-1 effector T-cells are important in the development of efficient antitumor activity [5], tumors secrete numerous type-2 and immunosuppressive cytokines that promote tumor proliferation and immune escape [6–8]. Tumor microenvironment also induces regulatory T-cells, which suppress antitumor immune responses resulting in promotion of tumor growth [9]. Type-17 T-cells play an important role in the clearance of pathogens [10], however, the antitumor effects of type-17 T-cells remains controversial [11]. Thus, the strategic skewing type-1 immunity is critical for the development of more effective cancer immunotherapy.
MicroRNAs (miRNAs) are endogenous small noncoding RNAs that regulate the expression of target genes by binding to the complementary element of target messenger RNAs [12]. miRNAs in the miRNA-17-92 (miR-17-92) cluster are elevated in various tumor types and promote tumor progression [13–16], while transgenic mice with miR-17-92 overexpressed in lymphocytes develop lymphoproliferative disorder and autoimmunity but not cancer [17]. Furthermore, miR-17-92-overexpressing T-cells increase IFN-γ production [17, 18], and we have recently demonstrated that transgene-mediated overexpression of miR-17-92 in T-cells provides long-term protection against gliomas when co-expressed with a chimeric antigen receptor [19]. These findings suggest that miR-17-92 promotes type-1 T-cell skewing and antitumor immunity.
Tumors produce a variety of immunosuppressive factors, such as transforming growth factor (TGF)-β, which may disturb antitumor immune responses [20]. TGF-β type II receptor (TGFBR2) has a high affinity for TGF-β1 [21], and is a verified target of miR-17-92 in solid cancers [22, 23]. miR-17-92 expression in CD34+ hematopoietic stem cells inversely correlated with the level of TGFBR2 transcript [24]. We therefore hypothesized that engineering of tumor antigen-specific CD8+ T-cells to overexpress miR-17-92 would confer resistance to immunosuppressive effects of TGF-β via decreased TGFBR2 expression.
Taken together, we sought to determine whether overexpression of miR-17-92 in tumor antigen-specific CD8+ T-cells confers type-1 activation and resistance to TGF-β1 suppression.
2. Materials and methods
2.1. Mice
C57BL/6 background wild-type (WT), transgenic (Tg) for the human gp10025-33-specific TCR (Pmel-1), miR-17-92 (C57BL/6-Gt [ROSA]26Sortm3(CAG-MIR17-92,-EGFP)Rsky/J) or Lck-Cre (B6.Cg-Tg[Lck-cre]548Jxm/J) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6-background mice that express transgene-derived miR-17-92 as well as the human gp10025-33-specific TCR were created by crossing miR-17-92 Tg mice and Pmel-1 mice, followed by further crossing with C57BL/6 background Lck-Cre mice. These mice overexpress miR-17-92 cluster in hgp100 25–33-specific T cells (miR-17-92/Pmel-TG cells hereafter). All mice were maintained and handled in accordance with the Animal Facility at the University of Pittsburgh per an Institutional Animal Care and Use Committee-approved protocol.
2.2. Reagents
The following antibodies were purchased from BioLegend (San Diego, CA): anti-CD8a (53-6.7), anti-CD44 (IM7) and anti-CD62L (MEL-14). H-2Db restricted gp100 tetramer was provided from The National Institutes of Health (NIH) Tetramer Core Facility. Goat anti-mouse TGFBR2, goat IgG isotype control and recombinant mouse TGF-β1 were purchased from R&D Systems (Minneapolis, MN). Rabbit anti-goat IgG was purchased from Abcam (Cambridge, MA). Recombinant human IL-2 was purchased from PeproTech (Rocky Hill, NJ). Human gp10025-33 (KVPRNQDWL) peptide was synthesized by the University of Pittsburgh Peptide Synthesis Facility.
2.3. Flow cytometry
Cells were stained with fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, PE combined with a cyanine dye (PE-Cy7)-, or allophycocyanin (APC)-conjugated mAbs. The samples were collected and analyzed using BD Accuri C6 flow cytometer and software (BD Biosciences, San Jose, CA).
2.4. RNA isolation and quantification of gene expression
Total RNA was extracted using RNeasy Mini Kit (Qiagen Inc., Valencia, CA), and subjected to RT-PCR analysis using TaqMan microRNA Reverse Transcription Kit and microRNA Assays (Applied Biosystems). Quantitative real-time PCR analysis was performed by StepOne™ Real-Time PCR Systems and Software (v2.3) (Life Technologies, Grand Island, NY). SNO202 was used as a housekeeping small RNA reference gene and to normalize another microRNA expression level. Relative expression of microRNA compared with control samples was calculated by the ddCt method.
2.5. ELISA
The amount of IFN-γ in the supernatant was measured by BD OptEIA ELISA sets (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions.
2.6. CTL analysis
Cytotoxicity was conducted using 6 h 51Cr-release assay as described previously [25]. In brief, CD8+ T-cells were isolated from Pmel-Tg and miR-17-92/Pmel-Tg mice and incubated with GL261 cells loaded with or without human gp10025-33 peptide before and after priming in vitro.
2.7. Cell sorting
CD8+ T-cells were purified using CD8a MicroBeads and magnetic cell sorting system (Miltenyi Biotec Inc., San Diego, CA). CD8+CD44+ and CD8+CD44− T-cells were sorted by MoFlo (Beckman Coulter, Inc., Brea, CA) at the Cytometry Facility of the University of Pittsburgh Cancer Institute.
2.8. Statistical analysis
Mean values between two groups were compared using Student’s t-test. Data are presented as mean+/− SD. P < 0.05 was considered significant.
3. Results
3.1. Expression of miR-17-92 conferred activation of type-1 CD8+ T-cells
To evaluate the effects of miR-17-92 expression in tumor antigen-specific CD8+ T-cells, we generated mice whose CD8+ T-cells express transgene-derived hgp10025-33-specific TCR (Pmel-1) as well as miR-17-92 (miR-17-92/Pmel-Tg). The control cells are obtained from mice transgenic for Pmel-1 and miR-17-92 alleles but not bred with the Lck-cre mice so the transgene miR 17-92 is not expressed. There was no difference between the two groups in terms of frequency of the hgp10025-33-tetramer-positive CD8+ T-cells (Fig. 1A). As expected, miR-17-5p as a miR-17-92 family member was expressed at higher levels in miR-17-92/Pmel-Tg than in control CD8+ T-cells (P < 0.001; Fig. 1B).
Figure 1. Transgene-mediated overexpression of miR-17-92 in CD8+ T-cells enhanced IFN-γ production and cytotoxicity.

CD8+ T-cells were isolated from control and miR-17-92/Pmel-Tg mice. (A) Frequencies of gp100 tetramer+ cells were analyzed by flow cytometry. Flow data are representative of three independent experiments. (B) Expression levels of miR-17-5p were measured by qRT-PCR. (n=3, ***P < 0.001, t-test) (C) CD8+ T-cells were stimulated with hgp10025-33 (0.125 μg/ml) in the presence of feeder cells for 46 hours and evaluated for IFN-γ production by ELISA. (*P < 0.05, t-test) (D) Antigen-specific cytotoxicity of CD8+ T-cells without in vitro stimulation was evaluated against GL261 glioma cells loaded with or without hgp10025-33 peptide by a 6-h 51Cr-release assay. (**P < 0.01, t-test) (E) CD8+ T-cells were cultured with IL-2 (100 IU/ml) and hgp10025-33 peptide (0.125 μg/ml) in the presence of feeder cells. After 6 days, these cells were harvested and evaluated for antigen-specific cytotoxic activities. (*P < 0.05, t-test) Graphs are presented as mean+/−SD from at least two independent experiments.
We next determined whether miR-17-92 overexpression in antigen-specific CD8+ T-cells would enhance IFN-γ production and cytotoxic activity in response to the cognate antigen, hgp10025-33. As shown in Fig. 1C, CD8+ T-cells isolated from miR-17-92/Pmel-Tg mice produced higher levels of IFN-γ compared with those from control mice when stimulated with hgp10025-33 peptide (P < 0.05). None of those cells expressed detectable levels of IFN-γ without the peptide stimulation (Fig. 1C). miR-17-92/Pmel-Tg mouse-derived CD8+ T-cells showed significantly higher levels of antigen-specific cytotoxicity than control CD8+ T-cells even without in vitro stimulation with the cognate peptide (P < 0.05 or P < 0.01; Fig. 1D). After culture with hgp10025-33 peptide and IL-2 for 6 days, both T-cell types exhibited increased cytotoxicity levels against the cognate antigen, while miR-17-92/Pmel-Tg CD8+ T-cells showed a higher cytotoxic activity than control CD8+ T-cells (Fig. 1E). The transgene-derived overexpression of miR-17-92 in CD8+ T-cell enhanced the antigen-specific cytotoxic activity even without in vitro stimulation.
3.2. Transgene-derived miR-17-92 overexpression in CD8+ T-cells increased the frequency of CD8+CD44+ memory T-cells that produce IFN-γ
CD8+ T-cells can be divided into three subsets: naïve, central memory, and effector memory cells using CD44 and CD62L cell-surface marker [26], and CD8+CD44+ memory-phenotype CD8+ T-cells exist even in mice which did not receive apparent stimulations with the cognate antigen [27–29]. To find whether transgene-mediated overexpression of miR-17-92 influences the frequency of naïve and memory CD8+ T-cells, we evaluated expression of CD44 and CD62L on CD8+ T-cells in splenocytes. Non-stimulated CD8+ T-cells from miR-17-92/Pmel-Tg mice demonstrated significantly increased proportions of both CD44+CD62L+ central and CD44+CD62L− effector memory cells compared to those from control mice (P < 0.001; Fig. 2A and 2B). Next, we isolated CD8+, CD8+CD44− naïve and CD8+CD44+ memory T-cells from miR-17-92/Pmel-Tg and control mice, and compared IFN-γ production levels following stimulation with hgp10025-33 peptide-pulsed GL261 glioma cells for 24 hours (Fig. 2C). Consistent with data in Fig. 1C, miR-17-92/Pmel-Tg mouse-derived CD8+ T-cells produced higher levels of IFN-γ than control CD8+ T-cells (P < 0.01). We observed a strikingly high level of IFN-γ in miR-17-92/Pmel-Tg mouse-derived CD8+CD44+ T-cells compared with corresponding cells from control mice (P < 0.001). Similarly, miR-17-92/Pmel-Tg CD8+CD44+ memory T-cells produced a higher levels of IFN-γ than control CD8+ T-cells when exposed to Quad-GL261 glioma cells which endogenously express transgene-derived major histocompatibility (MHC) class I-restricted hgp10025-33 [30] (P < 0.001; Fig. 2D). These results indicate that overexpression of miR-17-92 in CD8+ T-cells in vivo enhanced not only the frequency of CD8+CD44+ memory T-cells but also IFN-γ secretion levels by memory cells.
Figure 2. CD8+ T-cells from miR-17-92/Pmel-Tg mice demonstrated increased the frequency and IFN-γ production of CD8+CD44+ T-cells.

Spleens were harvested from control and miR-17-92/Pmel-Tg mice. (A) Representative flow data for CD44 and CD62L expression on CD8+ T-cells. (B) Frequencies of CD44−CD62L+ (naive), CD44+CD62L+ (central memory) and CD44+CD62L− (effector memory) cells on CD8+ T-cells. Data are presented as mean+/−SD. (n = 6 in each strain; ***P < 0.001, t-test) (C) CD8+, CD8+CD44+ and CD8+CD44− T-cells were isolated from the spleens of control and miR-17-92/Pmel-Tg mice. These cells were cocultured with hgp10025-33 peptide-pulsed or non-pulsed GL261 cells for 24 hours and evaluated for IFN-γ production by ELISA. (ND, not detected; **P < 0.01, ***P < 0.001, t-test) (D) CD8+CD44+ T-cells from control and miR-17-92/Pmel-Tg mice were cocultured with Quad-GL261 cells for 24 hours and evaluated for IFN-γ production by ELISA. (ND, not detected; ***P < 0.001, t-test) Bars and error bars indicate the mean and SD, respectively, from three independent experiments.
3.3. miR-17-92/Pmel-Tg-derived CD8+ T-cells are resistant to TGF-β1-mediated suppression
TGF-β type II receptor (TGFBR2) is a verified target of mir-17-92 in solid cancers [22, 23], and miR-17-92 expression in CD34+ hematopoietic stem cells inversely correlates with the level of TGFBR2 transcript [24]. Hence, we investigated whether overexpression of miR-17-92 in CD8+ T-cells would lead to down-regulation of TGFBR2, thereby rendering CD8+ T-cells more resistant to the suppressive effects of TGF-β1. As shown in Fig. 3A, while CD8+ T-cells from control mice expressed detectable levels of TGFBR2, ones from miR-17-92/Pmel-Tg mice were negative for TGFBR2. Furthermore, presence of recombinant TGF-β1 significantly suppressed hgp10025-33 peptide-induced IFN-γ production levels in control CD8+ T-cells but not significantly in miR-17-92/Pmel-Tg CD8+ T-cells (Fig. 3B). These data suggest that miR-17-92 confers resistance of CD8+ T-cells against TGF-β1 via down-regulation of TGFBR2 expression.
Figure 3. miR-17-92 expression in CD8+ T-cells provided resistance to suppressive effect of TGF-β1.
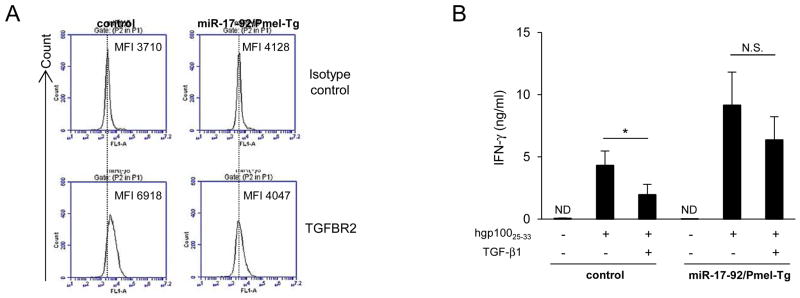
CD8+ T-cells were isolated from control and miR-17-92/Pmel-Tg mice. (A) Cell surface expression level of TGFBR2 was evaluated by flow cytometry. Flow data are representative of two independent experiments. (B) CD8+ T-cells were cultured with hgp10025-33 (0.125 μg/ml) and feeder cells in the presence or absence of TGF-β1 (4 ng/ml) for 46 hours. IFN-γ secretion was determined by ELISA. Bars and error bars indicate the mean and SD, respectively, from three independent experiments. (ND, not detected; N.S., no significant; *P < 0.05, t-test)
4. Discussion
In this study, we demonstrate that engineering of tumor antigen-specific CD8+ T-cells to overexpress miR-17-92 improves type-1 effector functions, such as enhanced IFN-γ production and cytotoxicity, in an antigen-specific manner and provides resistance to immunosuppressive effect of TGF-β1.
We have been dedicated to the development of effective immunotherapeutic strategies for central nervous system (CNS) tumors, such as gliomas. Gliomas account for approximately 30% of all primary central nervous system tumors and 80% of malignant brain tumors [31]. The current standard treatment for patients with malignant gliomas includes surgery, followed by chemotherapy and radiation therapy [32]. Despite recent advances in cancer therapy, prognosis is dismal. Previously, we have reported that CD49d (α4 integrin), which forms α4β1 and α4β7 integrin heterodimers at the cell surface [33], was preferentially expressed on type-1 CD8+ compared with type-2 CD8+ T-cells [34], and VLA-4 (the heterodimer of α4 and β1 integrins) expression on CTLs plays a significant role in the efficient CNS tumor infiltration of antigen-specific CTLs [35]. Although CD49d can form heterodimers with both β1 and β7 integrins, α4β7 complexes were not expressed by CD8+ T-cells [34], suggesting that CD49d is a suitable surrogate for VLA-4. We also found miR-17-92/Pmel-Tg CD8+ T-cells without in vitro stimulation expressed elevated levels of CD49d (data not shown) in addition to IFN-γ production (Fig. 1C) and cytotoxicity (Fig. 1D), demonstrating that miR-17-92 expression in tumor-antigen-specific CD8+ T-cells have the potential to improve not only antitumor immunity but also their infiltration into CNS tumor sites.
One of major barriers for successful ACT therapy is an immunosuppressive tumor microenvironment [36]. Malignant gliomas are heavily infiltrated by immature myeloid cells (i.e., tumor-associated macrophages), which play a role in promoting tumor growth and suppressing type-1 immunity by releasing the immunosuppressive factors such as IL-10 and TGF-β [37]. In the normal physiological state, TGF-β actively maintains T-cell homeostasis and regulates T-cell function [38]. However, in the tumor microenvironment, TGF-β creates an immunosuppressive milieu that inhibits antitumor immunity by suppressing or altering activation, maturation and differentiation of both innate and adaptive immune cells [39, 40]. miR-17-92 expression in antigen-specific CD8+ T-cells reduced the expression of TGFBR2 and conferred resistance to the immunosuppressive effects of TGF-β1 (Fig. 3). Moreover, IL-10 suppressed IFN-γ production from control CD8+ T-cells, but not miR-17-92/Pmel-Tg CD8+ T-cells, when stimulated with hgp10025-33 peptide (data not shown). Taken together, these data suggest that engineering of tumor antigen-specific CD8+ T-cells to express miR-17-92 may protect them from immunosuppressive effects in the tumor microenvironment.
Non-stimulated CD8+ T-cells from miR-17-92/Pmel-Tg mice demonstrated significantly increased proportions of CD44+ memory cells compared to those from control mice (Fig. 2A and 2B). Under lymphopenic conditions, naïve CD8+ T-cells undergo homeostatic proliferation and acquire phenotypic and functional characteristics of memory T-cells in the absence of stimulation with cognate antigens, but require recognition of MHC-bound self-peptides [27–29]. Given that gp100 is expressed in normal melanocytes [41], miR-17-92/Pmel-Tg CD8+ T-cells may interact with the endogenous gp100 peptide-MHC complex and convert to CD44+ memory-type T-cells.
We hypothesized that expression of miR-17-92 in CD8+ T-cells would have a lower threshold for TCR activation upon antigen-stimulation based on our own finding that the majority of miR-17-92/Pmel-Tg CD8+ T-cells are already primed upon isolation from host mice and a previous study showing that CD4+ T-cells with higher miR-17-92 expression have a lower threshold for activation with anti-CD3 antibody [17]. To test this, we used mouse (m)gp10025-33 peptide instead of human (h)gp 10025-33 to stimulate miR-17-92/Pmel-Tg-derived CD8+ T-cells because mgp10025-33 peptide is identical to the naturally existing peptide in mice, and binds to H-2Db with 100-fold lower efficiency than hgp10025-33 peptide [42], and thus we thought mgp10025-33 would be more relevant to cancer immunotherapy where endogenous autoantigens are targeted. Although stimulation with mgp10025-33 induced higher levels of IFN-γ from miR-17-92/Pmel-Tg CD8+ T-cells than from the control Pmel-Tg CD8+ T-cells, the threshold of mgp10025-33 peptide concentration required for detectable IFN-γ production was not different in the two T-cell types (data not shown). Another possibility of enhanced IFN-γ production and CTL activity in miR-17-92/Pmel-Tg CD8+ T-cells is that overexpression of miR-17-92 may enhance T-cell activation by influencing proximal TCR signals and/or by increasing the expression levels of any of these phosphorylated intermediates such as p56lck, ZAP-70 and linker for activation of T cells (LAT) [43]. Furthermore, miR-17-92 targets hypoxia-inducible factor (HIF)-1α in lung cancer cells [44]. Since HIF-1α negatively regulates T-cell functions [45–47] and T-cell-targeted disruption of HIF-1α a leads to increased IFN-γ production and improved effector functions [48–51], miR-17-92 expression in activated T-cells may promote type-1 function in part through down-regulation of HIF-1α.
miR-17-92/Pmel-Tg CD8+ T-cells showed cytotoxicity against hgp10025-33 peptide-pulsed, but not non-pulsed GL261 cells (Fig. 1D). In this regard, in contrary to our observations, Prins and colleagues have demonstrated that syngeneic T-cells primed with hgp10025-33 peptide can mediate cytotoxic effects on GL261 cells [52]. A possible reason for these inconsistent observations is that they used JAM cytotoxicy assays using 3H-thymidine labeled GL261 cells, whereas we performed 51Cr-release assays. The JAM assay has been demonstrated to be a more sensitive measurement of cell death than 51Cr-release assay because the JAM assay depends on DNA fragmentation which precedes loss of membrane integrity in most apoptotic cells [53].
Our findings in this study demonstrate that genetic engineering of tumor antigen-specific CD8+ T-cells with miR-17-92 confers both skewing type-1 immunity and resistance to suppressive effects in the tumor microenvironment. We also discovered that miR-17-92 expression in T-cells provided long-term protection against gliomas when co-transduced with the anti-EGFRvIII chimeric antigen receptor [19]. Although further investigations are required to determine the mechanisms, adoptive transfer of tumor antigen-specific CD8+ T-cells engineered to express miR-17-92 may have a potential to be a useful treatment strategy for patients with gliomas.
Highlights.
CD8+ T-cells from miR-17-92/Pmel-Tg mice show enhanced IFN-γ and cytotoxicity
miR-17-92/Pmel-Tg mouse-derived T-cells contained high numbers of memory cells
miR-17-92/Pmel-Tg-derived T-cells are resistant to TGF-β via decreased receptor
Acknowledgments
Grant Supports from: The National Institutes of Health (NIH) (2R01 NS055140) and Musella Foundation for Brain Tumor Research and Information. This project used University of Pittsburgh Cancer Institute (UPCI) shared resources (Animal Facility and Cytometry Facility) that are supported in part by NIH P30CA047904.
Abbreviations
- ACT
Adoptive cell transfer
- gp100
glycoprotein 100
- IFN-γ
interferon-gamma
- miR
microRNA
- Tg
transgenic
- TGF
transforming growth factor
- TGFBR2
transforming growth factor beta type II receptor
Footnotes
Conflicts of Interest
Hideho Okada and Gary Kohanbash are two of inventors on Pitt Ref. No. 01933: Title: Th1-Associated MicroRNAs and Their Use For Tumor Immunotherapy: Application No. 12/818,016, filed Country: United States of America. Because of this, interpretation of data are made by all authors but not solely by Dr. Okada and/or Kohanbash.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Qian X, Wang X, Jin H. Cell transfer therapy for cancer: past, present, and future. J Immunol Res. 2014;2014:525913. doi: 10.1155/2014/525913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mosmann TR, Cherwinski H, Bond MW, et al. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 3.Carter LL, Dutton RW. Type 1 and type 2: a fundamental dichotomy for all T-cell subsets. Curr Opin Immunol. 1996;8:336–342. doi: 10.1016/s0952-7915(96)80122-1. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. 2010;20:4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallett MA, Venmar KT, Fingleton B. Cytokine stimulation of epithelial cancer cells: the similar and divergent functions of IL-4 and IL-13. Cancer Res. 2012;72:6338–6343. doi: 10.1158/0008-5472.CAN-12-3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nitta T, Hishii M, Sato K, et al. Selective expression of interleukin-10 gene within glioblastoma multiforme. Brain Res. 1994;649:122–128. doi: 10.1016/0006-8993(94)91055-3. [DOI] [PubMed] [Google Scholar]
- 8.Wojtowicz-Praga S. Reversal of tumor-induced immunosuppression by TGF-beta inhibitors. Invest New Drugs. 2003;21:21–32. doi: 10.1023/a:1022951824806. [DOI] [PubMed] [Google Scholar]
- 9.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 10.McGeachy MJ. Th17 memory cells: live long and proliferate. J Leukoc Biol. 2013;94:921–926. doi: 10.1189/jlb.0313113. [DOI] [PubMed] [Google Scholar]
- 11.Yang B, Kang H, Fung A, et al. The role of interleukin 17 in tumour proliferation, angiogenesis, and metastasis. Mediators Inflamm. 2014;2014:623759. doi: 10.1155/2014/623759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang C. MicroRNomics: a newly emerging approach for disease biology. Physiol Genomics. 2008;33:139–147. doi: 10.1152/physiolgenomics.00034.2008. [DOI] [PubMed] [Google Scholar]
- 13.He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayashita Y, Osada H, Tatematsu Y, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- 15.Matsubara H, Takeuchi T, Nishikawa E, et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene. 2007;26:6099–6105. doi: 10.1038/sj.onc.1210425. [DOI] [PubMed] [Google Scholar]
- 16.Lawrie CH. MicroRNA expression in lymphoma. Expert Opin Biol Ther. 2007;7:1363–1374. doi: 10.1517/14712598.7.9.1363. [DOI] [PubMed] [Google Scholar]
- 17.Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sasaki K, Kohanbash G, Hoji A, et al. miR-17-92 expression in differentiated T cells - implications for cancer immunotherapy. J Transl Med. 2010;8:17. doi: 10.1186/1479-5876-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohno M, Ohkuri T, Kosaka A, et al. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer. 2013;1:21. doi: 10.1186/2051-1426-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu E, Chia PZ, Chen W. TGFbeta in T cell biology and tumor immunity: Angel or devil? Cytokine Growth Factor Rev. 2014;25:423–435. doi: 10.1016/j.cytogfr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin HY, Wang XF, Ng-Eaton E, et al. Expression cloning of the TGF-beta type II receptor, a functional transmembrane serine/threonine kinase. Cell. 1992;68:775–785. doi: 10.1016/0092-8674(92)90152-3. [DOI] [PubMed] [Google Scholar]
- 22.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mestdagh P, Bostrom AK, Impens F, et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Mol Cell. 2010;40:762–773. doi: 10.1016/j.molcel.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merkerova M, Vasikova A, Belickova M, et al. MicroRNA expression profiles in umbilical cord blood cell lineages. Stem Cells Dev. 2010;19:17–26. doi: 10.1089/scd.2009.0071. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura F, Dusak JE, Eguchi J, et al. Adoptive transfer of type 1 CTL mediates effective anti-central nervous system tumor response: critical roles of IFN-inducible protein-10. Cancer Res. 2006;66:4478–4487. doi: 10.1158/0008-5472.CAN-05-3825. [DOI] [PubMed] [Google Scholar]
- 26.Bannard O, Kraman M, Fearon D. Pathways of memory CD8+ T-cell development. Eur J Immunol. 2009;39:2083–2087. doi: 10.1002/eji.200939555. [DOI] [PubMed] [Google Scholar]
- 27.Murali-Krishna K, Ahmed R. Cutting edge: naive T cells masquerading as memory cells. J Immunol. 2000;165:1733–1737. doi: 10.4049/jimmunol.165.4.1733. [DOI] [PubMed] [Google Scholar]
- 28.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho BK, Rao VP, Ge Q, et al. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med. 2000;192:549–556. doi: 10.1084/jem.192.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Litterman AJ, Zellmer DM, Grinnen KL, et al. Profound impairment of adaptive immune responses by alkylating chemotherapy. J Immunol. 2013;190:6259–6268. doi: 10.4049/jimmunol.1203539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostrom QT, Gittleman H, Farah P, et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013;15(Suppl 2):ii1–56. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jovcevska I, Kocevar N, Komel R. Glioma and glioblastoma - how much do we (not) know? Mol Clin Oncol. 2013;1:935–941. doi: 10.3892/mco.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pribila JT, Quale AC, Mueller KL, et al. Integrins and T cell-mediated immunity. Annu Rev Immunol. 2004;22:157–180. doi: 10.1146/annurev.immunol.22.012703.104649. [DOI] [PubMed] [Google Scholar]
- 34.Sasaki K, Zhu X, Vasquez C, et al. Preferential expression of very late antigen-4 on type 1 CTL cells plays a critical role in trafficking into central nervous system tumors. Cancer Res. 2007;67:6451–6458. doi: 10.1158/0008-5472.CAN-06-3280. [DOI] [PubMed] [Google Scholar]
- 35.Zhu X, Nishimura F, Sasaki K, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007;5:10. doi: 10.1186/1479-5876-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gajewski TF, Woo SR, Zha Y, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 37.Chanmee T, Ontong P, Konno K, et al. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6:1670–1690. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malhotra N, Kang J. SMAD regulatory networks construct a balanced immune system. Immunology. 2013;139:1–10. doi: 10.1111/imm.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li MO, Flavell RA. TGF-beta, T-cell tolerance and immunotherapy of autoimmune diseases and cancer. Expert Rev Clin Immunol. 2006;2:257–265. doi: 10.1586/1744666X.2.2.257. [DOI] [PubMed] [Google Scholar]
- 40.Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-beta and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13:5262–5270. doi: 10.1158/1078-0432.CCR-07-1157. [DOI] [PubMed] [Google Scholar]
- 41.Kwon BS, Chintamaneni C, Kozak CA, et al. A melanocyte-specific gene, Pmel 17, maps near the silver coat color locus on mouse chromosome 10 and is in a syntenic region on human chromosome 12. Proc Natl Acad Sci U S A. 1991;88:9228–9232. doi: 10.1073/pnas.88.20.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nel AE. T-cell activation through the antigen receptor. Part 1: signaling components, signaling pathways, and signal integration at the T-cell antigen receptor synapse. J Allergy Clin Immunol. 2002;109:758–770. doi: 10.1067/mai.2002.124259. [DOI] [PubMed] [Google Scholar]
- 44.Taguchi A, Yanagisawa K, Tanaka M, et al. Identification of hypoxia-inducible factor-1 alpha as a novel target for miR-17-92 microRNA cluster. Cancer Res. 2008;68:5540–5545. doi: 10.1158/0008-5472.CAN-07-6460. [DOI] [PubMed] [Google Scholar]
- 45.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 46.Neumann AK, Yang J, Biju MP, et al. Hypoxia inducible factor 1 alpha regulates T cell receptor signal transduction. Proc Natl Acad Sci U S A. 2005;102:17071–17076. doi: 10.1073/pnas.0506070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eltzschig HK, Thompson LF, Karhausen J, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 48.Kojima H, Gu H, Nomura S, et al. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha -deficient chimeric mice. Proc Natl Acad Sci U S A. 2002;99:2170–2174. doi: 10.1073/pnas.052706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lukashev D, Klebanov B, Kojima H, et al. Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol. 2006;177:4962–4965. doi: 10.4049/jimmunol.177.8.4962. [DOI] [PubMed] [Google Scholar]
- 50.Guo J, Lu W, Shimoda LA, et al. Enhanced interferon-gamma gene expression in T Cells and reduced ovalbumin-dependent lung eosinophilia in hypoxia-inducible factor-1-alpha-deficient mice. Int Arch Allergy Immunol. 2009;149:98–102. doi: 10.1159/000189191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thiel M, Caldwell CC, Kreth S, et al. Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS One. 2007;2:e853. doi: 10.1371/journal.pone.0000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prins RM, Odesa SK, Liau LM. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res. 2003;63:8487–8491. [PubMed] [Google Scholar]
- 53.Usharauli D, Perez-Diez A, Matzinger P. The JAM Test and its daughter P-JAM: simple tests of DNA fragmentation to measure cell death and stasis. Nat Protoc. 2006;1:672–682. doi: 10.1038/nprot.2006.107. [DOI] [PubMed] [Google Scholar]