

Abstract
Inflammasomes are cytosolic protein complexes that respond to diverse danger signals by activating caspase-1. The sensor components of the inflammasome, often proteins of the nucleotide-binding oligomerization domain-like receptor (NLR) family, detect stress, danger stimuli, and pathogen-associated molecular patterns. We report that the eicosanoid 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and related cyclopentenone prostaglandins inhibit caspase-1 activation by the NLRP1 and NLRP3 inflammasomes. This inhibition was independent of 15d-PGJ2’s well characterized role as a peroxisome proliferator receptor-γ agonist, its activation of NRF2, or its anti-inflammatory function as an inhibitor of NF-κB. Instead, 15d-PGJ2 prevents the autoproteolytic activation of caspase-1 and the maturation of IL-1β through induction of a cellular state inhibitory to caspase-1 proteolytic function. The eicosanoid does not directly modify or inactivate the caspase-1 enzyme. Rather, inhibition is dependent on de novo protein synthesis. In a mouse peritonitis model of gout, using monosodium urate crystals to activate NLRP3,15d-PGJ2 caused a significant inhibition of cell recruitment and associated IL-1β release. Furthermore, in a murine anthrax infection model, 15d-PGJ2 reversed anthrax lethal toxin-mediated NLRP1-dependent resistance. The findings reported in this work suggest a novel mechanism for the anti-inflammatory properties of the cyclopentenone prostaglandins through inhibition of caspase-1 and the inflammasome.
INTRODUCTION
The inflammasome is a multi-protein complex that forms in response to a variety of danger signals. Upon activation, the sensor protein, often of the nucleotide-binding oligomerization domain-like receptor (NLR) family, recruits caspase-1 (formerly interleukin-cleavage enzyme, ICE). Caspase-1 undergoes auto-proteolytic activation and cleaves the zymogen forms of the pro-inflammatory cytokines IL-1β and IL-18 to their mature forms which are then secreted from the cell. Caspase-1 activation also leads to a rapid form of lytic cell death termed pyroptosis (1). Inflammasome function plays a key role in the innate immune response to infection (2) and the mechanisms by which pathogens activate and evade inflammasome detection are an active area of study. However, inflammasome signaling and the associated cytokine responses or tissue necrosis are also associated with numerous inflammatory disorders including type 2 diabetes, Alzheimer’s disease, arthritis, macular degeneration, and asthma (1). For this reason, discovery of novel inflammasome inhibitors is of clinical importance.
The prostaglandins are lipid signaling molecules derived from arachadonic acid that have diverse functions. Production of prostaglandins depends on the action of the cyclooxygenase (COX) enzymes of which there are two isoforms. COX-1 is expressed constitutively and predominantly produces prostanoids with housekeeping functions. COX-2 is induced following inflammatory stimuli and is the dominant source of prostaglandins during the inflammatory response. Most prostaglandins exert their effects through activation of specific transmembrane G protein-coupled receptors. These receptors activate a varied set of intracellular signaling pathways that lead to diverse biological activities (3). Many prostaglandins have demonstrated pro-inflammatory effects, but anti-inflammatory effects have been reported for cyclopentenone prostaglandins. Cyclopentenone prostaglandins contain a highly reactive α,β-unsaturated carbonyl that can form covalent attachments with thiol groups on proteins, thus altering their function (4). These prostaglandins inhibit pro-inflammatory NF-κB activity through covalent modification of IKK as well as direct inhibition of NF-κB binding to DNA (5–7). They also upregulate and activate anti-inflammatory heat shock proteins such as HSP70 (8).
15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) is the best-studied anti-inflammatory prostaglandin. It is formed as a dehydration product of PGD2 (9) and was the first identified endogenous ligand of the nuclear receptor PPARγ (10, 11). 15d-PGJ2 also activates the transcription factor NRF2 through formation of adducts with the NRF2 inhibitor KEAP1 (12). Both PPARγ and NRF2 are known to have anti-inflammatory roles (13, 14). It is through these actions that this lipid mediator can alter expression of cytokine, chemokine, and pro-inflammatory genes (15). A number of studies have shown that 15d-PGJ2 has therapeutic potential in animal models of inflammatory disease (for review see (15)).
In this work we report on a novel mechanism for the anti-inflammatory actions of 15d-PGJ2 and related cyclopentenone prostaglandins. Our studies show that these prostaglandins are potent inhibitors of the anthrax lethal toxin (LT) activation of the NLRP1 inflammasome and nigericin-mediated activation of the NLRP3 inflammasome. These prostaglandins inhibit inflammasome-mediated activation of caspase-1 and as a consequence, prevent maturation and release of IL-1β, both in vitro and in vivo, in a manner independent of effects on NF-κB. Our findings confirm the role of these lipid mediators as anti-inflammatory agents and supporting their development as therapeutic agents for the treatment of inflammatory diseases.
MATERIALS AND METHODS
Reagents
15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), CAY10410 (9,10-dihydro-15-deoxy-Δ12,14-prostaglandin J2), prostaglandin D2, prostaglandin E2, prostaglandin F2α, rosiglitazone, and T0070907 were purchased from Cayman Chemical (Ann Arbor, MI). Prostaglandin A1, prostaglandin J2, 4-cyclopentene-1,3-dione, 2-cyclopentenone, cycloheximide, actinomycin D, indomethacin, buthionine sulfoximine, and N-acetylcysteine and uric acid were from Sigma-Aldrich (St Louis, MO). Cyclopentanone and cyclopentene were obtained from Tokyo Chemical Industry (TCI, Portland, OR). Structures of these prostanoids are shown in Table I. Nigericin, lactacystin, and ultrapure lipopolysaccharide (LPS) were purchased from Calbiochem (San Diego, CA).
Table I.
EC50 values of structural variants of 15d-PGJ2
| Compound | EC50 (μM)1 | Structure |
|---|---|---|
| 15d-PGJ2 | ~20 |
|
| PGJ2 | ~40 |
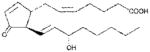
|
| PGA1 | ~240 |
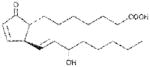
|
| PGD2 | ~400 |
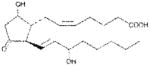
|
| PGE2 | >1000 |
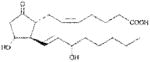
|
| PGF2α | >1000 |
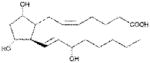
|
| Cyclopentene | >1000 |
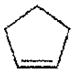
|
| Cyclopentanone | >1000 |
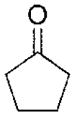
|
| 2-cyclopentenone | ~510 |
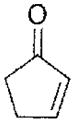
|
| 4-cyclopenten-1,3-dione | ~60 |
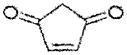
|
| CAY10410 | >1000 |
|
RAW264.7 cells were treated 30 min with variable concentrations of compound. Cells were challenged with LT (1 μg/mL, 1.5 h) and cell viability was measured by MTT staining. At least two experiments represented by a single shown study were used to generate an EC50 value for each compound’s ability to inhibit LT-induced pyroptosis.
Toxins
Protective antigen (PA), lethal factor (LF), and FP59 were purified from Bacillus anthracis as described previously (16, 17). FP59 is a fusion protein of the PA binding domain of LF to the ADP-ribosylation domain of Pseudomonas aeruginosa exotoxin A (18, 19). LFn-Fla, a toxin also delivered by PA, is a similar fusion of the first 254 amino acids of LF to full-length flagellin from Legionella pneumophila (kind gift of Dr. Russell Vance, University of California at Berkeley, Berkeley, CA) (20). FlaTox is the combination of LFn-Fla and PA. LT (lethal toxin) is the combination of LF and PA. Concentrations of LT correspond to the concentration of each toxin component (i.e., 1 μg/mL LT is 1 μg/mL PA + 1 μg/mL LF). Concentrations of FlaTox correspond to the concentration of LFn-Fla. Concentration of PA was always twice that of LFn-Fla in FlaTox experiments (i.e., 1 μg/mL of FlaTox is 2 μg/mL PA + 1 μg/mL LFn-Fla).
Bacillus anthracis spores
Spores were prepared from the nonencapsulated, toxigenic B. anthracis Ames 35 (A35) strain (21) by growing the bacteria on sporulation agar at 37°C for 1 day followed by 5 days at 30°C, and inspection by microscopy to verify >95% sporulation. Spores were purified from plates by four rounds of centrifugation and sterile water washes, followed by two heat treatments at 70°C for 30 min (to kill any vegetative bacteria). Spore quantification was performed using a Petroff Hausser counting chamber (Hausser Scientific, Horsham, PA) and verified by dilution plating.
Cell culture
RAW264.7 cells and L929 mouse fibroblast cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, and 50 μg/mL gentamicin (all purchased from Life Technologies, Grand Island, NY). Select studies used lower amounts of FBS as indicated in figure legends. Mouse bone marrow was cultured in complete DMEM (as above) supplemented with 30% L929 cell-conditioned supernatant and grown 7–9 days to allow time for differentiation to bone-marrow derived macrophages (BMDMs).
Animal Studies
Mice were used as source of bone marrow. Balb/cJ (harboring Nlrp1bS/S and LT-responsive macrophages), C57BL/6J (harboring Nlrp1bR/R) and mice deficient in nuclear factor erythroid 2-related factor 2 (NRF2) on the C57BL/6J background (N10) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice deleted for NLRP3 have been previously described (22). C57BL/6NTacNlrp1bS/S congenic mice carrying the LT-responsive Nlrp1bS/S have been previously described (23). These mice are resistant to spore infection relative to their congenic C57BL/6NTacNlrp1bR/R counterparts due to LT-mediated activation of the NLRP1 inflammasome (23). Cycloxygenase-1 (COX-1) knockout mice on the C57BL/6NTac were a gift from Katrin Mayer-Barber (National Institute of Allergy and Infectious Diseases, Bethesda, MD). These mice were backcrossed (four generations) in our laboratory to the C57BL/6NTacNlrp1bS/S congenic mice carrying the LT-responsive Nlrp1bS/S allele
For the mouse uric-acid induced peritonitis studies (used as a model for the arthritic disease gout) (24), C57BL/6J mice were injected IP with 125 μg of 15d-PGJ2 or vehicle (10% DMSO in PBS) at 5 min prior and 4 h after administration of monosodium urate (MSU) crystals. MSU crystals were prepared by crystallization of uric acid as previously described (25) and injected into mice (4 mg/250 μl PBS, IP). At 6 h after MSU administration, mice were euthanized and peritoneal lavage performed with PBS (6 ml/mouse). Infiltrating cells were counted following erythrocyte lysis using ACK buffer (Life Technologies, Grand Island, NY). IL-1β measurements in lavage fluids were made using an ELISA kit from R&D systems (Minneapolis, MN).
For sensitization of anthrax spore-resistant NLRP1bS-expressing mice, 15d-PGJ2 or vehicle was administered to Balb/cJ, C57BL/6J, C57BL/6NTacNlrp1bS/S or C57BL/6NTacNlrp1bR;R mice (100 μg, 200 μl, SC, at 5 min and 1 h post spore infection). Animals were infected with 2–4 ×107 A35 spores (200 μl, SC) and monitored for 7 days post infection for signs of malaise or death.
All animal experiments were performed in strict accordance with guidelines from the National Institutes of Health and the Animal Welfare Act, approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Cytotoxicity assays
RAW264.7 and Balb/cJ (LT-responsive) BMDM cells were grown in 96-well plates to 90% confluence and pre-treated with various drugs or vehicle at a range of doses or times (as described in figure legends). Cells were then incubated with LT or medium. Cell viability was assessed by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma-Aldrich) as previously described (26). In select experiments, cell death was assessed by staining cells with 5 μM propidium iodide (Sigma-Aldrich) in medium without phenol red. Fluorescence was measured on a Wallac 1420 Victor 3V (PerkinElmer, Waltham, MA) plate reader with excitation at 530 nm and emission at 615 nm.
MEK, caspase-1, and IL-1β cleavage
RAW264.7 and BMDM cells were exposed to LPS (1 μg/mL) and then various drugs or vehicle (at doses and timing indicated in figure legends), prior to addition of inflammasome activators (LT, FlaTox or nigericin, at indicated doses). Cells were then lysed in RIPA buffer containing LF inhibitor PT-168541-1 (kind gift of Alan Johnson, Panthera Biopharma) and processed for Western blotting using primary antibodies against MEK1 (444942, Calbiochem), IL-1β (AF-401-NA, R&D systems, Minneapolis, MN), MEK3 (sc-959) and caspase-1 p10 (sc-514, Santa Cruz Biotechnology, Santa Cruz, CA) as previously described (26). Secondary antibodies used in these studies were anti-goat infrared dye (800CW) (Rockland Immunochemicals, Gilbertsville, PA) and anti-rabbit infrared dye (800CW) (LI-COR Biosciences, Lincoln, NE). Immunoblots were visualized with the Odyssey infrared imaging system (LI-COR Biosciences).
In vitro caspase-1 assay
LPS (1 μg/mL for 2 h) was used to induce pro-IL-1β as a substrate for recombinant caspase-1. Approximately 107 cells were lysed per milliliter of sucrose buffer (250 mM sucrose, 10 mM HEPES, pH 7.3) by passage through a 29G needle. Lysates of LPS-treated RAW264.7 cells were incubated with 1 U active recombinant mouse caspase-1 (MBL International, Woburn, MA) per 50 μL of lysate in the presence or absence of 15d-PGJ2 or Boc-Asp-(OBzl)-chloromethylketone (Boc-D-CMK, Anaspec, San Jose, CA) for 3 h at 37°C. In other experiments cells were first pre-treated with 15d-PGJ2 for 1 h prior to preparation of lysates. Caspase-1-mediated cleavage of IL-1β was analyzed by Western blotting. Because 15d-PGJ2, an inhibitor of multiple signaling proteins in the NF-κB cascade (5–7), inhibits LPS-mediated upregulation of IL-1β, all LPS priming was performed prior to 15d-PGJ2 treatment.
Evaluation of caspase-1 sequestration in a high molecular weight complex
Sucrose buffer lysates of 15d-PGJ2-treated or heat shocked (42°C) RAW264.7 cells (as a positive control for caspase-1 sequestration, (27)), were centrifuged at 10,000 × g for 10 min at 4°C. Western blotting for caspase-1 was performed on the supernatant and pellet.
Evaluation of 15d-PGJ2 conjugation to NLRP3
C57BL/6J wild type or NLRP3-deficient BMDMs were stimulated with LPS (1 μg/mL, 4 h) to induce upregulation of NLRP3. Cells were then incubated with 15d-PGJ2 or biotinylated 15d-PGJ2 (Cayman Chemical) (50 μM, 1 h). Sucrose buffer lysates were made in the presence of Complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Approximately 200 μg of cell lysate was mixed with 5 μg of anti-NLRP3 antibody (MAB7578, R&D Systems) and placed on a rotary shaker at 4°C for 2 h. Protein A/G agarose slurry (20 μL, Santa Cruz Biotechnology) was added and the incubation was continued overnight. Bound immune complexes were washed with PBS four times prior to elution of proteins in 2X SDS loading buffer. Biotinylated adducts were detected with IR680-conjugated streptavidin (LI-COR Bioscienses) by Western blotting.
RESULTS
Cyclopentenone prostaglandins require an α,β-unsaturated carbonyl to prevent NLRP1-dependent pyroptosis
Anthrax LT contains a protease which cleaves the N-terminus of rodent NLRP1 proteins, leading to caspase-1 activation (28–30). Activation of caspase-1 leads to a rapid form of cell death known as pyroptosis (for review, see (31)). LT activation of the NLRP1 inflammasome in select inbred rodent strains that harbor LT-responsive alleles leads to pyroptosis, while strains with resistant NLRP1 alleles can undergo apoptosis (32–35). Our earlier studies showed that heat shock inhibits the inflammasome through sequestration of procaspase-1 in a large complex (27), preventing LT-mediated pyroptosis. We tested a number of cyclopentenone prostaglandins which induce heat shock proteins such as HSP70 (36, 37) along with cyclopentanone prostaglandins (PG) for the ability to inhibit the NLRP1 inflammasome-dependent pyroptosis of LT-sensitive macrophages. All the cyclopentenone prostanglandins tested inhibited the well characterized NLRP1-dependent LT-induced pyroptosis. Interestingly, most of the cyclopentanone prostanglandins that were tested failed to inhibit (Figure 1A, Table I). The exception was the cyclopentanone prostaglandin PGD2 which had low inhibitory activity (Figure 1A, Table I); however, it has been shown that this prostaglandin undergoes non-enzymatic dehydration to the cyclopentenone prostaglandin PGJ2 in aqueous media (38) and many of the purported physiological effects of PGD2 are actually mediated by PGJ2 and its downstream dehydration products such as 15d-PGJ2 (39).
Figure 1. Cyclopentenone prostaglandins protect cells from LT-induced pyroptosis.

RAW264.7 cells (A) or Balb/cJ BMDMs (B) were treated with varying concentrations of prostaglandins for 30 min in a dose range where the compounds were non-toxic. Cells were then challenged with 1 μg/mL LT and viability was measured by MTT staining at 1.5–2 h post-LT. All values are average from two wells in one representative experiment. Each curve is representative of at least two and up to ten similar experiments, depending on the particular compound shown. (C) RAW264.7 cells were incubated with 15d-PGJ2 (50 μM, 30 min) followed by LT (1 μg/mL) for indicated times. Western blotting of cell lysates was performed with antibodies against the N-terminus of MEK1 and MEK3. The * indicates a cross-reactive protein band which acts as an internal equal loading control. Results represent three similar Westerns.
The restriction of inhibitory activity to only those prostaglandins with cyclopentenone rings suggested a requirement for an α,β-unsaturated carbonyl. We further evaluated this requirement by examining the inhibitory activity of cyclopentene, cyclopentanone, 2-cyclopentenone, and various structural analogs (Table I). Cyclopentene and cyclopentanone, which have no α,β-unsaturated carbonyls, had no effect on LT-induced pyroptosis. However, 2-cyclopentenone, which has one carbonyl, did demonstrate inhibitory activity. Furthermore, 4-cyclopentene-1,3-dione which has two α,β-unsaturated carbonyls had a lower EC50 than 2-cyclopentenone. Finally, the 15d-PGJ2 analog CAY10410, which is identical to 15d-PGJ2 except for the absence of the α,β-unsaturated bond in the ring, had no inhibitory activity.
15d-PGJ2 inhibited LT-induced pyroptosis of both RAW264.7 cells (Figure 1A) and bone-marrow derived macrophages (Figure 1B) with an EC50 in the micromolar (20–40 μM) range. Reducing the amount of FBS to 1% or lower decreased the EC50 by up to ten-fold (Supplemental Figure 1A). This finding is in agreement with studies showing that electrophilic cyclopentenone prostaglandins such as 15d-PGJ2 are rapidly inactivated by FBS in cell culture medium (40).
15d-PGJ2 does not inhibit LT cleavage of MEKs
To determine whether 15d-PGJ2 protects against LT pyroptosis by inhibiting toxin translocation to the cytosol or enzymatic activity, we examined the cleavage of the cytosolic MEK substrates of LT. LT rapidly cleaves the N-terminus of multiple MEK proteins and this cleavage can be monitored by loss of an epitope (MEK1) and by altered mobility of the MEK3 protein (41, 42). We found cleavage of the MEK proteins was uninhibited by treatment of the cells with 15d-PGJ2, thus confirming that, in the presence of the drug, active LT translocated to the cytosol (Figure 1C). Thus 15d-PGJ2 does not impact the binding, endocytosis, translocation or activity of LF.
15d-PGJ2 inhibits cytokine processing of multiple inflammasomes
Inflammasome activation causes rapid autoproteolysis of caspase-1 which leads to processing and secretion of the proinflammatory cytokine IL-1β (1). 15d-PGJ2 inhibits NF-κB phosphorylation by IκB as well as binding of NF-κB to DNA (5–7). NF-κB signaling is important for priming of the inflammasome. For all studies of inflammasome activation and cytokine processing, cells were primed with LPS before exposure to 15d-PGJ2 to avoid any NF-κB signaling inhibitory effects which could lead to differences in priming and IL1β levels. Using this method, no differences in inflammasome priming, indicated by pro-IL1β levels, were observed in prostaglandin-treated cells in any experiments (as demonstrated by all Western blots). 15d-PGJ2 inhibited caspase-1 autoproteolysis (monitored by generation of the p10 fragment) as well as maturation and secretion of IL-1β in LT-treated cells at a step subsequent to NF-κB signaling (Figure 2A). Early studies have established that MEK signaling pathways that led to NF-κB activation (41, 42) are inactivated by LT, and furthermore, inhibitors of NF-κB such as BAY11-7082, BAY11-7085, SN-50 and knockdown of p65 have previously been shown to have no effect on LT-mediated activation of NLRP1 (data not shown). Thus the inhibitory effects of LT on caspase-1 were independent of effects on NF-κB. Interestingly, application of the drug up to 45 min after LT caused significant inhibition of cytokine processing, further confirming an effect independent of inhibition of NF-κB signaling. Inhibition of pyroptosis by 15d-PGJ2 required earlier application of the drug (Supplemental Figure 1B and C). Importantly, the ability of 15d-PGJ2 to inhibit the inflammasome was not limited to NLRP1 or linked to pyroptosis. 15d-PGJ2 also inhibited cytokine processing and secretion in response to treatment with nigericin, an ionophore that activates the NLRP3 inflammasome (Figure 2B). 15d-PGJ2 demonstrated only modest inhibition of the NLRC4/NAIP5 inflammasome after stimulation with FlaTox (Figure 2C). However, previous work has similarly found that FlaTox is more difficult to inhibit (43), possibly due to its ligand binding-based mechanism of activation (44, 45). These data indicate that 15d-PGJ2 is capable of inhibiting multiple inflammasomes with diverse activating stimuli.
Figure 2. 15d-PGJ2 inhibits caspase-1 autoproteolysis and cytokine maturation of multiple inflammasomes.

Balb/cJ BMDMs were primed with LPS (1 μg/mL, 2 h). Cells were then exposed to 15d-PGJ2 (50 μM, 30 min) followed by (A) LT (1 μg/mL), (B) nigericin (50 μM), or (C) FlaTox (1 μg/mL) for indicated times. Western blotting of cell lysates and culture supernatants was performed with antibodies against the p10 fragment of caspase-1 or IL-1β. Balb/cJ BMDMs were primed with LPS (1 μg/mL, 2 h). Cells were then treated with 15d-PGJ2 (50 μM, 30 min) followed by FlaTox (1 μg/mL) for indicated times. Western blotting of cell lysates and culture supernatants was performed with antibodies against IL-1β. The * indicates a cross-reactive protein band which acts as an internal equal loading control. Results shown in these Westerns are representative of between four and up to sixteen similar experiments per treatment.
15d-PGJ2 induces a cellular state inhibitory to caspase-1
Given the ability of this compound to inhibit multiple inflammasomes, we hypothesized that 15d-PGJ2 could inhibit caspase-1 directly. The α,β-unsaturated carbonyl makes cyclopentenone prostaglandins highly electrophilic and capable of conjugation to cysteine residues on various proteins, altering their functions (4). Caspase-1, a cysteine protease, contains catalytically important thiol groups that could be modified. However, using recombinant active caspase in a cell lysate assay where 15d-PGJ2 was added to lysates, we found no evidence of direct caspase-1 inhibition (Figure 3A). Interestingly, however, lysates made from cells pre-treated with 15d-PGJ2 were inhibitory to recombinant caspase-1 (Figure 3B). This indicated that a cellular state or protein that could inhibit caspase-1 enzymatic activity was induced by 15d-PGJ2 in intact cells.
Figure 3. 15d-PGJ2 does not inhibit caspase-1 enzymatic activity directly but induces a cellular state inhibitory to caspase-1.

(A) RAW264.7 cells were primed with LPS (1 μg/mL, 2h). Sucrose lysates were incubated with recombinant active caspase-1 (1 U/50 μL, 3 h, 37°C) in the presence or absence of 15d-PGJ2 at indicated concentrations or positive control caspase-1 inhibitor Boc-D-CMK (400 μM). (B) RAW264.7 cells were primed with LPS (1 μg/mL, 2 h) followed by treatment with 15d-PGJ2 (50 μM, 1 h). As above, sucrose lysates were incubated with recombinant active caspase-1. In all panels, IL-1β processing was observed by Western blot. The * indicates a cross-reactive protein band which acts as an internal equal loading control. Western results are representative of at least three similar experiments.
Inflammasome inhibition by 15d-PGJ2 is dependent on protein translation
We previously reported that both heat shock and arsenical compound inhibition of inflammasome activation did not require de novo protein synthesis (27, 43). In the case of heat shock, procaspase-1 is trapped in a high molecular weight complex that can be centrifuged away from the cytosolic cellular fraction (27). We found that 15d-PGJ2, a potent inducer of heat shock proteins (8), did not induce sequestration of caspase-1 into a high molecular weight complex (Supplemental Figure 2).
In contrast to the response to both heat shock and arsenical compounds, the transcription inhibitor actinomycin D and the translation inhibitor cycloheximide reversed the protective effects of 15d-PGJ2 in a dose-dependent manner (Figure 4A and B). This requirement for transcription further supported the fact that 15d-PGJ2 manifested its inhibitory effects on the inflammasome in a manner independent of the eicosanoid’s inhibitory effect on NF-κB transcription. Similar results were found upon an even more complete enzymatic inhibition of translation using FP59, a fusion protein of an ADP-ribosylating enzyme which targets elongation factor 2 (Figure 4C). Furthermore, inhibition of translation partially restored cytokine processing in 15d-PGJ2-treated cells (Figure 4D). This evidence indicated that inflammasome inhibition by 15d-PGJ2 required de novo protein translation, either to induce a cytoprotective protein or to maintain a certain threshold level of a caspase-1 inhibitory protein with normally rapid turnover (the levels of which diminish after inhibition of protein synthesis). Interestingly, inhibition of NLRP3-mediated cytokine processing was not affected by cycloheximide treatment (data not shown), suggesting the lipid mediator could have multiple inhibitory mechanisms.
Figure 4. 15d-PGJ2-mediated inhibition of the NLRP1 inflammasome requires protein synthesis.

(A) Balb/cJ BMDMs or (B) RAW264.7 cells were incubated with variable concentrations of cycloheximide or actinomycin D for 1 h. Cells were then exposed to 15d-PGJ2 (50 μM, 30 min) followed by LT (1 μg/mL, 1.5–2 h). (C) RAW264.7 cells were treated with 10 ng/mL PA with or without 100 ng/mL FP59 for 2 h. PA receptor saturation in this cell type occurs at doses 20-fold higher than that used here (69). Thus sufficient receptor remained on the cell surface to mediate LT-intoxication. Unbound toxin was then washed from the cells, and the cells were incubated with 15d-PGJ2 and LT as above. Cell viability was measured by MTT staining. Each point was assayed in triplicate. Shown results are for a representative of two identical experiments. In (C), error bars represent standard error of the mean. (D) Balb/cJ BMDMs were treated with LPS (1 μg/mL, 3 h). Cells were then allowed to rest in LPS-free media for 1 h before cycloheximide treatment (2 μg/mL, 1 h). 15d-PGJ2 (50 μM, 30 min) followed by LT (1 μg/mL, 75 min) were then applied. Cell lysates were analyzed for IL-1β maturation by Western blot. The * indicates a cross-reactive protein band which acts as an internal equal loading control. Western results are representative of three similar experiments.
Inhibition is independent of PPARγ, NRF2, or COX-1
15d-PGJ2 is a ligand of the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) (10, 11) which is known to have anti-inflammatory roles (14). We hypothesized that the eicosanoid could mediate its effects through this receptor. However, the PPARγ agonist rosiglitazone was unable to inhibit LT-induced pyroptosis (Figure 5A). Furthermore, despite loss of the α,β-unsaturated bond in the ring, the 15d-PGJ2 analog CAY10410 retains the ability to activate PPARγ (46). This drug does not protect cells from LT-induced pyroptosis (Table I). Similarly, these compounds were unable to prevent LT- (Figure 5B) or nigericin- (data not shown) induced cytokine processing. Additionally, the PPARγ inhibitor T0070907 did not reverse the ability of 15d-PGJ2 to inhibit the inflammasome (Figure 5C and D). Together, these data suggest that PPARγ signaling and transcriptional processes downstream of this receptor are not required for the effects of 15d-PGJ2 on the inflammasome.
Figure 5. 15d-PGJ2 inflammasome inhibition is independent of PPARγ and NRF2.

(A) RAW264.7 cells were incubated with variable concentrations of PPARγ agonist rosiglitazone for 1.5 h. Cells were exposed to LT (1 μg/mL, 2 h) and cell viability assessed by MTT staining. (B) Balb/cJ BMDMs were primed with LPS (1 μg/mL, 2 h) followed by 15d-PGJ2, CAY10410, or rosiglitazone (50 μM, 30 min) and LT (1 μg/mL, 75 min). IL-1β in cell lysates and supernatants was assessed by Western blot. (C) RAW264.7 cells were treated with variable concentrations of PPARγ antagonist T0070907 for 1 h before treatment with 15d-PGJ2 (50 μM, 30 min). Following LT treatment (1 μg/mL, 2 h), cell viability was assessed by MTT staining. (D) RAW264.7 cells were primed with LPS (1 μg/mL, 2 h) followed by treatment with T0070907 (30 μM, 1 h). Cells were then incubated with 15d-PGJ2 (50 μM, 30 min) followed by LT (1 μg/mL, 1.5 h). (E) C57BL6/J WT and NRF2−/− BMDMs were primed with LPS (1 μg/mL, 2 h) followed by 15d-PGJ2 (50 μM, 30 min) then with nigericin (50 μM, 30 min). Cell lysates and culture supernatants were analyzed for IL-1β maturation by Western blot. In (A) and (C), each condition was assayed in duplicate. Curves are representative of at least two similar experiments. The * indicates a cross-reactive protein band which acts as an internal equal loading control.
15d-PGJ2 also activates the Cap ‘n’ Collar family transcription factor NRF2 (nuclear factor erythroid 2-related factor 2) (12) by binding to cysteine residues of the NRF2-repressor KEAP1 (Kelch-like ECH associated protein 1). NRF2 controls the expression of a large and diverse group of cytoprotective and anti-inflammatory genes (13) which we hypothesized could be inhibitory to inflammasome function. However, this seemed unlikely as NRF2 is stabilized and activated by proteasome inhibition (47–50) and treatment of macrophages with the proteasome inhibitor lactacystin does not prevent nigericin-based NLRP3-dependent cytokine processing (data not shown and (35)). We tested the effects of 15d-PGJ2 in BMDMs lacking NRF2 and found the drug was capable of inhibiting nigericin-induced cytokine processing, indicating the effects of this eicosanoid on the inflammasome did not involve this transcription factor (Figure 5E).
Recent studies have linked inflammasome activation with release of a variety of lipid mediators described as an “eicosanoid storm” (20). We hypothesized that modulation of this prostanoid release by exogenous 15d-PGJ2 could alter inflammasome function. However, 15d-PGJ2 inhibited LT-mediated pyroptosis and cytokine processing independent of COX-1, a central enzyme in the prostaglandin biosynthetic pathway (Figure 6A and B). Additionally, the cyclooxygenase inhibitor indomethacin did not reverse 15d-PGJ2- mediated protection from pyroptosis (Supplemental Figure 3A).
Figure 6. 15d-PGJ2 inflammasome inhibition is independent of COX-1.

(A) BMDMs from Cox1−/− (on the C57BL/6NTacNlrp1bS/S background) mice were primed with LPS (1 μg/mL, 2h). Cells were then incubated with 15d-PGJ2 (50 μM, 30 min) followed by LT (1 μg/mL, 75 min). Cell lysates were analyzed for IL-1β maturation by Western blot. (B) COX-1−/− BMDMs were exposed to varying concentrations of 15d-PGJ2 for 30 min. Cells were challenged with 1 μg/mL LT and viability was measured by MTT staining at 1.5 h post-LT. Each condition was assayed in triplicate. Curves are representative of two or more similar experiments. The * indicates a cross-reactive protein band which acts as an internal equal loading control.
Glutathione modulation does not alter inhibition
Cyclopentenone prostaglandins form conjugates with cellular glutathione which can alter the physiological effectiveness of the prostaglandin (51). Increasing the levels of cellular glutathione using the glutathione precursor N-acetylcysteine or decreasing cellular levels of glutathione using the glutathione synthesis inhibitor buthionine sulfoximine had no effect on the protection of cells from LT-induced pyroptosis by 15d-PGJ2 (Supplemental Figure 3B and C).
Cyclopentenone prostaglandins are also potent inducers of intracellular reactive oxygen species (52), and oxidative stress is a potential mechanism of inflammasome regulation (53). However, the failure of N-acetylcysteine, a potent antioxidant, to alter 15d-PGJ2 protection suggests that induction of reactive oxygen species does not play a role in the effects of 15d-PGJ2 on the inflammasome. Furthermore, alterations of oxidative state by 15d-PGJ2 are usually coupled with direct modification of proteins. We utilized a biotin-labeled 15d-PGJ2 analog coupled with immunoprecipitation and did not find any evidence that 15d-PGJ2 directly bound to NLRP3 (data not shown), suggesting a novel inhibitory mechanism is operating that does not involve covalent adduct formation to NLRP3.
15d-PGJ2 inhibits the inflammasomes in mice
To investigate the effects of 15d-PGJ2 in vivo we utilized two models. The MSU-induced peritonitis model used to replicate the inflammation associated with gout requires NLRP3 activation and caspase-1 dependent release of IL-1β for cell recruitment (24). We used this model in mice treated with vehicle or 15d-PGJ2 to assess the relative cell numbers recruited to the peritoneum and associated levels of secreted IL-1β. MSU crystals induced a robust recruitment of cells to the peritoneum which was significantly inhibited by the prostaglandin treatment (Figure 7A). Similarly, the IL-1β release associated with injection of MSU was significantly lower in mice treated with 15d-PGJ2 (Figure 7B). To test the effects of the drug on the NLRP1 inflammasome in vivo, we took advantage of the finding that inbred mouse strains with the LT-responsive Nlrp1bS/S allele (such as Balb/cJ) are highly resistant to anthrax spore infection compared to those strains (such as C57BL/6 mice) which express the NLRP1bR variant (23). Similarly, C57BL/6JNTac congenic mice which are identical at >99% of loci but differ at the Nlrp1b locus have striking different susceptibility to spore infection which is linked to LT induced, NLRP1b-mediated caspase-1 dependent control of IL-1β responses and neutrophil recruitment (23). The mice in which LT can cleave and activate NLRP1b harbor myeloid cells which undergo pyroptosis in conjunction with an unprimed IL-1β response that leads to resistance. We found that inhibition of this activation of NLRP1b by treatment of spore-resistant Balb/cJ or C57BL/6JNTacNlrp1bS/S congenic mice with 15d-PGJ2 led to a rapid and striking sensitization to spore infection and a susceptibility comparable to mice harboring the Nlrp1bR/R allele (Figure 7C, 7D). Because the effects of LT on susceptibility to spore are uniquely independent of NF-κB signaling, the 15d-PGJ2 effects in this model, unlike the MSU cell recruitment model, are likely to be on a downstream inflammasome activation event required for IL-1β release.
Figure 7. 15d-PGJ2 inhibition of the inflammasome in mice.

(A, B) A well-established mouse monosodium urate induced peritonitis model was utilized to assess impact of 15d-PGJ2 on the NLRP3 inflammasome in vivo. C57BL/6J mice (n=9/group) were injected IP with 125 μg of 15d-PGJ2 or vehicle (10% DMSO in PBS) at 5 min prior and 4 h after administration of monosodium urate (MSU) crystals. Control mice were injected with vehicle (n=4) or drug (n=7) alone. MSU crystals were injected into mice (4 mg/250 ul PBS, IP) and after 6 h infiltrating cells were counted (A) and IL-1β levels assessed (B). P-values (unpaired t-test) comparing the MSU (+vehicle) groups to MSU (+15d-PGJ2) groups are <0.0001 in both panels. (C, D) Anthrax spore-resistant NLRP1bS/S-expressing Balb/cJ (n=5) and C57BL/6NTacNlrp1bS/S (n=5) mice were treated with 15d-PGJ2 (100 μg, 200 μl, SC, at 5 min and 1 h post spore infection) or with vehicle and infected with 2×107 A35 spores (C) or 4×107 A35 spores (D). Spore-sensitive NLRP1bR/R-expresssing C57BL/6J (n=5) and C57BL/6NTacNlrp1bR;R (n=4) mice infected with the same spore dose served as controls. For panel C, the P-value comparing vehicle-treated, spore-infected Balb/cJ mice to the 15d-PGJ2-treated infected Balb/cJ group or infected C57BL/6J mice is <0.007. There is no significant difference between the infection susceptibility for drug-treated Balb/cJ mice and genetically susceptible C57BL/6J mice infected with B. anthracis spores. For panel D, the P-value comparing vehicle-treated, spore-infected C57BL/6NTacNlrp1bS/S to 15d-PGJ2-treated infected group is 0.0065. The P-value comparing vehicle-treated, spore-infected C57BL/6NTacNlrp1bS/S to similarly infected C57BL/6NTacNlrp1bR;R is 0.00429. The susceptibility curve for the drug-treated and spore-infected C57BL/6NTacNlrp1bS/S mice is not significantly different from the spore-infected genetically susceptible C57BL/6NTacNlrp1bR/R mice. The Log-rank test was used for assessment of all P-values in panels C and D.
DISCUSSION
In this work we show that cyclopentenone prostaglandins inhibit activation of caspase-1 and maturation of IL-1β by the NLRP1 and NLRP3 inflammasomes. This inhibition is shown to be independent of the drugs’ inhibitory effects on NF-κB signaling and transcription and not due to interference with inflammasome-activating stimuli or direct modification of caspase-1. Inflammasome inhibition occurred instead through induction of a cellular state inhibitory to casapse-1 enzymatic activity. This cellular state was dependent on de novo protein translation but independent of the PPARγ or NRF2 signaling and transcription pathways often associated with the actions of these prostaglandins. Our results suggest a novel mechanism and transcription pathway are responsible for the anti-inflammatory effects of these prostaglandins.
There is substantial evidence that the reactive α,β-unsaturated carbonyl group is necessary for many of the biological activities of cyclopentenone prostaglandins (4). The unsaturated carbonyl imparts a reactive electrophilic character to carbon 9 of the molecule. This allows the compound to form adducts with cellular thiols such as glutathione or cysteine residues on proteins (4). The α,β-unsaturated carbonyl appears to be necessary for inflammasome inhibition as prostaglandins lacking the cyclopentenone ring were inactive. Similarly, 2-cyclopentenone, but not the related compounds cyclopentene and cyclopentanone, was able to inhibit the inflammasome. This requirement for a chemically reactive center suggests that the mechanism of inflammasome inhibition is through conjugation to exposed thiol residues on a key inflammasome regulatory component. However, we found no evidence of direct inactivation of caspase-1 or NLRP3. The finding that de novo protein synthesis is required for the compound’s effects suggests that direct modification of the inflammasome is not likely, but we cannot rule out the possibility of 15d-PGJ2-mediated modification of an unknown yet important inflammasome component. Our finding that inhibition of NLRP3 was not dependent on protein synthesis suggests that the mechanism of 15d-PGJ2-inhibition differs across inflammasomes. This may be due to the varied biochemical mechanisms of inflammasomes activation. NLRP1, for example, is activated in rodent macrophages through direct proteolytic cleavage by LT (28–30). NLRC4, in contrast, acts as an adapter for a number of NAIP proteins which directly bind to flagellin and flagellin-like proteins (44, 45), and NLRP3 responds to a wide range of danger signals through an as yet unresolved mechanism (1).
PPARγ, and NRF2 are the primary transcriptional pathways activated by 15d-PGJ2 (15), and we hypothesized that one of them may play a role in the ability of this prostaglandin to inhibit the inflammasome. 15d-PGJ2 was the first identified endogenous ligand of PPARγ, and many of the biological effects of this prostaglandin result from PPARγ activation (10, 11). However, the compound also induces PPARγ-independent reactions. In fact, prior studies have demonstrated that 15d-PGJ2 has anti-inflammatory effects on macrophages deficient in PPARγ (54), and we find that inflammasome inhibition is also independent of PPARγ. A recent study demonstrated that NRF2−/− BMDMs are deficient in inflammasome function (55). Although we also find a slight reduction of cytokine processing in NRF2−/− BMDMs relative to wild type BMDMs, this difference is far less pronounced than previously reported. In striking contrast, treatment with 15d-PGJ2 potently inhibits inflammasome function in a manner independent of NRF2.
The inducible cyclooxygenase COX-2 is believed to have a dual role in inflammation, both establishing the initial inflammatory response and leading to its resolution at later times. COX-2 is rapidly induced during inflammation and contributes to acute inflammation through production of pro-inflammatory signaling molecules (56). However, COX-2 induction during late stages of inflammation leads to production of anti-inflammatory prostaglandins such as 15d-PGJ2 (57). These prostaglandins along with additional lipid mediators called lipoxins, resolvins, and protectins thus constitute important endogenous molecules in the resolution of inflammation (58). In vivo concentrations of 15d-PGJ2 are believed to be in the nanomolar range (59). Thus, one may question the physiological relevance of the micromolar concentrations necessary for inflammasome inhibition in vitro. However, it has been demonstrated that upon dilution into culture media containing FBS a majority (97–99%) of the prostaglandin is inactivated and does not enter cells (40). Thus the levels actually available for the cells in vitro are likely at physiological concentrations. Additionally, it can be postulated that systemic measurements of free prostaglandin in vivo are an underestimation of true levels found within localized pockets of inflammation or in the cell vicinity, given the high reactivity of these eicosanoids with serum and intracellular proteins (60).
Exogenous administration of 15d-PGJ2 also has efficacy as an anti-inflammatory treatment (15). In this work we show that 15d-PGJ2 can inhibit MSU-mediated neutrophil recruitment in an NLRP3-dependent inflammatory peritonitis model. Due to the reactivity and resulting instability of cyclopentenone prostaglandins, most experimental studies rely on administration of relatively high doses of 15d-PGJ2. Thus, alternative delivery mechanisms for the prostaglandin may increase its therapeutic potential. Loading of polyglycolide nanocapsules with the prostaglandin increased serum availability and improved the anti-inflammatory activity of 15d-PGJ2 in a peritonitis model (61). Retroviral overexpression of the prostaglandin synthase responsible for 15d-PGJ2 production has also been successful in a murine air-pouch model of acute-inflammation, as well as in bleomycin-induced lung injury and scleroderma models (62–64). Synthetic derivatives of 15d-PGJ2 with similar biological activities have also been reported (65, 66).
Administration as an anti-inflammatory treatment in animal models of inflammation including ischemic brain injury, LPS-induced fever, spinal cord injury, chronic obstructive pulmonary disease, colitis, myocarditis, pancreatitis, and arthritis has been reported (15). The mechanism of action of the prostanoid in these diverse inflammatory models may not be uniform. For example, troglitazone, a synthetic PPARγ ligand was equally effective as 15d-PGJ2 at improving adjuvant-induced arthritis (67), while the protective effect of 15d-PGJ2 in a carrageenan-induced acute lung injury model was found to be NRF2-dependent (68). The inflammasome is known to play a role in a number of the diseases for which 15d-PGJ2 has shown therapeutic effects (1), suggesting that this compound may be acting through its ability to inhibit IL-1β processing via the inflammasome. In this study, in addition to establishing the inhibitory effects of 15d-PGJ2 in a NLRP3-dependent peritonitis model, we were also able to cause a striking reversal of NLRP1 and caspase-1 dependent resistance of mice to anthrax spore infection by treatment with a single dose of the prostaglandin. These findings demonstrate that the anti-inflammatory effects of 15d-PGJ2 in vivo may be partially mediated through its impact on caspase-1 and the inflammasome pathways.
In conclusion, we demonstrate that the cyclopentenone prostaglandins inhibit the inflammasome. Most studies to date have explained the anti-inflammatory effects of 15d-PGJ2 by its effects on transcriptional pathways such as PPARγ, NRF2, and NF-κB. We suggest that a full explanation of the anti-inflammatory effects of 15d-PGJ2 must include its modulation of IL-1β release through inhibition of the inflammasome.
Supplementary Material
Acknowledgments
We thank Devorah Crown for bone marrow extraction for all studies. We thank Dr. Russell Vance (UC Berkeley) for providing the LFn-Fla protein and Dr. Katrin Mayer-Barber (NIAID, NIH) for the gift of mice. Rasem Fattah (NIAID, NIH) performed the purification of proteins from B. anthracis.
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
Abbreviations used in this manuscript
- BMDM
bone marrow derived macrophages
- PG
prostaglandin
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin
- J2 PA
anthrax protective antigen
- LF
anthrax lethal factor
- LT
anthrax lethal toxin (combination of PA and LF)
- LFn-Fla
fusion protein of N-terminal domain of LF to flagellin
- FlaTox
combination of PA and LFn-Fla
- FP59
fusion protein of N-terminal domain of LF to the ADP-ribosylating domain of Pseudomonas aeruginosa exotoxin A
- NLR
nucleotide-binding oligomerization domain–like receptor
- NLRP1
NLR family leucine-rich repeat protein 1
- NLRP3
NLR family leucine-rich repeat protein 3
- NLRC4
NLR family caspase-1 recruitment domain–containing protein 4
- NAIP5
NLR family apoptosis inhibitory protein 5
- PPARγ
peroxisome proliferator-activated receptor γ
- NRF2
nuclear factor erythroid 2-related factor 2
- COX
cyclooxygenase
References
- 1.Lamkanfi M, Dixit Vishva M. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- 5.Cernuda-Morollón E, Pineda-Molina E, Cañada FJ, Pérez-Sala D. 15-deoxy-Δ12,14-prostaglandin J2 inhibition of NF-κB-DNA binding through covalent modification of the p50 subunit. J Biol Chem. 2001;276:35530–35536. doi: 10.1074/jbc.M104518200. [DOI] [PubMed] [Google Scholar]
- 6.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 7.Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang C-H, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc Natl Acad Sci USA. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amici C, Sistonen L, Santoro MG, Morimoto RI. Antiproliferative prostaglandins activate heat shock transcription factor. Proc Natl Acad Sci USA. 1992;89:6227–6231. doi: 10.1073/pnas.89.14.6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol. 2005;114:100–109. doi: 10.1016/j.clim.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Forman BM, Tontonoz P, Brun RP, Spiegelman BM, Evans RM. 15-deoxy-Δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 11.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 12.Kansanen E, Kivelä AM, Levonen A-L. Regulation of Nrf2-dependent gene expression by 15-deoxy-Δ12,14-prostaglandin J2. Free Radical Biol Med. 2009;47:1310–1317. doi: 10.1016/j.freeradbiomed.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 13.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 14.Clark RB. The role of PPARs in inflammation and immunity. J Leukocyte Biol. 2002;71:388–400. [PubMed] [Google Scholar]
- 15.Surh Y-J, Na H-K, Park J-M, Lee H-N, Kim W, Yoon I-S, Kim D-D. 15-deoxy-Δ12,14-prostaglandin J2, an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem Pharmacol. 2011;82:1335–1351. doi: 10.1016/j.bcp.2011.07.100. [DOI] [PubMed] [Google Scholar]
- 16.Liu S, Leung HJ, Leppla SH. Characterization of the interaction between anthrax toxin and its cellular receptors. Cell Microbiol. 2007;9:977–987. doi: 10.1111/j.1462-5822.2006.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park S, Leppla SH. Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr Purif. 2000;18:293–302. doi: 10.1006/prep.2000.1208. [DOI] [PubMed] [Google Scholar]
- 18.Arora N, Leppla SH. Residues 1–254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J Biol Chem. 1993;268:3334–3341. [PubMed] [Google Scholar]
- 19.Liu S, Netzel-Arnett S, Birkedal-Hansen H, Leppla SH. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000;60:6061–6067. [PubMed] [Google Scholar]
- 20.von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz Ba, Leppla SH, Gronert K, Vance RE. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pomerantsev AP, Sitaraman R, Galloway CR, Kivovich V, Leppla SH. Genome engineering in Bacillus anthracis using Cre recombinase. Infect Immun. 2006;74:682–693. doi: 10.1128/IAI.74.1.682-693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 23.Moayeri M, Crown D, Newman ZL, Okugawa S, Eckhaus M, Cataisson C, Liu S, Sastalla I, Leppla SH. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog. 2010;6:e1001222. doi: 10.1371/journal.ppat.1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 25.Martin WJ, Walton M, Harper J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. 2009;60:281–289. doi: 10.1002/art.24185. [DOI] [PubMed] [Google Scholar]
- 26.Newman ZL, Sirianni N, Mawhinney C, Lee MS, Leppla SH, Moayeri M, Johansen LM. Auranofin protects against anthrax lethal toxin-induced activation of the Nlrp1b inflammasome. Antimicrob Agents Chemother. 2011;55:1028–1035. doi: 10.1128/AAC.00772-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levin TC, Wickliffe KE, Leppla SH, Moayeri M. Heat shock inhibits caspase-1 activity while also preventing its inflammasome-mediated activation by anthrax lethal toxin. Cell Microbiol. 2008;10:2434–2446. doi: 10.1111/j.1462-5822.2008.01220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chavarría-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Path. 2013;9:e1003452. doi: 10.1371/journal.ppat.1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, Liu S, Leppla SH, Moayeri M. Anthrax lethal factor cleaves mouse Nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PloS one. 2012;7:e49741. doi: 10.1371/journal.pone.0049741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Path. 2012;8:e1002638. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243:206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 33.Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4312–4317. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muehlbauer SM, Evering TH, Bonuccelli G, Squires RC, Ashton AW, Porcelli SA, Lisanti MP, Brojatsch J. Anthrax lethal toxin kills macrophages in a strain-specific manner by apoptosis or caspase-1-mediated necrosis. Cell cycle. 2007;6:758–766. doi: 10.4161/cc.6.6.3991. [DOI] [PubMed] [Google Scholar]
- 35.Wickliffe KE, Leppla SH, Moayeri M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol. 2008;10:332–343. doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohno K, Fukushima M, Fujiwara M, Narumiya S. Induction of 68,000-Dalton heat shock proteins by cyclopentenone prostaglandins. J Biol Chem. 1988;263:19764–19770. [PubMed] [Google Scholar]
- 37.Santoro MG, Garaci E, Amici C. Prostaglandins with antiproliferative activity induce the synthesis of a heat shock protein in human cells. Proc Natl Acad Sci USA. 1989;86:8407–8411. doi: 10.1073/pnas.86.21.8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fitzpatrickz FA, Wynalda MA. Albumin-catalyzed metabolism of prostaglandin D2: identification of products formed in vitro. J Biol Chem. 1983;258:11713–11718. [PubMed] [Google Scholar]
- 39.Narumiya S, Fukushima M. Δ12-prostaglandin J2, an ultimate metabolite of prostaglandin D2 exerting cell growth inhibition. Biochem Biophys Res Commun. 1985;127:739–745. doi: 10.1016/s0006-291x(85)80005-x. [DOI] [PubMed] [Google Scholar]
- 40.Oh JY, Giles N, Landar A, Darley-Usmar V. Accumulation of 15-deoxy-Δ12,14-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochem J. 2008;411:297–306. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukusawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 42.Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- 43.Maier NK, Crown D, Liu J, Leppla SH, Moayeri M. Arsenic trioxide and other arsenical compounds inhibit the NLRP1, NLRP3, and NAIP5/NLRC4 inflammasomes. J Immunol. 2014;192:763–770. doi: 10.4049/jimmunol.1301434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kofoed EM, Vance RE. NAIPs: building an innate immune barrier against bacterial pathogens. Bioessays. 2012;34:589–598. doi: 10.1002/bies.201200013. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 46.Shiraki T, Kamiya N, Shiki S, Kodama TS, Kakizuka A, Jingami H. α,β-Unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor γ. J Biol Chem. 2005;280:14145–14153. doi: 10.1074/jbc.M500901200. [DOI] [PubMed] [Google Scholar]
- 47.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 48.McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen T, Sherratt PJ, Huang H-C, Yang CS, Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element: degradation of Nrf2 by the 26 S proteasome. J Biol Chem. 2003;278:4536–4541. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]
- 50.Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem. 2003;278:2396–2402. doi: 10.1074/jbc.M209195200. [DOI] [PubMed] [Google Scholar]
- 51.Atsmon J, Freeman ML, Meredith MJ, Sweetman BJ, Il LJR. Conjugation of 9-deoxy-Δ9, Δ12(E)-prostaglandin D2 with intracellular glutathione and enhancement of its antiproliferative activity by glutathione depletion. Cancer Res. 1990;50:1879–1885. [PubMed] [Google Scholar]
- 52.Kondo M, Oya-Ito T, Kumagai T, Osawa T, Uchida K. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J Biol Chem. 2001;276:12076–12083. doi: 10.1074/jbc.M009630200. [DOI] [PubMed] [Google Scholar]
- 53.Rubartelli A. Redox control of NLRP3 inflammasome activation in health and disease. J Leukocyte Biol. 2012;92:951–958. doi: 10.1189/jlb.0512265. [DOI] [PubMed] [Google Scholar]
- 54.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 55.Zhao C, Gillette DD, Li X, Zhang Z, Wen H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J Biol Chem. 2014;289:17020–17029. doi: 10.1074/jbc.M114.563114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajakariar R, Yaqoob MM, Gilroy DW. Cox-2 in inflammation and resolution. Mol Interventions. 2006;6:199–207. doi: 10.1124/mi.6.4.6. [DOI] [PubMed] [Google Scholar]
- 57.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 58.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40:315–327. doi: 10.1016/j.immuni.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-Δ12,14-PGJ2 and the ligation of PPARγ. J Clin Invest. 2003;112:945–955. doi: 10.1172/JCI18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Y, Morrow JD, Roberts LJ., II Formation of reactive cyclopentenone compounds in vivo as products of the isoprostane pathway. J Biol Chem. 1999;274:10863–10868. doi: 10.1074/jbc.274.16.10863. [DOI] [PubMed] [Google Scholar]
- 61.Alves C, de Melo N, Fraceto L, de Araújo D, Napimoga M. Effects of 15d-PGJ2-loaded poly(D,L-lactide-co-glycolide) nanocapsules on inflammation. Br J Pharmacol. 2011;162:623–632. doi: 10.1111/j.1476-5381.2010.01057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ando M, Murakami Y, Kojima F, Endo H, Kitasato H, Hashimoto A, Kobayashi H, Majima M, Inoue M, Kondo H, Kawai S, Hayashi I. Retrovirally introduced prostaglandin D2 synthase suppresses lung injury induced by bleomycin. Am J Respir Cell Mol Biol. 2003;28:582–591. doi: 10.1165/rcmb.2002-0162OC. [DOI] [PubMed] [Google Scholar]
- 63.Kohno S, Endo H, Hashimoto A, Hayashi I, Murakami Y, Kitasato H, Kojima F, Kawai S, Kondo H. Inhibition of skin sclerosis by 15-deoxy-Δ12,14-prostaglandin J2 and retrovirally transfected prostaglandin D synthase in a mouse model of bleomycin-induced scleroderma. Biomed Pharmacother. 2006;60:18–25. doi: 10.1016/j.biopha.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 64.Murakami Y, Akahoshi T, Hayashi I, Endo H, Hashimoto A, Kono S, Kondo H, Kawai S, Inoue M, Kitasato H. Inhibition of monosodium urate monohydrate crystal-induced acute inflammation by retrovirally transfected prostaglandin D synthase. Arthritis Rheum. 2003;48:2931–2941. doi: 10.1002/art.11271. [DOI] [PubMed] [Google Scholar]
- 65.Ciucci A, Gianferretti P, Piva R, Guyot T, Snape TJ, Roberts SM, Santoro MG. Induction of apoptosis in estrogen receptor-negative breast cancer cells by natural and synthetic cyclopentenones: role of the IκB kinase/nuclear factor-κB pathway. Mol Pharmacol. 2006;70:1812–1821. doi: 10.1124/mol.106.025759. [DOI] [PubMed] [Google Scholar]
- 66.Fukushima S, Kishimoto S, Takeuchi Y, Fukushima M. Preparation and evaluation of o/w type emulsions containing antitumor prostaglandin. Adv Drug Del Rev. 2000;45:65–75. doi: 10.1016/s0169-409x(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 67.Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K-i, Kohno M, Yamada R, Hla T, Sano H. 15-deoxy-Δ12,14-PGJ2 induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106:189–197. doi: 10.1172/JCI9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mochizuki M, Ishii Y, Itoh K, Iizuka T, Morishima Y, Kimura T, Kiwamoto T, Matsuno Y, Hegab AE, Nomura A, Sakamoto T, Uchida K, Yamamoto M, Sekizawa K. Role of 15-deoxyΔ12,14 prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am J Respir Crit Care Med. 2005;171:1260–1266. doi: 10.1164/rccm.200406-755OC. [DOI] [PubMed] [Google Scholar]
- 69.Wickliffe KE, Leppla SH, Moayeri M. Killing of macrophages by anthrax lethal toxin: involvement of the N-end rule pathway. Cell Microbiol. 2008;10:1352–1362. doi: 10.1111/j.1462-5822.2008.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.