

Abstract
Many malignant cells release the NKG2D ligand ULBP2 from their cell surface to evade immunosurveillance by natural killer cells and CD8 T cells. Although the shedding mechanism remains unclear, various inhibitors of matrix metalloproteinases have been shown to efficiently block the release of soluble ULBP2. The clinical use of these inhibitors however is limited because of adverse side effects. Using high throughput screening technique, we identified a specific inhibitor of phosphatase of regenerating liver 3 (PRL-3) that could reduce the level of soluble ULBP2 in the culture supernatant of various cancer cell lines. Inhibition or gene knockdown of PRL-3 did not reduce ULBP2 shedding but rather suppressed post-translational maturation of ULBP2, resulting in intracellular retention of immature ULBP2. We then found that ULBP2 was constitutively associated with heat shock protein HSP60. Complete maturation of ULBP2 required tyrosine phosphorylation of HSP60 which was mediated by PRL-3.
Introduction
The activating receptor NKG2D expressed on human natural killer (NK) cells and T cells recognizes the members of two ligand families: MHC class I chain related molecules (MICA and MICB) and UL16 binding proteins (ULBP1-6). These NKG2D ligands (NKG2DLs) are self-proteins having restricted expression on normal tissues. Their expression, however, is upregulated in situations of stress and disease such as pathogen infection or tumor transformation (1). The interaction between NKG2D and NKG2DLs triggers NK cell cytokine secretion and degranulation which induce apoptosis of target cells. Many reports have suggested that the expression level of NKG2DLs on tumor cells directly correlates with tumor susceptibility to NK cytolysis (2) and with cancer patient survival rate (3).
Members from the two NKG2DL families vary considerably in protein structure. For example, MICA, MICB, ULBP4 and ULBP5 possess a transmembrane domain while ULBP1, 2, 3 and 6 are glycosylphosphatidylinositol (GPI)-anchored proteins (4). Despite these variations, the topography of different ligands with NKG2D, and also their efficacy of triggering NK cytolysis are similar (5, 6). The reason for the existence of multiple distinct NKG2DLs for a single receptor remains unclear. It has been suggested that the expression of multiple NKG2DLs on target cells may ensure efficient recognition by NK cells to avoid immune escape (7). This hypothesis is supported by the observations that different forms of stress or stimulation upregulate different NKG2DLs (1).
Gene transcription, microRNA regulation, and proteolytic cleavage from the cell membrane are some mechanisms that can regulate NKG2DL expression on tumor cells (1). Transcription of MICA and MICB has been shown to be inducible by heat shock, oxidative stress or cell proliferation (8–10), whereas ULBP1-3 expression may be suppressed by histone deacetylase 3 (HDAC3) in epithelial tumor cells (11). Furthermore, activation of the ataxia telangiectasia mutated (ATM)/ ATM-and Rad3-related (ATR) kinase-mediated DNA damage response has been shown to upregulate the expression of NKG2DLs in various tumor cell types (12, 13). Recently multiple microRNAs were found to be able to downregulate the mRNA level of MICA, MICB and ULBP2 by targeting their 3’-untranslated regions (14–16). In addition to mRNA regulation, the protein level of NKG2DLs on cell surface can be regulated by shedding mediated through metalloproteinases (17–19). For example, the membrane-bound ULBP2 can be proteolytically released from cancer cell lines such as HCT116 (colon cancer), HL60 and Jurkat (leukemia) cells and that the protein shedding is reducible by matrix metalloprotease (MMP) inhibitors (19). Several studies revealed a direct correlation between the patient serum level of soluble ULBP2 and the prognosis of cancer (2, 20). Various metalloproteinase inhibitors have been identified to block the release of NKG2DLs from tumor cells (18, 19). However, clinical trials with these broad-spectrum inhibitors only had limited success because of severe side effects related to musculoskeletal pain and inflammation (21, 22).
To investigate if the release of soluble ULBP2 can be blocked by other protease inhibitors that possess less toxicity, we established a high-throughput screening system using a protease inhibitor library. We screened simultaneously a phosphatase inhibitor library because the activity of many proteases is dependent on their phosphorylation status (23). A specific phosphatase inhibitor targeting the phosphatase of regenerating liver 3 (PRL-3), PRL-3 inhibitor I (PRL3-I), was identified by the high-throughput screening. PRL-3, also known as protein tyrosine phosphatase 4A3 (PTP4A3), is a metastasis-associated phosphatase that plays essential roles in cancer progression and metastasis (24–26). The expression of PRL-3 is restricted to normal skeletal muscle, pancreas, fetal heart, developing blood vessels and pre-erythrocytes, but is highly upregulated in malignant tissues, particularly in later stages during tumor progression. Inhibition of PRL-3 can block the migration and invasion of metastatic cancer cells both in vitro and in vivo (27, 28). Curcumin, a natural compound that reduces the expression of PRL-3, has been shown to be safe and exhibit therapeutic efficacy in patients with progressive advanced cancers in phase I and II clinical trials (28), highlighting the potential of PRL-3-targeting therapy in cancer treatment. We, therefore, studied the biologic mechanisms of PRL-3 regulation of ULBP2 levels.
Surprisingly, the reduction of ULBP2 release upon PRL3-I treatment was not associated with an increase but rather a decrease in surface level of ULBP2 on cancer cells. We found that the post-translational maturation of ULBP2 protein, which involved the formation of GPI-anchor and complete glycosylation, was the key determinant of its surface expression. ULBP2 was found to constitutively interact with the heat shock protein HSP60. PRL-3 regulated the tyrosine phosphorylation of HSP60, which was essential for the maturation of ULBP2. The present study revealed a novel post-translational regulatory mechanism in cancer cells for controlling the protein maturation and surface expression of ULBP2.
Materials and methods
Cell lines, antibodies, and reagents
HCT116, CAL27, 293T, HT29, SW480, Hela, and DU145 cell lines were obtained from American Type Culture Collection (ATCC). The protease and phosphatase inhibitor library were purchased from Enzo Life Sciences. Mouse anti-ULBP1 (AUMO2) (as primary antibody for flow cytometry) and anti-ULBP2 (BUMO1) (as capture antibody for ELISA) antibody were purchased from BAMOMAB; mouse anti-ULBP2 (IgG2a, as detection antibody for ELISA), allophycocyanin-conjugated mouse anti-ULBP2 (for flow cytometry), goat anti-ULBP2 and anti-ULBP1 (for Western blot) from R&D Systems; allophycocyanin-conjugated anti-mouse IgG antibody (as secondary antibody for flow cytometry) and HRP-conjugate goat anti-mouse IgG2a (as secondary antibody for ELISA) from Jackson ImmunoResearch; mouse anti-tubulin (DM1A+DM1B), rabbit anti-HSP60 from Abcam; HRP-conjugated anti-mouse and anti-rabbit secondary antibody, rabbit anti-CD73 and rabbit anti-Na,K-ATPase from Cell Signaling; HRP-conjugated anti-goat secondary antibody from Santa Cruz; HRP-conjugated anti-phosphotyrosine antibody (4G10 Platinum) and MMP inhibitor III from EMD Millipore; Mouse anti-Flag (clone M2) antibody, DMSO, PRL-3 inhibitor I, curcumin, nonactin, staurosporine and phosphatidylinositol-specific phospholipase C (PI-PLC) from Sigma; Src I1 and GM 6001 from TOCRIS Bioscience; TMB ELISA substrate solution from eBioscience; Cell Titer-Glo luminescent cell viability assay from Promega.
Flow cytometry
Cell lines were stained with isotype control or allophycocyanin-conjugated anti-ULBP2 for 20 min at 4°C. For ULBP1 staining, cells were first incubated with anti-ULBP1 and then with allophycocyanin-conjugated anti-mouse IgG antibody. Cells were washed and analyzed on a C6 flow cytometer (Accuri). Data were analyzed using FlowJo software (TreeStar).
ULBP2 ELISA
ULBP2 shed from cell surface to the culture supernatant was detected by ELISA method as described before (19). Briefly, 100 µl (triplicate) of cultured supernatant was added into a 96-well plate pre-coated with 1 µg/ml of mouse anti-ULBP2 (BUMO1) and incubated at 4°C overnight. The plate was washed and then incubated with mouse anti-ULBP2 (IgG2a, 100 µl/well at 1 µg/ml) for 1 h followed by incubation with HRP-conjugated goat anti-mouse IgG2a for 1 h at room temperature. TMB substrate was added and the plate was read using Victor2 plate reader (Perkin Elmer).
Quantitative real-time PCR (qRT-PCR) assay
Total RNA was extracted from DMSO or PRL3-I-treated (40 µM) tumor cells by using the RNA Clean & Concentrator (Zymo Research). cDNA was generated by using the SuperScript VILO cDNA Synthesis kit (Life Technologies) and diluted 10-fold for analysis using the 7900HT Fast Real-time PCR System (Applied Biosystems). The data were calculated as the CT of target genes normalized to the CT of GAPDH of each sample. Primer sequences of the target genes are: ULBP2-F: 5’-AAATGTCACAACGGCCTG-3’, ULBP2-R: 5’-TGAGGGGTTCCTTGGG-3’, GAPDH-F: 5’-ATGGGGAAGGTGAAGGTCG3’, GAPDH-R: 5’-GGGGTCATTGATGGCAA-CAATA-3’.
Cytotoxicity assay
NK cells were isolated from health donors by autoMACS Pro Seperator using NK cell isolation kit (Miltenyi) and cultured overnight with 10 U/ml of IL-2 (eBioscience). Cell lines treated for 18 h with DMSO or PRL3-I (40 µM) were subjected to BATDA release assays (Perkin Elmer) according to the manufacturer’s instructions. The results were calculated as follows:
Immunoprecipitation
Total cell lysates from ~ 1 × 107 cells (lysed in cell lysis buffer: 50mM Tris (pH 8.0), 10 mM EDTA, 125 mM NaCl, 1% Triton X-100, proteinase inhibitor cocktail (Thermo Scientific) for 10 min at 4°C) were incubated with 1 µg antibody (anti-ULBP2, anti-HSP60, anti-phosphotyrosine or anti-Flag) for 2 h at 4 °C, followed by 1 h incubation with 20 µl Protein G microbeads (Miltenyi). Target proteins were separation by MultiMACS M separator (Miltenyi) according to the manufacturer’s instructions.
Western blot analysis
Protein samples prepared in NuPAGE LDS sample buffer (Life technologies) were run onto NuPAGE 4–12% Bis-Tris gels and then transferred onto nitrocellulose membranes using the iBlot transfer system (Life technologies). After incubation with 5% nonfat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) for 15 min, the membranes were incubated with antibodies against ULBP2 (1:1000), HSP60 (1:2000), Flag-tag (1:2000), phosphotyrosine (1:800), or tubulin (1:5000) at 4 °C for 16 h. Membranes were washed three times for 15 min and incubated with a 1:2500 dilution of HRP-conjugated anti-mouse, anti-goat or anti-rabbit secondary antibodies for 1 h (except for phosphotyrosine detection because the antibody, 4G10 has already been HRP-conjugated). The membranes were washed with TBST five times and developed with the Western Lightning Plus ECL system (Perkin Elmer) according to the manufacturer’s instructions.
Endo H, PNGase F and PI-PLC digestion
Twenty µg of total proteins from the PRL3-I-treated or untreated HCT116-ULBP2 cell lysates were digested with 1500U of Endo H or PNGase F (NEB) at 37°C for 2 h according to the manufacturer’s instruction. For experiments with PI-PLC and PNGase F double digestion, cell lysates were first incubated with 0.2U PI-PLC at 37°C for 1 h before the addition of PNGase F. Digested samples were subjected to Western blot analysis.
Pulse-chase experiment
HCT116-ULBP2 cells treated with DMSO or 40 µM PRL3-I for 18 h were washed once with PBS and then starved in DMEM without methionine, cysteine, and glutamine for 1 h at 37 °C. A final concentration of 50 µM of L-Azidohomoalanine (AHA) (Life Technologies), 5 mM of L-cysteine (Sigma) and 1× GlutaMAX (Life Technologies) were added for 1 h additional incubation. Cells were washed and replaced with complete DMEM supplemented with 10% FBS. Cell lysates were collected at different time points for Click-it biotinylation according to the manufacturer’s instructions. Biotinylated proteins were isolated using streptavidin microbeads (Miltenyi) for Western blot analysis to detect the protein synthesis of ULBP2.
Subcellular fractionation
Subcellular Protein fractions (plasma membrane, organelle and cytosol) were separated using the Minute plasma membrane isolation kit (Invent Biotechnologies) according to the manufacturer’s instructions.
Lentiviral transduction
Lentiviral vector was cotransfected with the ViraPower lentiviral packaging mix (Life Technologies) into 293T cells according to the manufacturer’s manual. The culture supernatant, which contained the lentiviral particles, was collected 72 h post-transfection and concentrated by ultracentrifugation. Lentiviral titers were determined (based on GFP+ cells detection by flow cytometry 48 h after transduction) by transducing 293T cells with serial dilutions of the concentrated lentiviral culture supernatant. Stable lentiviral transduced HCT116 cell lines were established by culturing 1×106 cells with the concentrated lentiviral supernatant (multiplicity of infection = 10) containing 8 µg/ml of polybrene. Positive transduced cells were isolated by GFP-based cell sorting 1 week after transduction.
siRNA knockown experiments
siRNA targeting PRL-3 (D-006859-04), HSP60 (D-010600-02) and Src (D-003175-05) were purchased from ThermoFisher Scientific. Negative knockdown control siRNA (4390843) was from Life Technologies. For siRNA transfection, 2 × 105 cells were transfected with 60 pmol of siRNA molecules using lipofectamine RNAiMAX (Life Technologies) according to the manufacturer’s instructions.
Confocal microscopy
2×105 HCT116-ULBP2 cells were seeded on glass slides and treated with DMSO or PRL3-I (40 µM) for 18 h. Cells were fixed with 4% paraformaldehyde and permeabilized by PBS containing 0.5% Triton X-100 and then stained with anti-ULBP2 (clone 165903 from R&D Systems), DAPI and either anti-calnexin (clone C5C9 from Cell Signaling) or anti-GM130 (ab31561 from abcam). Cellular localization of ULBP2 was analyzed by using the Nikon C2plus confocal microscope. Fluorescence images were taken using 60× 1.45NA Plan Apo oil objective (pinhole radius = 40 µm, 1 Airy unit) with the depth of the scanned images about 500 nm. Images were analyzed using NIS-Elements Advanced Research software.
Triton X-114 partitioning experiment
DMSO or PRL3-I (40 µM) treated HCT116-ULBP2 cells were lysed in 2% Triton X-114 in TBS (prepared from the 10% Triton X-114 stock solution from ThermoFisher Scientific). Extraction was performed by incubating the cell lysate at 4°C for 1 h with occasional mixing. Insoluble cell debris was removed by centrifugation at 10000 g at 4°C for 10 min. Supernatant was collected and incubated at 37°C for 5 min (solution became cloudy after the incubation) and phases were separated by centrifugation at 1000 g at 25°C for 10 min. The upper aqueous phase and the lower detergent phase were analyzed by Western Blotting.
PRL-3 activity assay
HCT116 cells stably transduced with a control lentiviral vector (control) or a PRL-3 overexpression construct (HCT116-PRL-3) were treated with DMSO, 40 µM PRL3-I or 40 µM Src I1 for 5 h. One million cells were lysed in 80 µl cell lysis buffer (PBS-1% Triton X-100) and then diluted 20 fold with 20 mM of Tris-HCl (pH 8.0). One hundred microliter of the diluted lysate were incubated with 100 µl of the phosphatase substrate, DiFMUP (16µM) (Life Technologies) for 30 min. The fluorescence intensity was measured using the Victor2 plate reader (Perkin Elmer). PRL-3 activity in HCT116-PRL-3 cells was calculated as follow:
Results
High throughput screening identified a PRL-3 inhibitor that reduced the release of soluble ULBP2 from tumor cell lines
We generated a lentiviral construct that contained an ULBP2-knockdown shRNA expression cassette targeting the 5’ untranslated region (UTR) of human ULBP2. The vector also contained a CMV promoter-driven recombinant ULBP2 which lacked the natural 5’-UTR and was fused to two successive GFP at the N-terminus (ULBP2-2GFPN-term) (Fig. 1A). 293T cells transduced with this construct expressed a significantly lower level of endogenous ULBP2 (determined by quantitative real-time polymerase chain reaction (qRT-PCR) using primers detecting the 5’-UTR; data now shown) but a high level of ULBP2-2GFPN-term on the cell surface (determined by flow cytometry and fluorescence microscopy; unpublished observations). With the coexpression of two GFP molecules at the N-terminus, the release of the recombinant ULBP2 from the cell surface could be detected by the measurement of fluorescence intensity in the cultured supernatant. In line with the previous studies (19), the release of ULBP2-2GFPN-term from 293T cells could be blocked by a MMP inhibitor, MMP-I III, in a dose dependent manner (Fig. 1B). The subsequent high-throughput screening procedure is illustrated in Fig. 1C. Culture supernatants were collected 5 days after the treatment of different inhibitors (14 µM) for fluorescence detection. The viability of the cells was determined by analyzing the production of ATP in the cells. Compounds that reduced ≥ 20% of ULBP2 release but affected ≤ 5% of cell viability were considered potential candidates. Among 53 protease inhibitors and 33 phosphatase inhibitors that we screened, 6 compounds fulfilled these criteria, including the broad-spectrum MMP inhibitor GM 6001 that has been reported to inhibit ULBP2 shedding (29) (Fig. 1D). Another candidate was a specific phosphatase inhibitor targeting PRL-3 (PRL3-I). Confirmatory experiments showed that this PRL3-I could reduce the release of endogenous ULBP2 from cancer cell lines HCT116 and CAL27 (Fig. 1E).
Figure 1.

PRL3-I inhibits soluble release of ULBP2 from tumor cells. (A) Schematic representation of the lentiviral vector (named ULBP2-2GFPN-term) containing a U6 promoter-driven ULBP2-shRNA construct and a CMV promoter-driven recombinant ULBP2 construct. 293T cells transfected with this vector expressed reduced level of endogenous ULBP2 but higher level of recombinant ULBP2 with two GFP molecules tagged at the N-terminal. (B) Stably transduced 293T cells (293T-ULBP2-2GFPN-term) were cultured with different concentration of MMP-I III (0–100 µM) for 18 h. Culture supernatant was collected for the detection of fluorescence intensity (n = 3). (C) Schematic representation of the high-throughput library screening procedure. (D) Relative fluorescence intensity of the cultured supernatant and cell viability of the 293T-ULBP2-2GFPN-term cells treated with 14 µM of GM 6001 and PRL3-I in the high throughput screening were compared with those treated with DMSO (solvent control; considered 100% for both fluorescence intensity and cell viability) and staurosporine (Stauro; cytotoxic reagent control) (n = 3). (E) The release of soluble ULBP2 from HCT116 and CAL27 cells treated with 40 µM of PRL3-I for 18 h was determined by ELISA (n = 4). * P < 0.01, ** P < 0.0005, *** P < 0.0001.
Surface expression of ULBP2 unexpectedly decreased in cancer cell lines upon PRL3-I treatment
Effective blockage of ULBP2 shedding by GM 6001 or MMP-I III in HCT116 cells resulted in an increase in ULBP2 surface expression (Fig. 2A). Surprisingly, the inhibition of ULBP2 released from cancer cell lines upon PRL3-I treatment was not associated with an increase but rather a decrease in ULBP2 surface expression (Fig. 2B; Supplemental Fig. 1A and B), even though the mRNA level of ULBP2 was increased in PRL3-I treated cells (Fig. 2C). These results suggested that the inhibition of ULBP2 release by PRL3-I was a result of downregulation of ULBP2 surface expression rather than blockage of shedding. The surface expression of other NKG2D ligands such as ULBP1 (Fig. 2B) or MICA (unpublished observations) remained unchanged with PRL3-I treatment; thus the specific reduction of surface ULBP2 in PRL3-I-treated HCT116 and CAL27 cells resulted in only a moderate decrease in susceptibility to NK cell cytolysis (Fig. 2D).
Figure 2.

PRL3-I reduces the surface expression of ULBP2 on tumor cells. (A) Cell surface expression of ULBP2 was analyzed by flow cytometry in HCT116 cells treated with DMSO (solvent control), 40 µM of GM 6001 or MMP-1 III for 18 h. Data are representative of two independent experiments. (B) Cell surface expression of ULBP1 and ULBP2 were analyzed by flow cytometry in different cancer cell lines treated with DMSO or PRL3-I (40 µM) for 18 h. Data are representative of three independent experiments. (C) ULBP2 mRNA expression in HCT116 and CAL27 cells treated with DMSO or PRL3-I (40 µM) for 18 h was analyzed by qRT-PCR analysis. Data are normalized to GAPDH mRNA levels and are presented as fold-change relative to the expression in DMSO-treated cells (n = 4). (D) NK cell cytotoxicity on HCT116 and CAL27 cells treated with DMSO or PRL3-I (40 µM) for 18 h was determined by a BATDA release assay using IL-2 (10 U/ml)-primed primary NK cells isolated from healthy donors at various E:T ratios (n = 4). * P < 0.05, ** P < 0.01, *** P < 0.005, **** P < 0.0001.
PRL3-I suppressed ULBP2 protein maturation, resulting in retention in endoplasmic reticulum
To investigate how PRL3-I affected the surface expression of ULBP2, we first performed Western blot analysis on cell lysate prepared from HCT116 cells treated with various concentrations of PRL3-I (Fig. 3A). A 37 kDa band representing the mature form of ULBP2 was detected in the lysate from HCT116 cells without PRL3-I treatment. An additional band approximately 32 kDa began to appear in samples treated with 5 µM of PRL3-I. Increasing the concentration of PRL3-I (up to 40 µM) increased the abundance of the 32 kDa band, alongside with a gradual reduction of the 37 kDa mature ULBP2. Similar results were observed in CAL27 cells upon PRL3-I treatment. To confirm the ULBP2 identity of the 32 kDa band, we generated a stable transduced HCT116 cell line that specifically overexpressed ULBP2 (HCT116-ULBP2). In the absence of PRL3-I, a strong 37 kDa band and a faint 32 kDa band were detected in the HCT116-ULBP2 cell lysate. Consistent with the results of endogenous ULBP2, PRL3-I treatment increased the abundance of ULBP2 in the 32 kDa form in HCT116-ULBP2 cells (Fig. 3B). This particular switch in molecular size of ULBP2 accompanied a decrease in surface expression of ULBP2 (Fig. 3C). In contrast, there was no difference in protein size and surface expression of ULBP1 in PRL3-I treated HCT116 cells overexpressing ULBP1 (HCT116-ULBP1).
Figure 3.
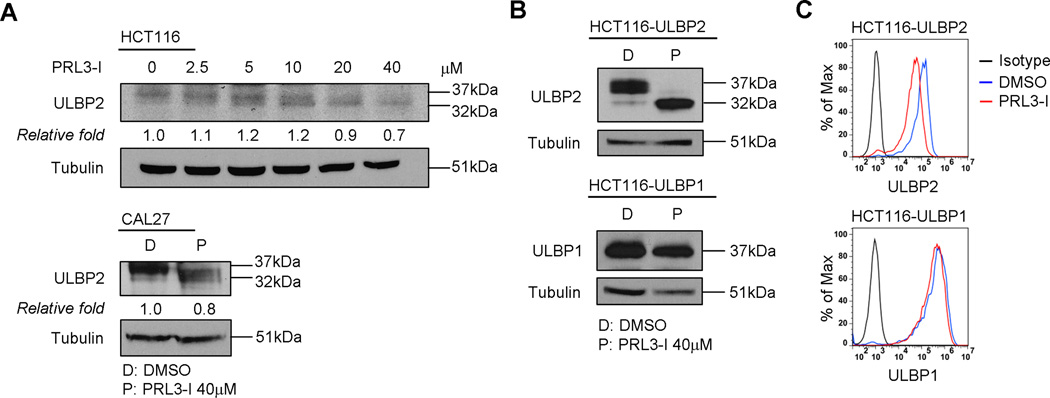
Treatment with PRL3-I associated with the appearance of low-molecular-weight ULBP2. (A) HCT116 cells were treated with increasing concentration (0 – 40 µM) of PRL3-I while CAL27 cells were treated with either DMSO or 40 µM of PRL3-I for 18 h. Total cell lysates were collected for Western blot analysis to detect ULBP2 expression. Data shown are representative of two independent experiments. (B) HCT116 cells overexpressing ULBP2 (HCT116-ULBP2) or ULBP1 (HCT116-ULBP1) were treated with DMSO or PRL3-I (40 µM) for 18 h. Total cell lysates were collected for Western blot analysis to detect ULBP2 or ULBP1 expression. Data shown are representative of three independent experiments. (C) HCT116-ULBP2 and HCT116-ULBP1 cells were treated with DMSO or PRL3-I (40 µM) for 18h. Cell surface expression of ULBP2 or ULBP1 was analyzed by flow cytometry. Results are representative of three independent experiments.
Newly synthesized ULBP2 consists of 246 amino acids, which has a molecular weight of 27 kDa. The protein is then post-translationally modified by glycosylation to become a mature 37 kDa glycoprotein (30). The 32 kDa ULBP2 appeared upon PRL3-I treatment might therefore, represent either an immature form of ULBP2 or a degradation product from the mature protein. We treated the cell lysates from HCT116-ULBP2 cells cultured overnight with DMSO (solvent control) or 40 µM PRL3-I with either endoglycosidase H (Endo H) or peptide -N-glycosidase F (PNGase F). Endo H preferentially cleaves immature high mannose-containing N-linked oligosaccharides while PNGase F removes all N-linked glycans regardless of their level of processing as immature or mature proteins. We found that the 32 kDa bands in both DMSO and PRL3-I treated cell lysate were sensitive to Endo H digestion while PNGase F treatment converted both 32 and 37 kDa bands to the 27–28 kDa unglycosylated ULBP2 (the appearance of slightly different sizes of ULBP2 upon DMSO and PRL3-I treatment was further investigated in the following section) (Fig. 4A). Similar results were also observed in PRL3-I-treated HCT116 cell lysates (Supplemental Fig. 2). In addition, we also performed a pulse-chase experiment to track ULBP2 synthesis in HCT116-ULBP2 cells (Fig. 4B). In the absence of PRL3-I, both 32 kDa and 37 kDa bands appeared during early ULBP2 synthesis. The abundance of the 37 kDa mature ULBP2 gradually increased, which eventually predominated over the 32 kDa immature form. Upon PRL3-I treatment, the synthesis of ULBP2 was completely halted at the immature stage. Collectively, these results suggest that the 32 kDa band corresponded to an immature form, rather than a degradation product of ULBP2 and that the complete maturation of ULBP2 required the activity of PRL-3.
Figure 4.

PRL3-I suppressed ULBP2 maturation, resulting in the retention of ULBP2 proteins in ER. (A) Total cell lysates collected from DMSO- or PRL3-I-treated HCT116-ULBP2 cells (18 h) were untreated or treated with Endo H or PNGase F for Western blot analysis to detect ULBP2 expression. Results are representative of two independent experiments. (B) Pulse-chase experiments were performed to detect the synthesis of AHA-labeled ULBP2 in HCT116-ULBP2 cells upon DMSO or PRL3-I treatment. Data shown are representative of two independent experiments. (C) Total cell lysates of HCT116-ULBP2 cells treated with DMSO or PRL3-I (18 h) were subjected to fractionation for the detection of the subcellular localization of ULBP2 by Western blot analysis. Subcellular fractions were validated using anti-Na,K-ATPase (plasma membrane), anti-Calnexin (ER) and anti-tubulin (cytosol). Data shown are representative of three independent experiments. (D) HCT116-ULBP2 cells were seeded on glass slides and treated with DMSO or PRL3-I (40 µM) for 18 h. Cells were fixed (with or without permeabilization) and stained with anti-ULBP2 and DAPI for confocal microscopy analysis. Results are representative of two independent experiments. (E) HCT116-ULBP2 cells were seeded on glass slides and treated with DMSO or PRL3-I (40 µM) for 18 h. Cells were fixed, permeabilized and stained with anti-ULBP2, DAPI and either anti-calnexin (ER staining) or anti-GM130 (Golgi staining) for the detection of ULBP2 localization by confocal microscopy. Results are representative of two independent experiments.
We next performed subcellular fractionation of PRL3-I-treated or -untreated HCT116-ULBP2 cells for Western blot analysis. The 37 kDa mature ULBP2 located primarily in the plasma membrane fraction, whereas the 32 kDa immature form largely remained in the organelle fraction (Fig. 4C). Similarly, confocal microscopy confirmed that the majority of ULBP2 proteins expressed on the cell surface of DMSO-treated control cells (Fig. 4D). Upon PRL3-I treatment, large amounts of ULBP2 were retained in the endoplasmic reticulum (ER) (Fig. 4E). Collectively, these results suggest that PRL3-I suppresses the post-translational maturation of ULBP2 proteins, which in turn, affected their cell surface expression.
PRL3-I inhibited GPI-anchor formation of ULBP2
It has been suggested that the level of ULBP2 surface expression could be influenced by the GPI-anchor formation of the proteins (31). We generated a mutated ULBP2 construct (Fig. 5A) which had the amino acid sequence 216SSG218 substituted with 216TPV218 (ULBP2-ΔGPI) so that it lacked the GPI-anchor binding site (Supplemental Fig. 3A). Consistent with previous findings, the surface expression of ULBP2 was lower in HCT116-ULBP2-ΔGPI cells compared with that in HCT116-ULBP2 cells (Fig. 5B). Importantly, we found that the overexpressed ULBP2-ΔGPI proteins were in the 32 kDa immature form (Fig. 5C). Treatment with PRL3-I did not further affect the protein level (Fig. 5C) and surface expression of ULBP2-ΔGPI (Fig. 5D). Furthermore, the ULBP2-ΔGPI proteins demonstrated an intracellular ER retention pattern that was the same as the wild-type ULBP2 proteins upon PRL3-I treatment (Fig. 4E and Fig. 5E).
Figure 5.

PRL3-I suppressed cell surface expression of ULBP2 by the inhibition of GPI-anchor formation. (A) Schematic representation of the overexpression construct of wild-type (ULBP2) and mutant ULBP2 that is not able to form GPI-anchor (ULBP2-ΔGPI). (B) Flow cytometry was performed to compare the surface expression of ULBP2 between the untransduced and transduced HCT116 cells with either ULBP2 (HCT116-ULBP2) or ULBP2--ΔGPI (HCT116-ULBP2-ΔGPI) expression construct. Results are representative of three independent staining. (C) Total cell lysates from HCT116-ULBP2 and HCT116-ULBP2-ΔGPI cells treated with DMSO or PRL3-I for 18 h were collected for the detection of ULBP2 expression by Western blot analysis. Data shown are representative of three independent experiments. (D) The surface expression of ULBP2 in DMSO- or PRL3-I-treated HCT116-ULBP2-ΔGPI cells was determined by flow cytometry. Results are representative of three independent experiments. (E) HCT116-ULBP2-ΔGPI cells seeded on glass slides were fixed, permeabilized and stained with anti-ULBP2, DAPI and either anti-calnexin (ER staining) or anti-GM130 (Golgi staining) for the detection of ULBP2 localization by confocal microscopy. Results are representative of two independent experiments. (F) Schematic representation of the lentiviral construct expressing a chimeric GFP protein (GFP-ULBP2210-246) that was fused with the signal sequence (N-terminal) and the cytoplasmic tail of ULBP2 (C-terminal). (G) HCT116-GFP-ULBP2210-246 cells were treated with DMSO or 40 µM of PRL3-I for 18h. Flow cytometry was performed to detect the total GFP proteins and the cell surface expressed GFP proteins that were stained with the Alexa Fluor-647 conjugated anti-GFP antibody. Results are representative of three independent experiments. (H) Triton X-114 partitioning experiments were performed to detect the GPI-anchor formation on ULBP2 with or without PRL3-I treatment. The GPI-anchored protein CD73 was detected for the partitioning control. Results are representative of two independent experiments. (I) Total cell lysates collected from DMSO- or PRL3-I-treated HCT116-ULBP2 cells (18 h) or from HCT116-ULBP2-ΔGPI cells were untreated or treated with Endo H or PNGase F (with or without PI-PLC) for Western blot analysis to detect ULBP2 expression. Results are representative of two independent experiments.
To gain more insight into the correlation between GPI-anchor formation of ULBP2 and the effect of PRL3-I on ULBP2 expression, we generated a lentiviral construct expressing a chimeric GFP protein that possessed the ULBP2 signal sequence at the N-terminal and the cytoplasmic tail of ULBP2 carrying the GPI anchor sequence at the C-terminal (GFP-ULBP2210-246) (Fig. 5F). Because GFP is not a glycoprotein, its expression, protein folding and fluorescent property are not affected by the posttranslational glycosylation. With the GPI anchor sequence of ULBP2, GFP-ULBP2210-246 was able to express on the cell surface of HCT116 cells (detected by flow cytometry using Alexa Fluor-647-conjugated anti-GFP antibody; Fig. 5 G). Upon PRL3-I treatment, the surface expression of GFP-ULBP2210-246 was greatly reduced, but the total GFP protein level was not affected (Fig. 5G). We also performed Triton X-114 phase partitioning experiments to detect the soluble immature ULBP2 proteins appeared upon PRL3-I treatment. With this method, membrane proteins and GPI-anchored proteins are partitioned into the detergent phase whereas soluble proteins remain in the aqueous phase (32). As shown in Fig. 5H, mature ULBP2 was preferentially partitioned into the detergent phase. However, in the presence of PRL3-I, a significant amount of immature form of ULBP2 (but not the control GPI-protein CD73) appeared in the aqueous phase, indicating that some of the immature ULBP2 proteins became soluble upon PRL3-I treatment.
Because we observed a molecular weight difference of PNGase F digested ULBP2 proteins between DMSO (28kDa) and PRL3-I (27kDa) treated samples (Fig. 4A), we speculated that the slightly larger ULBP2 from DMSO control was due to the presence of GPI-anchor. To verify the existence of GPI-anchor in the 28kDa deglycosylated ULBP2 proteins, we performed PNGase F digestion with or without PI-PLC treatment. Previous studies have shown that PI-PLC treated GPI-proteins migrate more slowly on SDS-PAGE (33, 34). As shown in Fig. 5I, the 28kDa deglycosylated ULBP2 only appeared in DMSO control but not in PRL3-I treated sample or ULBP2-ΔGPI upon PNGase F digestion. With the PI-PLC treatment, the 28kDa band migrated more slowly on SDS-PAGE, which became about 29kDa. These results suggested that ULBP2 protein in DMSO- but not PRL3-I-treated cells contained GPI-anchor.
We further examined the surface expression of other transmembrane (Na,K-ATPase and HLA-E) or GPI-anchored proteins (CD73) on HCT116 cells. Unlike ULBP2, none of them was affected upon PRL3-I treatment (Supplemental Fig 3B). Collectively, we conclude that GPI anchor formation is essential for the complete maturation and surface expression of ULBP2 and that this process is specifically inhibited by PRL3-I.
PRL-3 was directly involved in protein maturation and cell surface expression of ULBP2
It has been reported that PRL3-I can inhibit other protein tyrosine phosphatases in addition to PRL-3 (35). To confirm that the suppression of PRL-3 by PRL3-I directly gave rise to the altered ULBP2 expression, we treated HCT116 cells with curcumin or siRNA targeting PRL-3 (PRL-3-KD). As shown in Fig. 6A, both curcumin and PRL-3-KD treatment reduced the surface expression of ULBP2 on HCT116 cells. In line with the results of PRL3-I treatment, the reduction of PRL-3 expression by curcumin or PRL-3-KD in HCT116-ULBP2 cells was associated with predominant expression of 32 kDa ULBP2 (Fig. 6B).
Figure 6.

PRL-3 directly regulated the protein maturation and cell surface expression of ULBP2. (A) HCT116 cells were treated with DMSO or 40 µM of curcumin for 18 h or transfected with either control-KD or PRL-3-KD siRNA and then cultured for 3 days. The cell surface expression of ULBP2 was determined by flow cytometry. Results are representative of three independent experiments. (B) HCT116-ULBP2 cells were treated with DMSO or 40 µM of curcumin for 18 h or transfected with either control or PRL-3-KD siRNA and then cultured for 3 days. Total cell lysates were collected for Western blot analysis to detect PRL-3 and ULBP2 expression. Data shown are representative of three independent experiments. (C) HCT116 cells stably transduced with a PRL-3 expression construct (HCT116-PRL-3) were treated with DMSO, PRL3-I (40 µM) or Src I1 (40 µM) for 18 h. The relative phosphatase activity of PRL-3 from the cell lysates was determined by the reaction with a fluorescence substrate, DiFMUP (n = 3). (D) The surface expression of ULBP2 in HCT116 and CAL27 cells treated with DMSO or Src I1 (40 µM) for 18 h was determined by flow cytometry. Results are representative of three independent experiments. (E) HCT116-ULBP2 cells were transfected with either control-KD or Src-KD siRNA and then cultured for 3 days. Total cell lysates were collected for the Western blot analysis to detect Src and ULBP2 expression. Data shown are representative of three independent experiments. (F) HCT116 cells were transfected with either control-KD or Src-KD siRNA and then cultured for 3 days. The cell surface expression of ULBP2 was determined by flow cytometry. Results are representative of three independent experiments.
Previous reports suggested that Src mediates the tyrosine phosphorylation of PRL-3, which is required for PRL-3 functions (36). Treatment of HCT116 cells overexpressing PRL-3 (HCT116-PRL-3) with a Src kinase inhibitor, Src I1, significantly reduced the phosphatase activity of PRL-3, which was comparable to the effect of PRL3-I (Fig. 6C). Moreover, Src I1 treatment reduced the surface expression of ULBP2 in HCT116 and CAL27 cells (Fig. 6D). Likewise, siRNA-mediated knockdown of Src in HCT116 cells resulted in predominant expression of 32 kDa ULBP2 (Fig. 6E) and decreased ULBP2 surface expression (Fig. 6F). Collectively, these results suggest an absolute requirement of Src-dependent PRL-3 activity in modulating the protein maturation and cell surface expression of ULBP2.
HSP60 interacted constitutively with ULBP2 and PRL-3 regulated HSP60 tyrosine phosphorylation
Recent studies have shown that PRL-3 regulates the tyrosine phosphorylation of various molecules such as ERK1/2 and integrin β1 (37). Because ULBP2 is not a tyrosine-phosphorylated protein and there is no direct protein-protein interaction between ULBP2 and PRL-3 (unpublished observations), we speculated that PRL-3 controls ULBP2 maturation through the regulation of phosphorylation in other tyrosine-phosphoproteins. Lysates of HCT116-ULBP2 cells treated with different concentration of PRL3-I were subjected to immunoprecipitation (IP) with anti-ULBP2 antibody and were analyzed by Western blotting using anti-phosphotyrosine antibody (4G10). In the absence of PRL3-I, a 60 kDa tyrosine-phosphorylated protein was co-immunoprecipitated with ULBP2. However, the intensity of the tyrosine-phosphorylated protein band gradually decreased when the concentration of PRL3-I increased (Fig. 7A). Using 4G10 for IP of cell lysates from DMSO- or PRL3-I-treated HCT116-ULBP2 cells, we observed a bias in favor of the mature ULBP2 (37 kDa) over the immature form (32 kDa) to be co-immunoprecipitated by the antibody (Fig. 7B).
Figure 7.

ULBP2 associated with HSP60 and that the tyrosine phosphorylation of HSP60 was regulated by PRL-3. (A) HCT116-ULBP2 cells were treated with increasing concentration of PRL3-I for 18 h. Total cell lysates were collected for immunoprecipitation of ULBP2. Western blot analysis was performed to detect tyrosine phosphorylated proteins in the immunoprecipitated fractions. Data shown are representative of three independent experiments. (B) Total cell lysates from HCT116-ULBP2 cells treated 18 h with DMSO or PRL3-I (40 µM) were collected for immunoprecipitation of tyrosine phosphorylated proteins or ULBP2. Western blot analysis was performed to detect ULBP2 in the immunoprecipitated fractions. Results are representative of three independent experiments. (C) Reciprocal immunoprecipitation of ULBP2 and HSP60 was performed from the total cell lysates of HCT116-ULBP2 cells treated with DMSO or PRL3-I for 18 h. Western blot analysis was performed to detect tyrosine phosphorylated proteins, HSP60 and ULBP2. Results are representative of three independent experiments. (D) Schematic representation of the overexpression construct of the Flag-tagged wild-type (F-HSP60) or the tyrosine phosphorylation mutant HSP60 (F-HSP60-YY-mu). (E) HCT116 cells stably transduced with either of these constructs were treated with DMSO or PRL3-I for 18 h. The Flag-tagged HSP60 proteins were immunoprecipitated for the detection of their tyrosine phosphorylation status. Results are representative of two independent experiments.
The 60 kDa member of the heat shock protein family, HSP60 has been known to act as a chaperone to regulate post-translational modification (38). It has also been shown that tyrosine phosphorylation is required for HSP60 to be expressed on cell membrane (39). To examine if the 60 kDa tyrosine-phosphoprotein was HSP60, we performed IP of both ULBP2 and HSP60 from the HCT116-ULBP2 cell lysates. As shown in Fig. 7C, anti-ULBP2 antibody could pull-down HSP60 while anti-HSP60 antibody co-immunoprecipitated both HSP60 and ULBP2. In both cases, the HSP60 bands were colocalized with the 60 kDa tyrosine-phosphoproteins on the immunoblots and that PRL3-I treatment consistently reduced the intensity of the 60 kDa tyrosine-phosphoprotein but did not affect the co-IP of HSP60 and ULBP2 (both mature 37 kDa and immature 32 kDa forms). It has been suggested that HSP60 can be tyrosine phosphorylated at position 227 and 243 (40, 41). Accordingly, we generated lentiviral constructs expressing Flag-tagged HSP60 with or without the substitution of tyrosine with phenylalanine at both position 227 and 243 (Fig. 7D). As shown in Fig. 7E, the level of tyrosine phosphorylation was lower in the HSP60 mutant (F-HSP60-YY-mu) compared to that in the wild-type protein (F-HSP60). PRL3-I treatment caused a reduction (~ 2.8 fold) of phosphorylation intensity on F-HSP60, but exerted minimal effect on F-HSP60-YY-mu. These results suggested that the tyrosine phosphorylation of HSP60 was regulated by PRL-3.
Protein maturation and cell surface expression of ULBP2 required tyrosine-phosphorylated HSP60
We next investigated on whether tyrosine phosphorylation of HSP60 is required for the maturation and surface expression of ULBP2. We transduced HCT116-ULBP2 cells with F-HSP60 or F-HSP60-YY-mu. Cell lysates were subjected to IP with anti-Flag-tag antibody for western blot analysis. A much higher amount of mature ULBP2 was co-immunoprecipitated with F-HSP60 than with F-HSP60-YY-mu (Fig. 8A). We further validated the role of HSP60 in ULBP2 maturation by treating HCT116 cells with nonactin or a siRNA specific targeting HSP60 (HSP60-KD). Nonactin is an ionophore that blocks the mitochondrial import and consequently inhibits the maturation of HSP60 (42). Consistent with previous studies, nonactin treatment inhibited the surface expression of HSP60 in HCT116 cells (Fig. 8B). Tyrosine phosphorylation of HSP60 was also reduced upon nonactin treatment (Fig. 8C). As expected as a consequence of reduced HSP60 phosphorylation, the maturation (Fig. 8D), together with the surface expression (Fig. 8E), of ULBP2 were inhibited by nonactin treatment. Similar results were observed by direct HSP60-KD (Fig. 8F and G). However, neither knockndown nor overexpression of HSP60 affected the mRNA level of ULBP2 (data not shown). Also, there was no change in the surface expression of ULBP2 in HSP60 overexpressing HCT116 cells (Supplemental Fig. 4), suggesting that the gene expression of ULBP2 was not affected by the expression level of HSP60. Collectively, these data suggest that the association with tyrosine-phosphorylated HSP60 is essential for the protein maturation and surface expression of ULBP2.
Figure 8.

The presence of tyrosine-phosphorylated HSP60 was required for ULBP2 maturation and cell surface expression. (A) Anti-Flag immunoprecipitation was performed to pull-down F-HSP60 or F-HSP60-YY-mu from the stable transduced HCT116 cells. Western blot analysis was performed to detect ULBP2 in the immunoprecipitated fractions. Data shown are representative of two independent experiments. Total cell lysates from HCT116-ULBP2 cells treated with DMSO or nonactin (40 µM) for 18 h were subjected to (B) subcellular fractionation for the detection of HSP60 localization, (C) anti-HSP60 immunoprecipitation to detect the tyrosine phosphorylation of HSP60, and (D) Western blot analysis for the total protein expression of ULBP2 and HSP60. Results are representative of two independent experiments. (E) Flow cytometry was performed to compare the surface expression of ULBP2 between DMSO and nonactin (40 µM) treated HCT116 and CAL27 cells. Results are representative of three independent experiments. (F) HCT116-ULBP2 cells were transfected with either control-KD or HSP60-KD siRNA and then cultured for 3 days. Total cell lysates were collected for the Western blot analysis to detect HSP60 and ULBP2 expression. Data shown are representative of three independent experiments. (G) HCT116 and CAL27 cells were transfected with either control-KD or HSP60-KD siRNA and then cultured for 3 days. The cell surface expression of ULBP2 was determined by flow cytometry. Results are representative of three independent experiments.
Discussion
This study reveals a novel role of PRL-3 in regulating the post-translational maturation of ULBP2. Either decreased expression or functional inhibition of PRL-3 reduced both the soluble release and the cell surface expression of ULBP2. While there was no significant reduction in the total protein level of ULBP2, inactivation of PRL-3 significantly affected the glycosylation and GPI anchor formation of the protein, rendering the production of an immature form of ULBP2 that did not favor cell surface expression, which in turn, reduced the soluble release of ULBP2.
Although PRL-3 possesses tyrosine phosphatase activity, inhibition of PRL-3 greatly decreased, rather than increased the tyrosine phosphorylation of HSP60. In fact, several studies have showed that PRL-3 expressing cells exhibit a pronounced increase in protein tyrosine phosphorylation (43). Src kinase has been shown to regulate PRL-3 activity (36). However, the tyrosine phosphorylation of Src is indirectly dependent on PRL-3 through its down-regulation of Csk, a negative regulator of Src (44). Little is known about the mechanism controlling the tyrosine phosphorylation of HSP60. Here we showed that treatment with PRL3-I or nonactin significantly reduced the level of tyrosine phosphorylation of HSP60 and that both of the treatments gave rise to a very similar alteration in the expression of ULBP2. While nonactin is well known to affect the mitochondrial transportation and maturation of HSP60 (42), it is possible that PRL-3 may directly regulate the tyrosine phosphorylation mechanism of HSP60 or indirectly through the regulation of HSP60 maturation process. Further investigation is necessary to understand the regulatory role of PRL-3 in the tyrosine phosphorylation of HSP60.
Although we have demonstrated an absolute requirement of PRL-3 to produce the 37 kDa complete mature form of ULBP2, treatment with PRL3-I in either HCT116 or HCT116-ULBP2 cells could never completely inhibit the surface expression of the endogenous or the overexpressed ULBP2 proteins respectively. Moreover, overexpression of ULBP2-ΔGPI in HCT116 cells could also moderately enhance the surface expression of ULBP2 (Fig. 5B), suggesting that the immature form of ULBP2 can somehow inefficiently express on the cell surface in the absence of a GPI anchor. These results are consistent with a previous report in which the authors showed that ULBP2, could also be expressed on the surface of K562 cells without the formation of GPI anchor despite in a much lower level (31). While structural analysis revealed no predictable transmembrane domain presented in the ULBP2 protein, the low-level existence of immature ULBP2 on cell surface might be the result of an interaction between ULBP2 and other membrane-associated proteins such as HSP60. Further studies are required to understand the mechanism of ULBP2 surface expression in the absence of GPI anchor.
The effect of PRL-3 on ULBP2 expression was specific. Treatment with PRL3-I only affected the surface expression of ULBP2 but not the other NKG2DLs such as ULBP1 and MICA (Fig. 2B and unpublished observations) or other GPI-anchored proteins (such as CD73) or transmembrane proteins (such as Na,K-ATPase and HLA-E) (Supplemental Fig. 3B) on cancer cells. The expression of other NKG2DLs on HCT116 and CAL27 cells may therefore, explain the fact that treatment with PRL3-I only moderately affected the susceptibility of the cancer cells to NK cytolysis (Fig. 2D). These results strengthen the clinical potential of targeting PRL-3 for reducing the level of soluble ULBP2, without causing a great impact on tumor clearance.
NKG2DLs have long been described as stress ligands because they are rarely expressed in normal tissues, but can be highly induced by different cellular stress pathways arisen during tumorigenesis or viral-infection (1). Heat shock stress pathways, for example, have been shown to mediate the gene transcription of MICA and MICB (45). However, there is no direct evidence showing that the expression of ULBPs is regulated by heat shock pathways. HSP60 was first identified as a chaperone which resides in mitochondria to regulate mitochondrial protein folding and transportation (38). The protein was later found in cytoplasm, on cell surface, and even in the extracellular environment, in which tyrosine phosphorylation is required for its surface expression (39). Because of its broad localization, HSP60 has been associated with diverse physiological functions, such as apoptosis and T cell activation (38). There is also increasing evidence that HSP60 plays essential roles in tumor development and metastasis (42, 46). Here we demonstrate for the first time that HSP60 interacts with and mediates the protein maturation and cell surface expression of ULBP2, suggesting the existence of independent heat shock stress pathways for post-translational regulation of ULBP2.
The fact that patient serum level of soluble ULBP2 directly correlates with the prognosis of various types of cancer suggesting that blockade of NKG2DL release might be a potential immunological approach for cancer therapy. An understanding of the mechanisms involved in the protein expression and processing of NKG2DLs from tumor cells, therefore, is crucial for the development of effective therapeutic strategies to reduce the release of these proteins. Here, we discovered a novel PRL-3-dependent regulatory mechanism that controls the protein maturation of ULBP2 which in turn, indirectly mediates the soluble release of ULBP2. With a restricted expression profile in normal tissues and a thus far safety record of several clinical trials (28), PRL-3 may serve as an alternative therapeutic target for the suppression of ULBP2 release in addition to MMPs.
Supplementary Material
Acknowledgements
We thank Barbara Rooney, James Houston and Martha Holladay for assistance with flow cytometry and cell sorting and Jimmy Cui for high-throughput library screening.
This work was supported in part by research grants from the National Institutes of Health P30 CA021765, Press On Fund, the Assisi Foundation of Memphis, and the American Lebanese Syrian Associated Charities.
Abbreviations used in this article
- GPI-anchor
glycosylphosphatidylinositol-anchor
- NKG2DL
NKG2D ligands
- PRL-3
phosphatase of regenerating liver 3
- PRL3-I
PRL-3 inhibitor I
- ULBP2
UL16 binding protein 2
Footnotes
Disclosures
The authors declare no competing financial interests.
References
- 1.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annual review of immunology. 2013;31:413–441. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hilpert J, Grosse-Hovest L, Grunebach F, Buechele C, Nuebling T, Raum T, Steinle A, Salih HR. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. Journal of immunology. 2012;189:1360–1371. doi: 10.4049/jimmunol.1200796. [DOI] [PubMed] [Google Scholar]
- 3.McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, Trowsdale J, Durrant LG. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:6993–7002. doi: 10.1158/1078-0432.CCR-09-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nature reviews. Immunology. 2007;7:737–744. doi: 10.1038/nri2144. [DOI] [PubMed] [Google Scholar]
- 5.Strong RK, McFarland BJ. NKG2D and Related Immunoreceptors. Advances in protein chemistry. 2004;68:281–312. doi: 10.1016/S0065-3233(04)68008-9. [DOI] [PubMed] [Google Scholar]
- 6.Radaev S, Sun PD. Structure and function of natural killer cell surface receptors. Annual review of biophysics and biomolecular structure. 2003;32:93–114. doi: 10.1146/annurev.biophys.32.110601.142347. [DOI] [PubMed] [Google Scholar]
- 7.Cerwenka A. New twist on the regulation of NKG2D ligand expression. The Journal of experimental medicine. 2009;206:265–268. doi: 10.1084/jem.20090225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:12445–12450. doi: 10.1073/pnas.93.22.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto K, Fujiyama Y, Andoh A, Bamba T, Okabe H. Oxidative stress increases MICA and MICB gene expression in the human colon carcinoma cell line (CaCo-2) Biochimica et biophysica acta. 2001;1526:10–12. doi: 10.1016/s0304-4165(01)00099-x. [DOI] [PubMed] [Google Scholar]
- 10.Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. The Journal of experimental medicine. 2012;209:2409–2422. doi: 10.1084/jem.20120565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez-Soto A, Folgueras AR, Seto E, Gonzalez S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: potential implications for the immunosurveillance of cancer. Oncogene. 2009;28:2370–2382. doi: 10.1038/onc.2009.117. [DOI] [PubMed] [Google Scholar]
- 12.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A, Foa R, Santoni A. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113:3503–3511. doi: 10.1182/blood-2008-08-173914. [DOI] [PubMed] [Google Scholar]
- 14.Yadav D, Ngolab J, Lim RS, Krishnamurthy S, Bui JD. Cutting edge: down-regulation of MHC class I-related chain A on tumor cells by IFN-gamma-induced microRNA. Journal of immunology. 2009;182:39–43. doi: 10.4049/jimmunol.182.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, Mandelboim M, Mandelboim O. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nature immunology. 2008;9:1065–1073. doi: 10.1038/ni.1642. [DOI] [PubMed] [Google Scholar]
- 16.Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, Schadendorf D, Paschen A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer research. 2012;72:460–471. doi: 10.1158/0008-5472.CAN-11-1977. [DOI] [PubMed] [Google Scholar]
- 17.Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. Journal of immunology. 2002;169:4098–4102. doi: 10.4049/jimmunol.169.8.4098. [DOI] [PubMed] [Google Scholar]
- 18.Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, Stevanovic S, Rammensee HG, Steinle A. Tumor-associated MICA is shed by ADAM proteases. Cancer research. 2008;68:6368–6376. doi: 10.1158/0008-5472.CAN-07-6768. [DOI] [PubMed] [Google Scholar]
- 19.Waldhauer I, Steinle A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer research. 2006;66:2520–2526. doi: 10.1158/0008-5472.CAN-05-2520. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi K, Chikumi H, Shimizu A, Takata M, Kinoshita N, Hashimoto K, Nakamoto M, Matsunaga S, Kurai J, Miyake N, Matsumoto S, Watanabe M, Yamasaki A, Igishi T, Burioka N, Shimizu E. Diagnostic and prognostic impact of serum-soluble UL16-binding protein 2 in lung cancer patients. Cancer science. 2012;103:1405–1413. doi: 10.1111/j.1349-7006.2012.02330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 22.Skiles JW, Gonnella NC, Jeng AY. The design, structure, and clinical update of small molecular weight matrix metalloproteinase inhibitors. Current medicinal chemistry. 2004;11:2911–2977. doi: 10.2174/0929867043364018. [DOI] [PubMed] [Google Scholar]
- 23.Sariahmetoglu M, Crawford BD, Leon H, Sawicka J, Li L, Ballermann BJ, Holmes C, Berthiaume LG, Holt A, Sawicki G, Schulz R. Regulation of matrix metalloproteinase-2 (MMP-2) activity by phosphorylation. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:2486–2495. doi: 10.1096/fj.06-7938com. [DOI] [PubMed] [Google Scholar]
- 24.Bessette DC, Qiu D, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer metastasis reviews. 2008;27:231–252. doi: 10.1007/s10555-008-9121-3. [DOI] [PubMed] [Google Scholar]
- 25.Al-Aidaroos AQ, Zeng Q. PRL-3 phosphatase and cancer metastasis. Journal of cellular biochemistry. 2010;111:1087–1098. doi: 10.1002/jcb.22913. [DOI] [PubMed] [Google Scholar]
- 26.Semba S, Mizuuchi E, Yokozaki H. Requirement of phosphatase of regenerating liver-3 for the nucleolar localization of nucleolin during the progression of colorectal carcinoma. Cancer science. 2010;101:2254–2261. doi: 10.1111/j.1349-7006.2010.01651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo K, Tang JP, Tan CP, Wang H, Zeng Q. Monoclonal antibodies target intracellular PRL phosphatases to inhibit cancer metastases in mice. Cancer biology & therapy. 2008;7:750–757. doi: 10.4161/cbt.7.5.5764. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Shen Y, Song R, Sun Y, Xu J, Xu Q. An anticancer effect of curcumin mediated by down-regulating phosphatase of regenerating liver-3 expression on highly metastatic melanoma cells. Molecular pharmacology. 2009;76:1238–1245. doi: 10.1124/mol.109.059105. [DOI] [PubMed] [Google Scholar]
- 29.Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, Weller M, Friese MA. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain : a journal of neurology. 2006;129:2416–2425. doi: 10.1093/brain/awl205. [DOI] [PubMed] [Google Scholar]
- 30.Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123–133. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez-Messina L, Ashiru O, Aguera-Gonzalez S, Reyburn HT, Vales-Gomez M. The human NKG2D ligand ULBP2 can be expressed at the cell surface with or without a GPI anchor and both forms can activate NK cells. Journal of cell science. 2011;124:321–327. doi: 10.1242/jcs.076042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doering TL, Englund PT, Hart GW. Detection of glycophospholipid anchors on proteins. Chapter 12: Unit 12 15. Current protocols in protein science / editorial board, John E. Coligan …[et al.] 2001 doi: 10.1002/0471140864.ps1205s02. [DOI] [PubMed] [Google Scholar]
- 33.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 34.Nishina KA, Supattapone S. Immunodetection of glycophosphatidylinositol-anchored proteins following treatment with phospholipase C. Analytical biochemistry. 2007;363:318–320. doi: 10.1016/j.ab.2007.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onda H, Ohkubo S, Shintani Y, Ogi K, Kikuchi K, Tanaka H, Yamamoto K, Tsuji I, Ishibashi Y, Yamada T, Kitada C, Suzuki N, Sawada H, Nishimura O, Fujino M. A novel secreted tumor antigen with a glycosylphosphatidylinositol-anchored structure ubiquitously expressed in human cancers. Biochemical and biophysical research communications. 2001;285:235–243. doi: 10.1006/bbrc.2001.5149. [DOI] [PubMed] [Google Scholar]
- 36.Fiordalisi JJ, Dewar BJ, Graves LM, Madigan JP, Cox AD. Src-mediated phosphorylation of the tyrosine phosphatase PRL-3 is required for PRL-3 promotion of Rho activation, motility and invasion. PloS one. 2013;8:e64309. doi: 10.1371/journal.pone.0064309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng L, Xing X, Li W, Qu L, Meng L, Lian S, Jiang B, Wu J, Shou C. PRL-3 promotes the motility, invasion, and metastasis of LoVo colon cancer cells through PRL-3-integrin beta1-ERK1/2 and-MMP2 signaling. Molecular cancer. 2009;8:110. doi: 10.1186/1476-4598-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cappello F, Conway de Macario E, Marasa L, Zummo G, Macario AJ. Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer biology & therapy. 2008;7:801–809. doi: 10.4161/cbt.7.6.6281. [DOI] [PubMed] [Google Scholar]
- 39.Asquith KL, Baleato RM, McLaughlin EA, Nixon B, Aitken RJ. Tyrosine phosphorylation activates surface chaperones facilitating sperm-zona recognition. Journal of cell science. 2004;117:3645–3657. doi: 10.1242/jcs.01214. [DOI] [PubMed] [Google Scholar]
- 40.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R, Macneill J, Ren JM, Yuan J, Bakalarski CE, Villen J, Kornhauser JM, Smith B, Li D, Zhou X, Gygi SP, Gu TL, Polakiewicz RD, Rush J, Comb MJ. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 41.Gu TL, Deng X, Huang F, Tucker M, Crosby K, Rimkunas V, Wang Y, Deng G, Zhu L, Tan Z, Hu Y, Wu C, Nardone J, MacNeill J, Ren J, Reeves C, Innocenti G, Norris B, Yuan J, Yu J, Haack H, Shen B, Peng C, Li H, Zhou X, Liu X, Rush J, Comb MJ. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PloS one. 2011;6:e15640. doi: 10.1371/journal.pone.0015640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barazi HO, Zhou L, Templeton NS, Krutzsch HC, Roberts DD. Identification of heat shock protein 60 as a molecular mediator of alpha 3 beta 1 integrin activation. Cancer research. 2002;62:1541–1548. [PubMed] [Google Scholar]
- 43.Al-Aidaroos AQ, Yuen HF, Guo K, Zhang SD, Chung TH, Chng WJ, Zeng Q. Metastasis-associated PRL-3 induces EGFR activation and addiction in cancer cells. The Journal of clinical investigation. 2013;123:3459–3471. doi: 10.1172/JCI66824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang F, Liang J, Wang WQ, Sun JP, Udho E, Zhang ZY. PRL3 promotes cell invasion and proliferation by down-regulation of Csk leading to Src activation. The Journal of biological chemistry. 2007;282:5413–5419. doi: 10.1074/jbc.M608940200. [DOI] [PubMed] [Google Scholar]
- 45.Fionda C, Soriani A, Malgarini G, Iannitto ML, Santoni A, Cippitelli M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. Journal of immunology. 2009;183:4385–4394. doi: 10.4049/jimmunol.0901797. [DOI] [PubMed] [Google Scholar]
- 46.Tsai YP, Yang MH, Huang CH, Chang SY, Chen PM, Liu CJ, Teng SC, Wu KJ. Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis. 2009;30:1049–1057. doi: 10.1093/carcin/bgp087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.