

Abstract
Macrophage migration inhibitory factor (MIF) enhances activation of leukocytes, endothelial cells and fibroblast-like synoviocytes (FLS), thereby contributing to the pathogenesis of rheumatoid arthritis (RA). A MIF promoter polymorphism in RA patients resulted in higher serum MIF concentration and worsens bone erosion; controversially current literature reported an inhibitory role of MIF in osteoclast formation. The controversial suggested that the precise role of MIF and its putative receptor CD74 in osteoclastogenesis and RA bone erosion, mediated by locally formed osteoclasts in response to receptor activator of NF-κB ligand (RANKL), is unclear. We reported that in an in vivo K/BxN serum transfer arthritis, reduced clinical and histological arthritis in MIF-/- and CD74-/- mice were accompanied by a virtual absence of osteoclasts at the synovium-bone interface and reduced osteoclast-related gene expression. Furthermore, in vitro osteoclast formation and osteoclast-related gene expression were significantly reduced in MIF-/- cells via decreasing RANKL-induced phosphorylation of NF-κB-p65 and ERK1/2. This was supported by a similar reduction of osteoclastogenesis observed in CD74-/- cells. Furthermore, a MIF blockade reduced RANKL-induced osteoclastogenesis via deregulating RANKL-mediated NF-κB and NFATc1 transcription factor activation. These data indicate that MIF and CD74 facilitate RANKL-induced osteoclastogenesis, and suggest that MIF contributes directly to bone erosion, as well as inflammation, in RA.
Keywords: Bone, Inflammation, cell signals, transcription factors, cytokines
Introduction
Macrophage migration inhibitory factor (MIF) is a 37.5kDa homotrimeric protein that plays a significant role in the development of chronic inflammatory conditions such as rheumatoid arthritis (RA)[1]. MIF is produced by many cell types, where its expression increased by inflammatory stress. The absence of MIF is protective in models of septic and endotoxic shock, due to reduced production of pro-inflammatory cytokines[2], and MIF also amplifies T cell-dependent adaptive immune responses[3]. In addition, MIF regulates leukocyte trafficking, increasing chemokine expression[4] and amplifying signals entrained by chemokine actions[5]. In RA synovial fibroblasts, MIF reduces apoptosis[6], and increases expression of cycloxygenase-2[7] and matrix metalloproteinases (MMPs)[8]. Thus, MIF expression in synovial tissue, which correlates with inflammation severity in RA patients[9], exerts important effects on the development of inflammation in RA. Indeed, in several animal models of RA that have distinct immunological mechanisms, MIF deficiency or neutralization markedly reduces disease severity[3,10,11]. However, an important consequence of RA is destruction of bone adjoining affected joints. While inflammation indirectly drives osteoclast formation, the latter process also involves specific cellular signals in osteoclast progenitors regulated by inflammatory cytokines. The role of MIF in such events is unknown.
The mechanisms that mediate MIF cellular actions are controversial. The extracellular domain of CD74 was the first identified as a MIF binding protein, and was shown to be required for MIF induced ERK1/2 mitogen activated protein kinase (MAPK) phosphorylation[12]. CD74 lacks intracellular signaling domains, so requires adaptor molecules such as CD44[13]. CD74-dependent MIF actions in alveolar macrophages increase neutrophil accumulation during lung inflammation[14]. We have previously demonstrated that CD74-/- macrophages exhibit reduced MAPK phosphorylation, RhoA GTPase activity, and actin polymerization[5]. Thus, while CD74 mediates some cellular actions of MIF, which is potentially important in MIF actions.
Bone structure is maintained by a tightly regulated balance between bone formation and bone resorption, mediated by osteoblasts and osteoclasts respectively. Formation of osteoclasts, which are multinucleated cells derived from the fusion of haemopoietic-derived progenitors, is controlled by local levels of receptor activator of nuclear factor kappa B ligand (RANKL)[15]. RANKL elicits activation of several intracellular pathways, such as MAPKs, and activation of the transcription factors NF-κB and NFATc1[16]. These in turn elicit expression of osteoclast-associated factors such as tartrate resistant acid phosphatase (TRAP) and calcitonin receptor (CTR)[16]. In RA, osteoclasts are observed at bone erosion sites at the synovial-bone interface[17]. RANKL-/- mice are susceptible to inflammatory synovitis but lack osteoclasts and do not display periarticular bone erosion[18], indicating that bone destruction secondary to inflammation requires RANKL-dependent differentiation of osteoclasts. Indeed, pro-inflammatory cytokines, such as TNF and IL-1, enhance local production of RANKL by osteoblasts and may amplify RANKL effects on common intracellular signalling events[19]. The hypothesis that MIF also exerts such amplifying effects is supported by numerous studies. For example, carriage of a high expression MIF allele is associated with accelerated erosive disease in RA patients[20]. MIF-/- mice exhibit protection from bone loss induced by ovariectomy[21] and periodontal disease[22]. MIF-/- mice show lower osteoclast numbers at fracture callus[23], and elevated bone resorption is seen in MIF transgenic mice[24]. However, Jacquin et al[25] have reported the converse, that MIF reduced osteoclastogenesis in vitro, and that MIF-/- mice have significantly reduced trabecular bone mass. The same group also reported decreased trabecular volume in the long bones of CD74-/- mice[26]. Up to date, limited mechanismic studies in human cells and controversial reports in murine models indicate that the role of MIF in osteoclastogenesis and arthritic bone erosion is not fully explored. Moreover, the influence of CD74 in arthritis models, and arthritic bone erosion is unknown.
To properly address the processes that cause RA-associated bone loss, better understanding of the influence of MIF in bone erosion is needed. We report here that in K/BxN serum transfer-induced arthritis, reduced inflammation in MIF-/- and CD74-/- mice was accompanied by markedly reduced bone damage. RANKL-induced osteoclast formation was impaired in the absence of MIF or CD74, with accompanying impairment of key osteoclast signalling pathways. These data suggest that MIF and CD74 are factors that enhance RANKL-induced osteoclastogenesis and, consequently, contribute to bone loss in RA.
MATERIALS AND METHODS
Mice
All animal experiments were performed in accordance with the approval of the Monash University Animal Research Ethics Committee.
MIF-/- and CD74-/- mice were generated on the C57Bl/6 background[5,27] and wild-type (WT) C57Bl/6 mice were used as controls. Mice were kept in the animal facility of Monash Medical Centre and fed ad libitum.
Cell culture reagents
Tissue culture medium used in all cultures was minimal essential medium-alpha (MEM) (Life Technology, NSW Australia) supplemented with fetal bovine serum 10% (In Vitro Technologies, Noble Park North, Australia) and penicillin 50 U/mL, streptomycin 50 μg/mL and L-glutamine 2 mM (all Sigma-Aldrich, Castle Hill, NSW, Australia). K/BxN serum was generously provided by Professor John Hamilton (University of Melbourne, Parkville, Australia)[28]. Human recombinant MIF (rMIF) was produced as described[29]. Human recombinant RANKL was obtained from Peprotech (Rocky Hill, NJ) and mouse recombinant M-CSF from R&D Systems (Minneapolis, MN). The anti-MIF monoclonal antibody (mAb) was a kind gift from Dr Jie Tang, (Chinese Academy of Science, Beijing, China) and the appropriate control IgG1 antibody was obtained from R&D Systems (Minneapolis, MN).
Induction of K/BxN serum transfer arthritis
WT, MIF-/- and CD74-/- male mice were injected intraperitoneally (i.p.) with K/BxN sera (4 μl/g body weight) on days 0 and 2 to induce arthritis and clinical scores were assessed from day 0-8 as described previously[11]. Briefly, each limb was scored daily on a scale of 0 to 5, resulting in a maximum total clinical score of 20. For histological analysis, 4 μm sections were stained using Safranin-O and counterstained with fast green (giving blue-green colour staining in our sections), and scored for severity of synovitis and bone damage on a scale of 0-3, as described[11]. Osteoclasts were detected by histochemical staining for TRAP. Briefly, paraffin sections were dewaxed and rehydrated as described previously[11], and incubated at 4 °C overnight in freshly prepared TRAP substrate solution (pH 5.0) containing 50mM sodium Acetate, 40 mM potassium sodium tartrate, 5 mg/50 ml of Naphthol AS-MX phosphate and 20 mg/50 ml fast red violet LB salt (Sigma, Castle Hill, Australia).
In vitro osteoclastogenesis cultures
Bone marrow cells were flushed from the femora and tibiae of 6-12 week old WT MIF-/-, and CD74-/- mice. Cells were stimulated with 30 ng/ml M-CSF and RANKL for 7 days (except where indicated) with a change of medium and mediators at day 3; RANKL concentrations are indicated in individual experiments. Cells were then fixed with formaldehyde (4%, in phosphate buffered saline), followed by permeablization with acetone/methanol (50:50), and TRAP histochemical staining performed. In separate experiments, bone marrow cells obtained from WT and MIF-/- mice were stimulated with M-CSF (30 ng/ml) and RANKL (50 ng/ml), and RNA was collected as described previously[11]. Osteoclast-relevant gene expression was examined by real time PCR.
Osteoclastogenesis was also induced in RAW264.7 cells, a macrophage/pre-osteoclast cell line, by treatment with 100 ng/ml RANKL for 7 days. Extracellular MIF in RAW264.7 cultures was blocked by addition of a neutralising anti-MIF monoclonal antibody (100 ng/ml); purified murine IgG was employed as a control in these experiments.
RT-PCR analysis of mRNA expression
RNA from whole ankle joints was extracted as described previously [11]. Complementary DNA (cDNA) was made from total RNA using superscript III reverse transcriptase and either random primers or Oligo-dt (all from Invitrogen, Carlsbad, CA). Real-time PCR analysis was performed on Light Cycler Rotor-Gene 3000 (Corbett Research, Mortlake, NSW, Australia) using Power Sybr green PCR Master Mix (Applied Biosystems, Foster City, CA), following manufacturer instructions. The level of target gene expression was normalized against ß-actin (for oligo-dT generated cDNA, Fig. 6) or 18S (for random primers generated cDNA, Fig. 1 to Fig. 5) and results expressed as fold difference in expression relative to control. Primers used were as indicated in Table 1.
Figure 1.
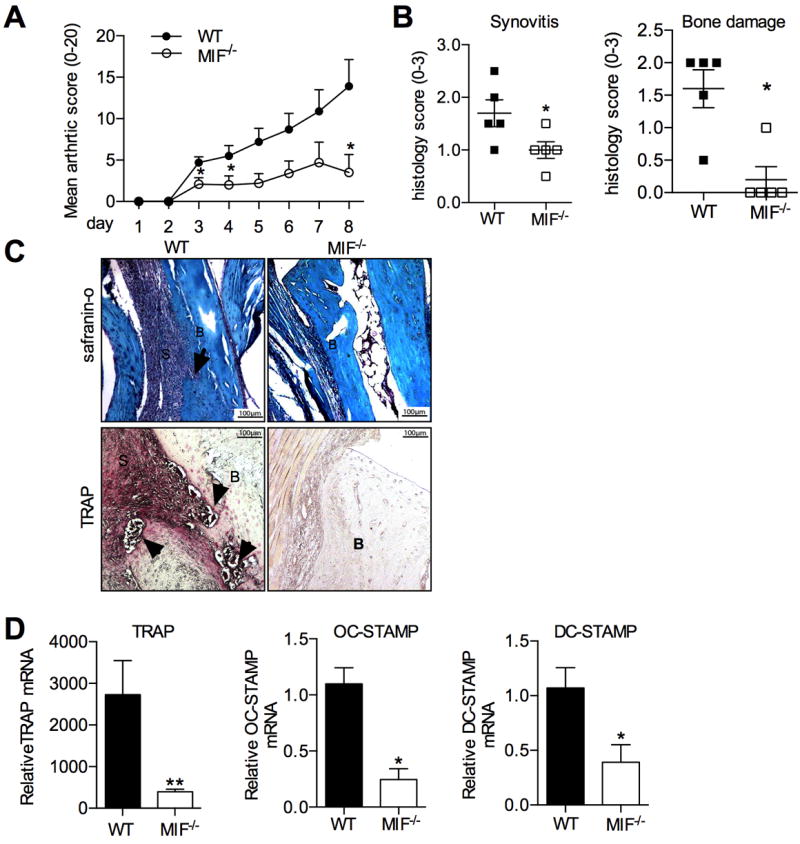
Inflammatory arthritis in WT and MIF-/- mice with K/BxN serum transfer arthritis. A: Clinical scoring of joint inflammation in K/BxN serum-injected WT mice (closed circles) and MIF-/- (open circles). B: Histological scores for synovitis (left) and bone damage (right) in WT and MIF-/- mice. C: Top: Safranin O stained sections of ankles WT and MIF-/- mice with K/BxN serum transfer arthritis, showing synovitis (S) and bone erosion (arrows) (B:bone). Bottom: TRAP histochemical staining (pink) in ankle sections of WT and MIF-/- mouse (‘B’=bone, ‘S’=synovium and TRAP+ cells indicated by arrows). Original magnification x200. C: Osteoclast-associated gene expression determined using RNA extracted from ankles of WT and MIF-/- mice with K/BxN serum transfer arthritis. All values were expressed as mean ±SEM; each group contains 5 mice. *p<0.05, **p<0.01, MIF-/- compared to WT.
Figure 5.
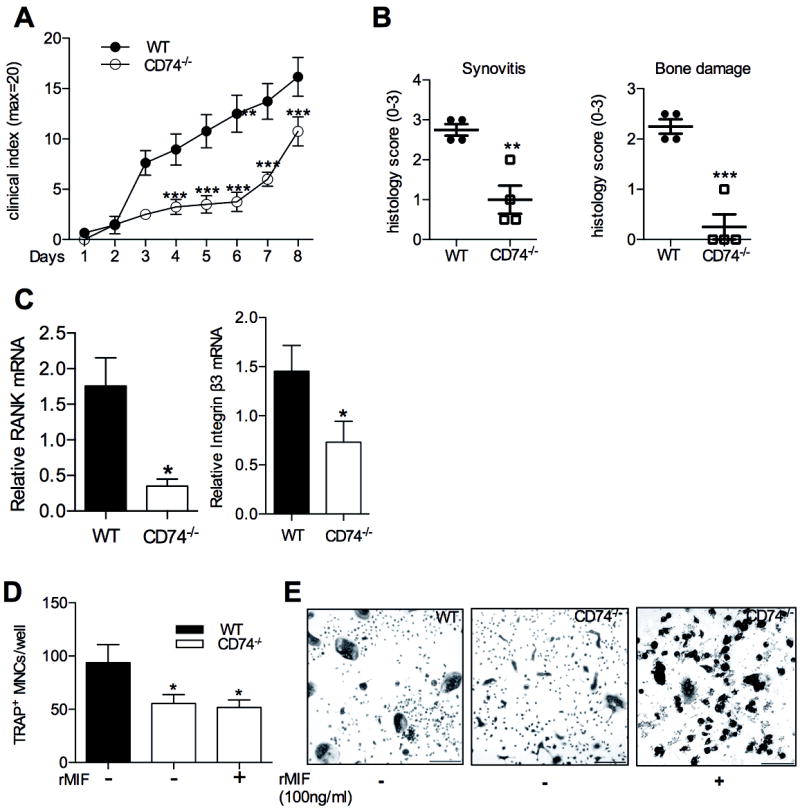
Inflammatory arthritis in WT and CD74-/- mice with K/BxN serum transfer arthritis. A: Clinical scoring of joint inflammation in K/BxN serum treated WT (closed circles) and CD74-/- (open circles) mice. B: Histological scores for synovitis (left) and bone damage (right) in WT and CD74-/- mice. C: Osteoclast-associated gene expression determined using RNA extracted from ankles of WT and CD74-/- mice with K/BxN serum transfer arthritis D: Osteoclastogenesis in WT and CD74-/- bone marrow cells in response to M-CSF (30 ng/ml) and RANKL (50 ng/ml). E: Representative TRAP histochemical staining of osteoclast cultures quantified in D. bars =20μm. Original magnification x200. All values are expressed as mean ±SEM; A&B: each group contains 3 mice. C: Experimental results were pooled from 5 independent experiments. *p<0.05, **p<0.01, *** p<0.001, CD74-/- compared to WT.
Table 1.
Real time PCR primers
| Primer Name | Forward primer | Reverse Primer |
|---|---|---|
| MIF | 5’-TGACTTTTAGCGGCACGAAC-3’ | 5’-GACTCAAGCGAAGGTGGAAC-3’ |
| CTR | 5’-TGTCCAGGGTATAAGCAA-3’ | 5’-GTTCCCACTGCATTGTCCACA-3’ |
| DC-STAMP | 5’-CTAGCTGGCTGGACTTCATCC-3’ | 5’-TCATGCTGTCTAGGAGACCTC-3’ |
| OC-STAMP | 5’-TGGGCCTCCATATGACCTCGAGTAG-3’ | 5’-TCAAAGGCTTGTAAATTGGAGGAGT-3’ |
| TRAP | 5’-TCCTGGCTCAAAAAGCACGTT-3’ | 5’-ACATAGCCCACACCGGTTCTC-3’ |
| DUSP 5 | 5’-AGGAGGAGCGTGGTCTCTC-3’ | 5’-GTGGAGGGCAGGATCTCA-3’ |
| DUSP 6 | 5’-GGCAAAAACTGTGGTGTCCT-3’ | 5’-CATCGTTCATGGACAGGTTG-3’ |
| DUSP 9 | 5’-TCCTGTACGACCAGGGTAGC-3’ | 5’-GTTCTCCGCTTCAGCCTTAG-3’ |
| βactin | 5’-TGTCCCTGTATGCCTCTGGT-3’ | 5’-GATGTCACGCACGATTTCC-3’) |
| 18s | 5’-GGATCCATTGGAGGGCAAGT-3’ | 5’-CGAGCTTTTTAACTGCAGCAACT-3’ |
Western blotting
Cell lysates were prepared and protein concentration determined as described[5]. Samples containing equivalent amounts of cellular proteins (30-50 μg/sample) were fractionated in 10% pre-cast gels (Perbio Science, Mordialloc, Australia) then transferred to hybond C extra nitrocellulose membranes (Millipore, Bedford, MA). Membranes were blocked with 2.5% bovine serum albumin (BSA) in Tris buffered saline containing 0.1% Tween (TBST). Immunoblotting was performed as described[5] using primary antibodies directed against ERK1/2, phospho-ERK1/2, p38, phospho-p38, phospho-serine 536 NF-κB p65, and ß-actin (Cell Signalling, Danvers, MA). Densitometry ratios were normalized to appropriate loading control content and results expressed as relative density.
NF-κB and NFAT Luciferase Reporter assays
The NF-κB-RAW cell line and NFAT-RAW cell lines are derived from the RAW264.7 macrophage cell line, stably transfected with NF-κB[30] and NFAT luciferase reporter constructs[31], respectively. Transcriptional activity was examined in response to stimulation with RANKL 100 ng/ml for 6 (NFκB) or 24 (NFAT) hours, after which cells were lysed with reporter lysis buffer (Promega, Madison, Wisconsin, USA) and luciferase activity measured using a luminometer (Wallac/Perkin Elmer, Turku, Finland). Results are expressed as fold change compared to untreated controls in the respective experimental group. In some experiments, NF-κB-RAW cells and NFAT-RAW cells were treated with anti-MIF mAb or control Ig (100 ng/ml) 1 hour before addition of RANKL.
Statistical analysis
Clinical and histological score data were analysed by Mann-Whitney U test for comparisons of group mean values. All other data were analysed using Student’s t-tests. Results are expressed as the mean ± SEM. For each test, p values less than 0.05 were considered significant.
RESULTS
MIF deficiency reduced inflammation and bone damage in the K/BxN serum transfer arthritis
We first sought to confirm the reported effect of MIF deficiency on inflammation in the K/BxN serum transfer arthritis model[11]. Consistent with our previous report, WT mice developed severe clinical joint inflammation and histological synovitis (Fig. 1A), accompanied by extensive local bone damage (Fig. 1B). Vehicle-injected mice did not develop any signs of clinical arthritis, or any histological evidence of joint inflammation or bone destruction (data not shown). In contrast, MIF-/- mice exhibited significantly less severe synovitis than WT mice, as determined by clinical (Fig. 1A) and histological scores (Fig. 1B). Most notably, histological evidence of bone damage was markedly and significantly reduced in MIF-/- mice (Fig. 1B&C), and was in fact undetectable in all but one MIF-/- mouse. Osteoclasts (identified by TRAP histochemical staining) and bone erosions were clearly evident at the synovial-bone interface in the joints of WT mice, but no TRAP+ osteoclasts were identified at the synovial-bone interface in any MIF-/- mouse (Fig. 1C). Consistent with this, expression of TRAP mRNA was significantly lower in MIF-/- joints (Fig. 1D). To further characterise the consequences of MIF deficiency on osteoclasts in these tissues, we also analysed the expression of osteoclast-associated markers in RNA extracted from ankle joints. Expression of other osteoclast fusion relevant gene, dendritic cell-specific transmembrane protein (DC-STAMP) and osteoclast-STAMP (OC-STAMP), mRNA was significantly lower in MIF-/- joints (Fig. 1D).
MIF deficiency reduced osteoclastogenesis in vitro
The arthritic erosion is due to the imbalance of osteoclast and osteoblast activity, therefore, we next examined the expression of MIF in osteoclasts and osteoblasts in vitro. In mouse BMM stimulated with M-CSF, osteoclast formation was dose-dependently induced by recombinant RANKL (Fig. 2A), as previously described. Accompanying this effect, the supernatant MIF concentration was significantly increased in response to RANKL (Fig. 2B). Accordingly, MIF mRNA was also increased in M-CSF plus RANKL treated bone marrow cells (Fig. 2C). No significant increase in MIF mRNA levels was seen prior to time-points when TRAP+ cells first became evident (data not shown). These data suggest that osteoclast maturation in response to prolonged RANKL stimulation is accompanied by enhanced MIF production.
Figure 2.
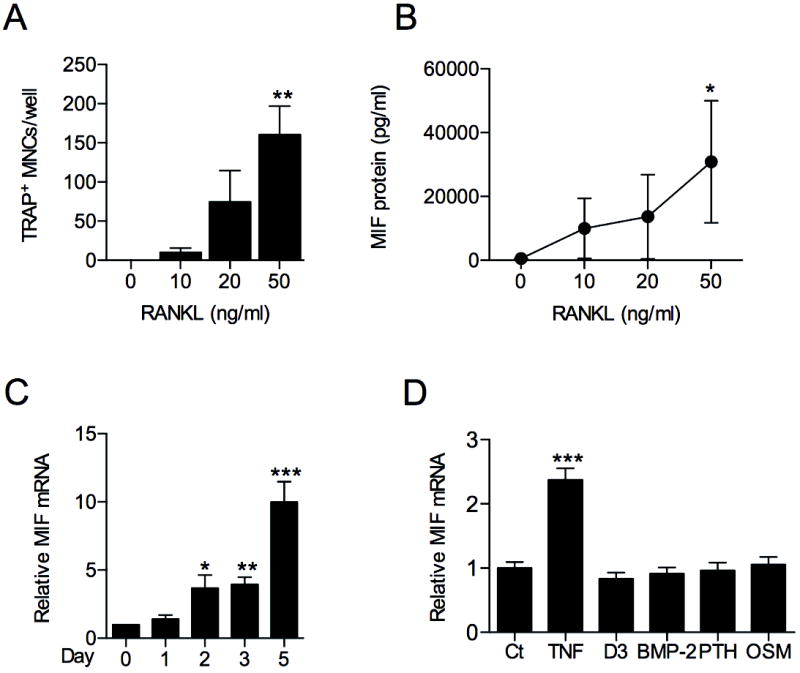
MIF expression in osteoclasts and osteoblasts. A: BMM from WT mice were expanded in culture with M-CSF (100ng/ml for 3 days), then stimulated with RANKL for 4 days and osteoclast formation analysed by TRAP histochemical staining. B: MIF concentration in supernatants obtained from cell cultures described in A, quantified by ELISA. C: MIF mRNA expression in WT bone marrow cells treated with M-CSF (30ng/ml) and RANKL (50ng/ml) analysed by real time RT-PCR. D: Primary osteoblasts isolated from WT mice were treated with osteogenic stimuli (1,25(OH)2D3 10-8 M or BMP2 (200 ng/ml) or osteolytic stimuli (PTH 10 nM, TNF 10 ng/ml or OSM 50 ng/ml) for 24 hours and MIF mRNA expression measured by real time PCR. All values were expressed as mean ±SEM. Experimental results were pooled from 3 independent experiments. *p<0.05, **p<0.01, *** p<0.001 for treated versus untreated or ‘day 0’ control as appropriate.
Since osteoclasts and their progenitors closely interact with osteoblasts, we also investigated the expression of MIF in mouse primary calvarial osteoblasts. No effect of BMP2, OSM, dihydroxy vitamin D3 (1,25(OH)2D3) or PTH on MIF mRNA expression was observed (Fig. 2D). In contrast, osteoblast MIF mRNA was significantly increased by TNF (Fig. 2D), suggesting elevated osteoblast MIF expression under conditions of inflammation.
We next examined the effects of MIF deficiency on in vitro osteoclast formation. Osteoclasts were quantified after 7 days culture of MIF-/- and WT bone marrow cells with M-CSF and RANKL. Compared to WT, significantly fewer osteoclasts were found in cultures of MIF-/- bone marrow cells (Fig. 3A&B), and there were also significantly reduced numbers of giant osteoclasts (osteoclasts with more than 10 nuclei, data not shown). More interestingly, addition of rMIF resulted in significantly increased osteoclast formation compared to untreated MIF-/- cells (Fig. 3A&B). Furthermore, in controversial to observation reported by Jacquin et al[25], rMIF did not alter RANKL-induced osteoclast formation in cultures of WT cells or RAW 264.7 cells (Supp. Fig. 1A&B). In keeping with this, RANKL-induced NF-κB- and NFAT-dependent transcriptional activity in RAW264.7 cells were not affected by the addition of rMIF (Supp. Fig. 1C&D), suggesting that endogenous MIF is sufficient to support osteoclastogenesis in these cells and exogenous MIF only can restore osteoclastogenesis in MIF depleted cells.
Figure 3.
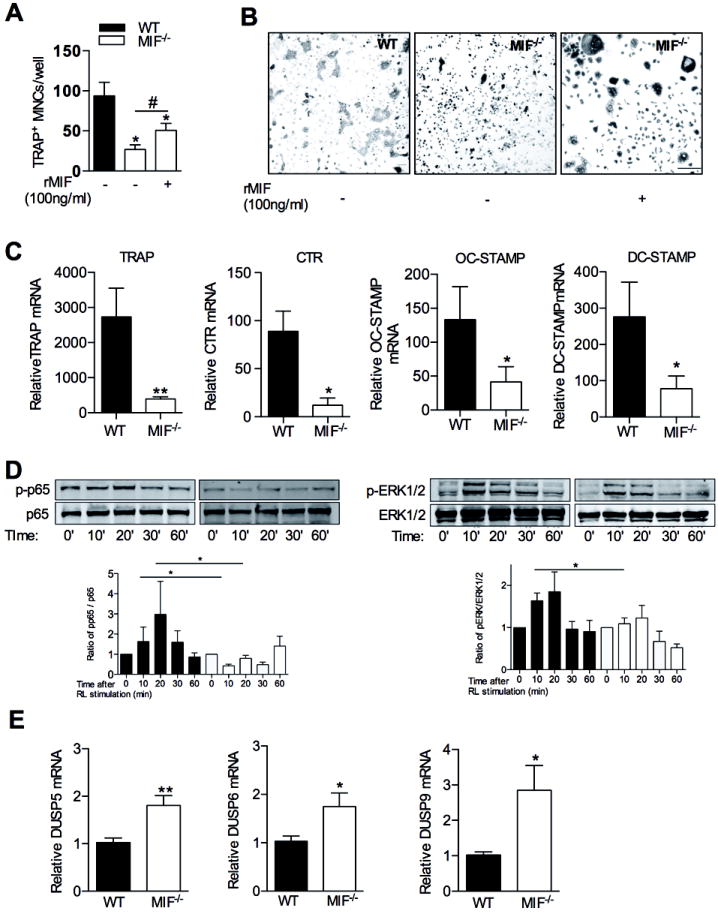
In vitro osteoclastogenesis in WT and MIF-/- cultured bone marrow cells. A: Osteoclastogenesis in WT and MIF-/- bone marrow cells in response to M-CSF (30 ng/ml) and RANKL (50 ng/ml) with or without rMIF (100 ng/ml) treatment. B: Representative TRAP histochemical staining of osteoclast cultures quantified in A. bars =20μm. Original magnification x200. C: Osteoclast-associated gene expression in M-CSF (30 ng/ml) plus RANKL (50 ng/ml) stimulated bone marrow cells after 5 days, measured by real time PCR. The results are expressed as fold change relative to bone marrow cells treated with M-CSF (30 ng/ml) alone. D: Analysis of M-CSF treated WT and MIF-/- BMM, showing their responses to RANKL (100 ng/ml) treatment over the time course indicated. Western blots (representative of 4 independent experiments) show phospho-ser536 NF-κB p65 (pp65), total p65 levels, phospho-ERK1/2 MAP kinase and total ERK1/2 levels as well as β-actin. Densitometry analysis of Western blots showing ratio of phospho-p65 (left) and phospho-ERK1/2 (right) to their appropriate loading controls (Time: time after RANKL treatement). E: mRNA expression of the indicated DUSP genes were measured by real time PCR in WT and MIF-/- BMM. All values are expressed as mean±SEM. All quantitative data were pooled from 4 independent experiments. The Western blot are representative for 3 independent experiments *p<0.05, **p<0.01 relative to WT cultures. #p<0.05, compared to MIF-/- with no rMIF treated culture.
Since the lack of MIF has been associated with reduced actin polymerization in BMM[5] and osteoclasts form distinctive actin rings, we also assessed whether the reduction in osteoclast formation in MIF-/- cells was associated with alterations in actin ring formation. There were indeed fewer multinucleated cells with actin rings in MIF-/- cultures (Supp. Fig. 2A), although this was concordant with degree of reduced osteoclastogenesis in MIF-/- cells; typical osteoclast-associated actin rings were observed in both WT and MIF-/- cultures, suggesting that actin ring formation was retained in the absence of MIF.
MIF deficiency also results in reduced survival in several cell types, including macrophages[32]. Since higher M-CSF levels can boost osteoclast survival, we determined whether the reduction in osteoclast numbers in MIF-/- cultures could be reversed with higher concentrations of M-CSF. However, in dose-response studies, the reduction in osteoclastogenesis in MIF-/- cells compared to WT cells remained statistically significant across the M-CSF concentration range 30 to 200 ng/mL (Sup Fig. 2B). Longer durations of RANKL-treated osteoclast cultures (up to 9 days) were also not associated with restoration of osteoclast formation in MIF-/- cells (data not shown). We also examined the effect of MIF deficiency on in vitro osteoclasts when RANKL and M-CSF was withdrawn, as each individually is an osteoclast survival factor. WT osteoclasts survived withdrawal of RANKL; fewer survived withdrawal of M-CSF, and none survived withdrawal of both RANKL and M-CSF (Supp. Fig. 2C). Similar effects of growth factor withdrawal were seen in MIF-/- osteoclasts (Supp. Fig. 2D), suggesting little or no influence of MIF deficiency on osteoclast survival.
MIF Deficiency reduced osteoclastogenesis relevant gene expression via down-regulating RANKL-induced NF-κB and MAP kinase phosphorylation
We also examined the mRNA expression of osteoclastogenesis relevant genes, including TRAP, CTR, OC-STAMP and DC-STAMP mRNA, which was also significantly reduced in RANKL-stimulated MIF-/- cells (Fig. 3C), while the expression of RANK and cathepsin K was not significantly different between WT and MIF-/- cells (data not shown). The reduction of these osteoclastogenesis relevant genes suggested an effect of MIF on RANKL-mediated signalling events. To investigate the effects of MIF on signal transduction events upstream of these RANKL-induced transcription factors, we studied the phosphorylation of NF-κB-p65 (serine 536), ERK1/2 and p38 MAP kinases, in WT and MIF-/- BMM. In WT BMM, RANKL induced p65 phosphorylation was significantly higher in WT cells (1.2 – 6 fold increase at 20 minutes after RANKL stimulation) compared to MIF-/- cells (0.67 - 1.09 fold increase at 20 minutes after RANKL stimulation) (Fig. 3D). In WT cells, RANKL stimulation also induced ERK1/2 phosphorylation at 10 and 20 minutes (Fig. 3D), but this was markedly diminished in MIF-/- cells (Fig. 3D). RANKL-elicited p38 phosphorylation was similar in WT and MIF-/- cells (data not shown). These findings indicate that MIF expression in BMM facilitates NF-κB-p65 and ERK phosphorylation signals in response to RANKL.
Since these data suggests an influence on ERK MAPK, we next investigated whether this pathway might influence osteoclast formation in MIF deficient mouse cells. We investigated this using the MEK (MAPK kinase 1) inhibitor PD98059, which blocks ERK1/2 activation. We found this compound indeed significantly reduced osteoclastogenesis in WT bone marrow cells (Supp. Fig. 3), indicating that this process is at least partially dependent on ERK MAPK phosphorylation.
DUSPs are a large and diverse group of phosphatases, 11 of which are known to de-phosphorylate activated MAPKs, decreasing their activity. We have previously reported increased MKP-1 (also known as DUSP-1) expression in MIF deficient cells[5,29]. To determine whether such an effect might underlie the effects of MIF on osteoclastogenesis, mRNA expression of those DUSPs that regulate ERK1/2 activity (DUSP-5, 6, 9) was examined in WT and MIF-/- BMM. Expression of DUSP-5, 6, and 9 mRNA was significantly higher in MIF-/- BMM cultures than WT (Fig. 3E). These findings suggest that MIF mediated ERK-DUSP signaling pathway at least is partially involved in RANKL-induced osteoclastogenesis.
MIF blockade decreased osteoclast formation and NF-κB and NFAT transcriptional activity in RAW264.7 cell
To determine whether neutralisation of extracellular MIF reduces osteoclast formation, as does genetic MIF deficiency, we examined the effects of a neutralising anti-MIF monoclonal antibody on osteoclastogenesis. Anti-MIF antibody treatment of RANKL-stimulated RAW264.7 cells resulted in significantly reduced osteoclast formation compared to control IgG-treated cultures (Fig. 4A,B), an effect seen at all concentrations of RANKL employed. Anti-MIF antibody treatment also significantly reduced RANKL-induced NF-κB (Fig. 4C) and NFAT (Fig. 4D) reporter activity in RAW264.7 cells stably transfected with these luciferase reporter constructs. Note that the latter is responsive to both NFATc1 and NFATc2, although NFATc2 is induced by RANKL-stimulated NFATc1, and both bind the same promoter motifs. These findings suggest that endogenous MIF is important for NF-κB and NFATc1 activation and osteoclast differentiation in RAW264.7 cells.
Figure 4.
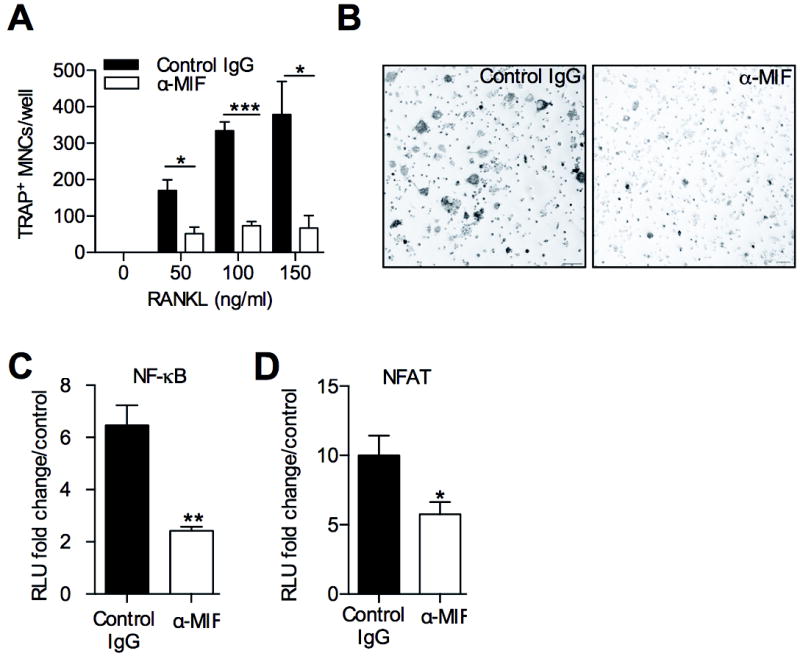
The effects of MIF blockade on osteoclastogenesis and transcription factors. A: RANKL-induced osteoclastogenesis, measured by the presence of TRAP+ MNCs, in RAW264.7 cells treated with anti-MIF monoclonal antibody (α-MIF) (100 ng/ml) or control IgG (100 ng/ml). B: Images of TRAP+ MNCs formed in cultures described in A; bars =200μm. Original magnification x100. C: Effect of α-MIF antibody (α-MIF) or control IgG on NF-κB reporter activity in RANKL (100 ng/ml, 6h)-treated stably transfected RAW cells. D: as C, but using RAW264.7 cells stably transfected with luciferase NFAT reporter. All values were expressed as mean±SEM. Experimental results were pooled from 3 independent experiments. *p<0.05, **p<0.01, ***p<0.001 relative to control antibody treated cultures.
CD 74 deficiency reduced inflammation and bone damage in K/BxN serum transfer arthritis in vivo and osteoclast formation in vitro
Since we observed that MIF deficiency reduced inflammation and bone erosion in K/BxN serum transfer arthritis, and as CD74 is reported as a mediator of MIF actions, we assessed the influence of CD74 on inflammation and inflammatory arthritic bone damage. The severity of K/BxN serum transfer-induced inflammatory arthritis was significantly reduced in CD74-/- mice compared to WT mice, as evidenced by lower clinical (Fig. 5A) and histological scores (Fig. 5B). Similar to the findings observed in MIF-/- mice, CD74-/- mice exhibited significantly lower levels of arthritic bone erosion (Fig. 5B), accompanied with reduced mRNA expression of RANK and integrin β3 (Fig. 5C). These data demonstrate that reduced arthritis in the absence of the MIF-binding protein CD74 is accompanied by greatly reduced bone erosion in the joint.
Similarly, osteoclasts were generated from WT and CD74-/- bone marrow cells using RANKL (50 ng/ml) and M-CSF (30 ng/ml). CD74-/- cultures yielded significantly fewer osteoclasts compared to WT (Fig. 5D&E). In contrast, rMIF did not enhance osteoclast formation in CD74-/- cells (Fig. 5D&E). These findings indicated that CD74 deficiency is associated with decreased in vitro osteoclastogenesis, and rMIF restores osteoclast formation only in cells lacking endogenous MIF (i.e., MIF-/- cells).
DISCUSSION
RA is a severe and disabling disease, characterised by chronic inflammation, the destruction of articular cartilage, and periarticular bone erosion. MIF participates in many pathological processes characteristic of RA, and is abundantly expressed in RA synovial macrophages and fibroblasts[5,33]. RA disease activity also correlates with synovial MIF expression[9]. Furthermore, MIF deletion or neutralisation significantly reduces the severity of arthritis in several animal arthritis models[10,34]. While much is known about its action in inflammation, the influence of MIF on bone, and specifically on mechanisms relevant to arthritic bone erosion, is still not fully understood. In the current study, we have explored the role of MIF and CD74 in RANKL-dependent osteoclast differentiation and activation in vitro and on arthritic bone damage in vivo, and conclude that MIF and CD74 significantly facilitate osteoclast formation. These findings have implications for the potential application of anti-MIF therapies as the treatment of RA, since our data suggests that such a therapy would reduce inflammation-associated bone erosion in addition to the effects on joint inflammation.
Among the strongest evidence suggesting a role of MIF in RA bone erosion comes from a longitudinal cohort study of RA patients, genotyped for a single nucleotide polymorphism (SNP) in the MIF promoter region that is associated with MIF overexpression[20], in which carriage of MIF-overexpression SNPs was associated with accelerated bone erosion. However, very little mechanism studies on the role of MIF on arthritic erosion in human have been done. In addition, current murine studies revealed a controversial effect of MIF on osteoclastogenesis. It has been reported that deficiency or neutralization of MIF is associated with protection from ovarectomy-induced bone loss[21,35] and bacterially-induced periodontal disease[22], both of which are mediated by osteoclasts. Accordingly, MIF transgenic mice were reported to exhibit high bone turnover[24]. Together these findings suggest that MIF plays a facilitatory role in osteoclastogenesis. In contrast, Jacquin et al. reported increased osteoclastogenesis in MIF-/- BMM, an inhibitory effect of exogenous rMIF in vitro, and reduced bone mass in vivo in MIF-/- mice, suggestive of an anti-resorptive effect of MIF[25]. The same group also reported that CD74-/- mice exhibited lower trabecular volume in vivo, accompanied with enhanced osteoclastogenesis in vitro[26]. These conflicting results make it difficult to determine whether MIF contributes directly to bone damage in RA. No previous studies have been undertaken in the setting of inflammation, as is seen in bone damage in RA, thus further studies are required.
While MIF is expressed in primary osteoblasts and osteoblastic cell lines[36] (in vivo, osteoblasts and closely related osteocytes are a major source of RANKL), we have yet to find a stimulus that elicits osteoblast MIF production other than TNF, an inducer of MIF in many cell types. This suggests that MIF is not a major modulator of osteoblast function, but that osteoblasts contribute to the production of MIF in the extracellular milieu during inflammation. Therefore, we focus on examine the role of MIF in osteoclastogenesis.
We determined MIF expression during RANKL-induced osteoclastogenesis, which revealed increased MIF expression in osteoclast cultures compared to the macrophages cultures, suggesting that expression of MIF is required during osteoclastogenesis. However, it is notable, that exogenous recombinant MIF had no effect on osteoclastogenesis in WT bone marrow cells or RAW264.7 cells. This can be explained by their constitutively high expression of MIF, suggesting that RANKL-induced MIF expression in these cells is sufficient to sustain osteoclastogenesis. This is consistent with our in vivo observation, where rMIF did not exacerbate arthritis and arthritic bone erosion in WT mice (unpublished data). Together, these observations suggested that endogenous MIF is critical in murine arthritis model and osteoclastogenesis.
We used the well-established K/BxN serum transfer model to investigate how a lack of endogenous MIF production would influence osteoclast-mediated bone damage in arthritis in vivo. Bone erosion in this model has been conclusively shown to depend on RANKL-mediated osteoclastogenesis[18]. We have recently reported that MIF-/- mice showed reduced clinical and histological synovitis in K/BxN serum transfer arthritis[11]. In the current study, we demonstrated that reductions in joint inflammation are accompanied by histological evidence of reduced bone erosion. It was striking that although reduced, MIF-/- mice still displayed synovitis, while the abrogation of bone erosion was near-complete. In addition, MIF deficiency was accompanied by marked reduction in the numbers of TRAP+ cells at the bone-synovium surface, and significantly reduced joint tissue expression of the osteoclastogenesis genes including TRAP, OC-STAMP and DC-STAMP. These data suggested the possibility that in addition to effects on bone erosion secondary to reductions in inflammation, MIF deficiency was exerting a direct effect on mechanisms of arthritic bone erosion. In line with these observations, K/BxN serum treated CD74-/- mice also exhibited significantly reduced clinical and histological arthritis scores compared to WT mice, as well as significantly reduced histological bone damage. This suggested that the CD74 mediation of MIF effects in inflammation and bone biology.
Since our data strongly suggested an influence of MIF in osteoclastogenesis, we next performed in vitro osteoclastogenesis assays to determine whether deficiency of MIF or CD74 would affect this process directly using in vitro culture system. We found that osteoclastogenesis in RANKL-stimulated bone marrow cells was significantly reduced in the absence of either MIF or CD74. These observations were independent of M-CSF or RANKL concentration or duration of exposure, and suggest that the lack of MIF results in decreased osteoclast differentiation rather than reduced osteoclast survival; note that both M-CSF and RANKL are independent survival factors for osteoclasts. The reduction in osteoclast numbers forming from MIF-/- cells could arise either from lower RANKL responsiveness or lower osteoclast progenitor numbers. The latter possibility is difficult to evaluate directly, as many types of immature macrophage cells give rise to osteoclasts, however when we treated cultures with higher M-CSF concentrations, which would increase progenitor proliferation, this did not reverse the effects of MIF deficiency on osteoclast yield. Consistent with this, RANKL-induced osteoclast formation in RAW264.7 cells, a proliferating cell line, was significantly inhibited by MIF neutralization, which strongly suggests a direct influence of MIF in RANKL-driven differentiation responses.
The role of extracellular MIF in these responses is suggested by the observation of increased MIF release during RANKL-induced osteoclastogenesis in vitro. This is also suggested by the restoration of osteoclast formation in MIF-/- bone marrow cultures in response to exogenous MIF and by the ability of extracellular neutralising anti-MIF antibody treatment to inhibit osteoclastogenesis. However, exogenous MIF also did not restore reduced osteoclastogenesis in CD74-deficient cells to that seen in wild type cells, although the lack of response in these cells may reflect the fact that they are MIF-replete if MIF acts through receptors other than CD74. However, consistent with the reported role of CD74 in cellular responses to MIF, cells from MIF-/-xCD74-/- DKO mice were also resistant to exogenous MIF restoration of osteoclastogenesis (unpublished data), in contrast to MIF-/- cells that express normal CD74.
In contrast to reductions in IL-1 and TNF receptor expression described in MIF-/- fibroblasts[37], levels of RANK in MIF-/- BMM did not differ from WT (unpublished data), suggesting that MIF did not exert its effect on RANKL responsiveness through the regulation of receptor expression. Finally, no difference in osteoclast survival between WT and MIF-/- cells was observed, suggesting that reduced osteoclast survival does not contribute to lower osteoclast numbers seen in the absence of MIF.
Our data suggests that there are important autocrine actions of MIF on key signalling events involved in osteoclast differentiation in response to RANKL. NF-κB and NFATc1 are crucial transcriptional factors in RANKL-induced osteoclastogenesis, without either of which osteoclastogenesis does not proceed[16,28,38]. A number of studies have demonstrated effects of MIF on intracellular signalling pathway activation[5,37]. We observed RANKL-induced NF-κB p65 ser-536 phosphorylation was reduced in MIF-/- cells, suggesting this as an upstream effect of MIF influencing NF-κB activation. The ERK1/2 MAP kinase pathway is important for osteoclast function and survival[39], while p38 MAPK activity is also non-redundant in osteoclast formation[40]. RANKL-induced NF-κB and ERK1/2 signals strongly increase the expression and activity of NFATc1. NFATc1 expression was notably down-regulated by MIF neutralisation, making it a likely key downstream target of MIF, particularly since NFATc1 plays a crucial role in osteoclast commitment (Fig. 4D). Furthermore, NF-κB (with NFATc1) also controls expression of a number of proteins that are important in osteoclast formation and function. We showed reductions in the absence of MIF of TRAP, CTR, OC-STAMP and DC-STAMP (Fig. 3C), each of which has been shown to be regulated by ERK, NF-κB or both[41]. Here, we showed that RANKL-induced ERK activation was impaired by MIF deficiency. Such an effect of MIF via ERK is potentially mediated through its effects on MAPK phosphatases (MKPs). MKP-1 (also known as DUSP-1) belongs to the DUSP family, each member of which selectively de-phosphorylates various activated MAP kinases. DUSP-1 expression is inhibited by MIF in murine BMM and human synovial fibroblasts, facilitating MAPK activation[5,42]. Of note, multiple DUSPs are particularly involved in the regulation of ERK1/2 activity[43]. We noted that the mRNA expression of three DUSP relevant to the control of ERK phosphorylation, DUSP-5, -6, and -9 was significantly enhanced in MIF-/- cells, consistent with the previous finding that macrophage MKP-1/DUSP-1 expression was significantly reduced by MIF[5]. Of note, we did not observe an effect of CD74 deficiency on expression of these DUSP (unpublished data), consistent with the previous observation that CD74-deficiency had no effect on MKP-1[5]. Together, these results indicate that not all aspects of the effects of MIF on osteoclasts are mediated via CD74. Moreover, reduced RANKL-induced osteoclastogenesis in RAW264.7 cells in response to MIF blockade was accompanied by reduced activation of both NF-κB and NFATc1, shedding lights of developing a MIF blockade as a therapy for RA patients.
We sought to examine the role of MIF on arthritic bone erosion and osteoclast differentiation in the context of its well-documented effects in arthritis, which data presented in the current studies again confirm. MIF has been considered as a possible therapeutic target for many autoimmune diseases, including rheumatoid arthritis[44]. Some apparent contradictions have been seen in studies that have reported different actions of MIF in the same disease models[27,45], which strongly suggests that specific experimental conditions may influence the effect of MIF. In our study, we have observed reduced bone erosion in the absence of endogenous MIF, results opposite to those in the study reported by Jacquin et al. Importantly, the suggestion that MIF plays a generally pro-osteolytic role in bone is supported by human studies, such the association in RA patients of a MIF polymorphism with high expression of circulating MIF and severe bone erosion compared to low MIF expression patients[20].
Our findings are in line with those of all other papers on this area[21-23,46], other than Jacquin et al and Mun et al. There are key differences between the current study and that Jacquin et al. Firstly, Jacquin et al., examined bone structure in naïve mice, whereas we examined the role of MIF in bone erosion in an arthritic model. We demonstrated an anti-inflammatory effect of MIF deficiency, consistent with several previous reports with different arthritis models[11,47,48]. In our arthritis model, only one MIF-/- mouse out of five showed even low levels of bone damage compared to WT mice, an observation repeatable with different batches of mice and K/BxN serum, suggesting that the role of MIF is significant under stressed conditions. This is in coordinate with a recent findings that high serum MIF level was associated with reduced susceptibility to the autoimmune disease systemic lupus erythematosus (SLE) but in established disease high serum MIF levels are highly associated with fatal organ damage[49] – indicating a pro-inflammatory effect of MIF in the setting of autoimmune-mediated systemic inflammation skin to what we observed. Moreover, different housing environments may also contribute to the divergent observations reported by different research groups, which has been previously reported in various knockout strains[50-52].
With regard to Mun et al, who studied CD74-/- mice, while littermates were used, again in vivo osteoclast actions were not studied in the setting of inflammatory stress. With regard to in vitro studies, both Jacquin and Mun reported anti-osteoclastogenic effects of MIF, in contrast to our findings. There were differences in experimental conditions between the studies, with our studies using RANKL concentrations almost double those in both papers, which again may reflect the inflammatory milieu more associated with arthritis than physiological bone remodeling. Moreover, we consider that our use in parallel experiments of anti-MIF mAb, neutralizing endogenous MIF from WT cells, strongly supports the validity of our findings.
In conclusion, our observations suggest that MIF plays an important role in osteoclastogenesis induced by RANKL, which is the chief driver of bone erosion in human RA. In the K/BxN serum transfer arthritis model, the reduction in arthritis in the absence of MIF or CD74 was accompanied by greatly decreased osteoclast formation and bone damage, and in vitro studies demonstrate that MIF or CD74 are required for optimal responses to RANKL during osteoclastogenesis. The findings indicate a direct effect of MIF on osteoclastogenesis, and suggest that therapeutic MIF inhibition in RA could have beneficial effects on bone erosion as well as inflammation. Information provided in this study will contribute to the translational process of MIF to possible alternative therapy for any clinical trails.
Supplementary Material
Supplementary Figure 1. The effects of exogenous rMIF (100 ng/ml) addition on osteoclast formation in vitro in M-CSF (30 ng/ml) and RANKL (50 ng/ml) treated bone marrow cells. A: Osteoclast formation in WT cells with and without rMIF (100ng/ml) treatment. B: Osteoclast formation in RAW 264.7 cells with and without rMIF (concentration as indicated) treatment. C: Effect of rMIF on NF-κB reporter activity in RANKL (100 ng/ml, 6h)-treated stably transfected RAW cells. D: as C, but using RAW264.7 cells stably transfected with luciferase NFAT reporter. All values were expressed as mean ±SEM. Experimental results were pooled from 3 independent experiments.
Supplementary Figure 2. A: Actin ring formation in WT and MIF-/- bone marrow cells. Cells were stimulated with M-CSF (30 ng/ml) plus RANKL (50 ng/ml) for 7 days. Actin was visualised with phalloidin-rhodamine in cells counterstained with DAPI and ring formation was quantified. B: Dose-dependent effects of M-CSF on osteoclastogenesis in WT and MIF-/- bone marrow cells. Survival of in vitro generated osteoclasts from WT (C) and MIF-/- (D) mice, upon withdrawal of RANKL, M-CSF or both. Experimental results were pooled from 5 independent experiments. All values were expressed as mean ±SEM. *p<0.05, *** p<0.001 WT versus MIF-/-.
Supplementary Figure 3. WT bone marrow cells were treated with M-CSF (30 ng/ml) and RANKL (50 ng/ml) for 7 days, and with the MEK inhibitor PD98059 (PD; 10 μM) or DMSO vehicle (Veh).
Highlights.
MIF and CD74 deficiency is associated with reduced K/BxN serum transfer arthritis
MIF and CD74 facilitate RANKL-induced osteoclastogenesis in vitro
MIF and CD74 facilitate RANKL-induced osteoclastogenesis relevant signaling events in vitro
Acknowledgments
This work was supported by Australian National Health and Medical Research Council Project Grant 611805 (JQ), The Ulysses Club of Australia Arthritis Research Scholarship (RG), NIH Grant (AR049610 and AR050498) (RB) and by the Victorian Government Operational Infrastructure Support Program (JQ). The authors wish to thank Dr. Camden Lo and Ms Anqi Li for imaging and photographical assistance. The author would like to thank Dr Sarin especially for her suggetions and commons to imporve the manuscript.
Footnotes
The authors declare no conflict of interest.
References
- 1.Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. 2006;5:399–410. doi: 10.1038/nrd2029. [DOI] [PubMed] [Google Scholar]
- 2.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–9. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 3.Santos L, Hall P, Metz C, Bucala R, Morand EF. Role of macrophage migration inhibitory factor (MIF) in murine antigen-induced arthritis: interaction with glucocorticoids. Clin Exp Immunol. 2001;123:309–14. doi: 10.1046/j.1365-2249.2001.01423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, et al. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol. 2006;177:8072–9. doi: 10.4049/jimmunol.177.11.8072. [DOI] [PubMed] [Google Scholar]
- 5.Fan H, Hall P, Santos LL, Gregory JL, Fingerle-Rowson G, Bucala R, et al. Macrophage migration inhibitory factor and CD74 regulate macrophage chemotactic responses via MAPK and Rho GTPase. J Immunol. 2011;186:4915–24. doi: 10.4049/jimmunol.1003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lacey D, Sampey A, Mitchell R, Bucala R, Santos L, Leech M, et al. Control of fibroblast-like synoviocyte proliferation by macrophage migration inhibitory factor. Arthritis Rheum. 2003;48:103–9. doi: 10.1002/art.10733. [DOI] [PubMed] [Google Scholar]
- 7.Sampey AV, Hall PH, Mitchell RA, Metz CN, Morand EF. Regulation of synoviocyte phospholipase A2 and cyclooxygenase 2 by macrophage migration inhibitory factor. Arthritis Rheum. 2001;44:1273–80. doi: 10.1002/1529-0131(200106)44:6<1273::AID-ART219>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 8.Onodera S, Kaneda K, Mizue Y, Koyama Y, Fujinaga M, Nishihira J. Macrophage migration inhibitory factor up-regulates expression of matrix metalloproteinases in synovial fibroblasts of rheumatoid arthritis. J Biol Chem. 2000;275:444–50. doi: 10.1074/jbc.275.1.444. [DOI] [PubMed] [Google Scholar]
- 9.Morand EF, Leech M, Weedon H, Metz C, Bucala R, Smith MD. Macrophage migration inhibitory factor in rheumatoid arthritis: clinical correlations. Rheumatology (Oxford) 2002;41:558–62. doi: 10.1093/rheumatology/41.5.558. [DOI] [PubMed] [Google Scholar]
- 10.Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol. 1997;158:5514–7. [PubMed] [Google Scholar]
- 11.Santos LL, Fan H, Hall P, Ngo D, Mackay CR, Fingerle-Rowson G, et al. Macrophage migration inhibitory factor regulates neutrophil chemotactic responses in inflammatory arthritis in mice. Arthritis Rheum. 2011;63:960–70. doi: 10.1002/art.30203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–76. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi X, Leng L, Wang T, Wang W, Du X, Li J, et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity. 2006;25:595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi K, Koga K, Linge HM, Zhang Y, Lin X, Metz CN, et al. Macrophage CD74 contributes to MIF-induced pulmonary inflammation. Respir Res. 2009;10:33. doi: 10.1186/1465-9921-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakashima T, Takayanagi H. New regulation mechanisms of osteoclast differentiation. Ann N Y Acad Sci. 2011;1240:E13–8. doi: 10.1111/j.1749-6632.2011.06373.x. [DOI] [PubMed] [Google Scholar]
- 16.Blair HC, Robinson LJ, Zaidi M. Osteoclast signalling pathways. Biochem Biophys Res Commun. 2005;328:728–38. doi: 10.1016/j.bbrc.2004.11.077. [DOI] [PubMed] [Google Scholar]
- 17.Romas E, Bakharevski O, Hards DK, Kartsogiannis V, Quinn JM, Ryan PF, et al. Expression of osteoclast differentiation factor at sites of bone erosion in collagen-induced arthritis. Arthritis Rheum. 2000;43:821–6. doi: 10.1002/1529-0131(200004)43:4<821::AID-ANR12>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 18.Pettit AR, Ji H, Stechow von D, Müller R, Goldring SR, Choi Y, et al. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159:1689–99. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schett G. Effects of inflammatory and anti-inflammatory cytokines on the bone. Eur J Clin Invest. 2011;41:1361–6. doi: 10.1111/j.1365-2362.2011.02545.x. [DOI] [PubMed] [Google Scholar]
- 20.Radstake TRDJ, Sweep FCGJ, Welsing P, Franke B, Vermeulen SHHM, Geurts-Moespot A, et al. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 2005;52:3020–9. doi: 10.1002/art.21285. [DOI] [PubMed] [Google Scholar]
- 21.Oshima S, Onodera S, Amizuka N, Li M, Irie K, Watanabe S, et al. Macrophage migration inhibitory factor-deficient mice are resistant to ovariectomy-induced bone loss. FEBS Lett. 2006;580:1251–6. doi: 10.1016/j.febslet.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 22.Madeira MFM, Queiroz CM, Costa GM, Santos PC, Silveira EM, Garlet GP, et al. MIF induces osteoclast differentiation and contributes to progression of periodontal disease in mice. Microbes Infect. 2012;14:198–206. doi: 10.1016/j.micinf.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi T, Onodera S, Kondo E, Tohyama H, Fujiki H, Yokoyama A, et al. Impaired fracture healing in macrophage migration inhibitory factor-deficient mice. Osteoporos Int. 2011;22:1955–65. doi: 10.1007/s00198-010-1385-0. [DOI] [PubMed] [Google Scholar]
- 24.Onodera S, Sasaki S, Ohshima S, Amizuka N, Li M, Udagawa N, et al. Transgenic mice overexpressing macrophage migration inhibitory factor (MIF) exhibit high-turnover osteoporosis. J Bone Miner Res. 2006;21:876–85. doi: 10.1359/jbmr.060310. [DOI] [PubMed] [Google Scholar]
- 25.Jacquin C, Koczon-Jaremko B, Aguila HL, Leng L, Bucala R, Kuchel GA, et al. Macrophage migration inhibitory factor inhibits osteoclastogenesis. Bone. 2009;45:640–9. doi: 10.1016/j.bone.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 26.Mun SH, Won HY, Hernandez P, Aguila HL, Lee S-K. Deletion of CD74, a putative MIF receptor, in mice enhances osteoclastogenesis and decreases bone mass. J Bone Miner Res. 2013;28:948–59. doi: 10.1002/jbmr.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USa. 2003;100:9354–9. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cook AD, Turner AL, Braine EL, Pobjoy J, Lenzo JC, Hamilton JA. Regulation of systemic and local myeloid cell subpopulations by bone marrow cell-derived granulocyte-macrophage colony-stimulating factor in experimental inflammatory arthritis. Arthritis Rheum. 2011;63:2340–51. doi: 10.1002/art.30354. [DOI] [PubMed] [Google Scholar]
- 29.Aeberli D, Yang Y, Mansell A, Santos L, Leech M, Morand EF. Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett. 2006;580:974–81. doi: 10.1016/j.febslet.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 30.Wang C, Steer JH, Joyce DA, Yip KHM, Zheng MH, Xu J. 12-O-tetradecanoylphorbol-13-acetate (TPA) inhibits osteoclastogenesis by suppressing RANKL-induced NF-kappaB activation. J Bone Miner Res. 2003;18:2159–68. doi: 10.1359/jbmr.2003.18.12.2159. [DOI] [PubMed] [Google Scholar]
- 31.Singh PP, van der Kraan AGJ, Xu J, Gillespie MT, Quinn JMW. Membrane-bound receptor activator of NFκB ligand (RANKL) activity displayed by osteoblasts is differentially regulated by osteolytic factors. Biochem Biophys Res Commun. 2012;422:48–53. doi: 10.1016/j.bbrc.2012.04.103. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, et al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci USa. 2002;99:345–50. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leech M, Metz C, Hall P, Hutchinson P, Gianis K, Smith M, et al. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999;42:1601–8. doi: 10.1002/1529-0131(199908)42:8<1601::AID-ANR6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 34.Leech M, Metz C, Santos L, Peng T, Holdsworth SR, Bucala R, et al. Involvement of macrophage migration inhibitory factor in the evolution of rat adjuvant arthritis. Arthritis Rheum. 1998;41:910–7. doi: 10.1002/1529-0131(199805)41:5<910::AID-ART19>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 35.Onodera S, Oshima S, Nishihira J, Yasuda K, Tohyama H, Irie K, et al. Active immunization against macrophage migration inhibitory factor using a novel DNA vaccine prevents ovariectomy-induced bone loss in mice. Vaccine. 2008;26:829–36. doi: 10.1016/j.vaccine.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 36.Onodera S, Suzuki K, Matsuno T, Kaneda K, Kuriyama T, Nishihira J. Identification of macrophage migration inhibitory factor in murine neonatal calvariae and osteoblasts. Immunology. 1996;89:430–5. doi: 10.1046/j.1365-2567.1996.d01-751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toh M-L, Aeberli D, Lacey D, Yang Y, Santos LL, Clarkson M, et al. Regulation of IL-1 and TNF receptor expression and function by endogenous macrophage migration inhibitory factor. J Immunol. 2006;177:4818–25. doi: 10.4049/jimmunol.177.7.4818. [DOI] [PubMed] [Google Scholar]
- 38.Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest. 2008;118:2088–97. doi: 10.1172/JCI33392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura H, Hirata A, Tsuji T, Yamamoto T. Role of osteoclast extracellular signal-regulated kinase (ERK) in cell survival and maintenance of cell polarity. J Bone Miner Res. 2003;18:1198–205. doi: 10.1359/jbmr.2003.18.7.1198. [DOI] [PubMed] [Google Scholar]
- 40.Matsumoto M, Sudo T, Saito T, Osada H, Tsujimoto M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of NF-kappa B ligand (RANKL) J Biol Chem. 2000;275:31155–61. doi: 10.1074/jbc.M001229200. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Zhai Z, Liu G, Tang T, Lin Z, Zheng M, et al. Sanguinarine inhibits osteoclast formation and bone resorption via suppressing RANKL-induced activation of NF-κB and ERK signaling pathways. Biochem Biophys Res Commun. 2013;430:951–6. doi: 10.1016/j.bbrc.2012.12.051. [DOI] [PubMed] [Google Scholar]
- 42.Ralph JA, Ahmed AU, Santos LL, Clark AR, McMorrow J, Murphy EP, et al. Identification of NURR1 as a mediator of MIF signaling during chronic arthritis: effects on glucocorticoid-induced MKP1. Am J Pathol. 2010;177:2366–78. doi: 10.2353/ajpath.2010.091204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]
- 44.Greven D, Leng L, Bucala R. Autoimmune diseases: MIF as a therapeutic target. Expert Opin Ther Targets. 2010;14:253–64. doi: 10.1517/14728220903551304. [DOI] [PubMed] [Google Scholar]
- 45.Honma N, Koseki H, Akasaka T, Nakayama T, Taniguchi M, Serizawa I, et al. Deficiency of the macrophage migration inhibitory factor gene has no significant effect on endotoxaemia. Immunology. 2000;100:84–90. doi: 10.1046/j.1365-2567.2000.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohkawara T, Koyama Y, Onodera S, Takeda H, Kato M, Asaka M, et al. DNA vaccination targeting macrophage migration inhibitory factor prevents murine experimental colitis. Clin Exp Immunol. 2011;163:113–22. doi: 10.1111/j.1365-2249.2010.04277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herrero LJ, Nelson M, Srikiatkhachorn A, Gu R, Anantapreecha S, Fingerle-Rowson G, et al. Critical role for macrophage migration inhibitory factor (MIF) in Ross River virus-induced arthritis and myositis. Proc Natl Acad Sci USa. 2011;108:12048–53. doi: 10.1073/pnas.1101089108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santos LL, Dacumos A, Yamana J, Sharma L, Morand EF. Reduced arthritis in MIF deficient mice is associated with reduced T cell activation: down-regulation of ERK MAP kinase phosphorylation. Clin Exp Immunol. 2008;152:372–80. doi: 10.1111/j.1365-2249.2008.03639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bucala R. MIF, MIF alleles, and prospects for therapeutic intervention in autoimmunity. J Clin Immunol. 2013;33(Suppl 1):S72–8. doi: 10.1007/s10875-012-9781-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hertig A, Rondeau E. Role of the coagulation/fibrinolysis system in fibrin-associated glomerular injury. J Am Soc Nephrol. 2004;15:844–53. doi: 10.1097/01.asn.0000115400.52705.83. [DOI] [PubMed] [Google Scholar]
- 51.Quinn JMW, Sims NA, Saleh H, Mirosa D, Thompson K, Bouralexis S, et al. IL-23 inhibits osteoclastogenesis indirectly through lymphocytes and is required for the maintenance of bone mass in mice. J Immunol. 2008;181:5720–9. doi: 10.4049/jimmunol.181.8.5720. [DOI] [PubMed] [Google Scholar]
- 52.Chen L, Wei X-Q, Evans B, Jiang W, Aeschlimann D. IL-23 promotes osteoclast formation by up-regulation of receptor activator of NF-kappaB (RANK) expression in myeloid precursor cells. Eur J Immunol. 2008;38:2845–54. doi: 10.1002/eji.200838192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. The effects of exogenous rMIF (100 ng/ml) addition on osteoclast formation in vitro in M-CSF (30 ng/ml) and RANKL (50 ng/ml) treated bone marrow cells. A: Osteoclast formation in WT cells with and without rMIF (100ng/ml) treatment. B: Osteoclast formation in RAW 264.7 cells with and without rMIF (concentration as indicated) treatment. C: Effect of rMIF on NF-κB reporter activity in RANKL (100 ng/ml, 6h)-treated stably transfected RAW cells. D: as C, but using RAW264.7 cells stably transfected with luciferase NFAT reporter. All values were expressed as mean ±SEM. Experimental results were pooled from 3 independent experiments.
Supplementary Figure 2. A: Actin ring formation in WT and MIF-/- bone marrow cells. Cells were stimulated with M-CSF (30 ng/ml) plus RANKL (50 ng/ml) for 7 days. Actin was visualised with phalloidin-rhodamine in cells counterstained with DAPI and ring formation was quantified. B: Dose-dependent effects of M-CSF on osteoclastogenesis in WT and MIF-/- bone marrow cells. Survival of in vitro generated osteoclasts from WT (C) and MIF-/- (D) mice, upon withdrawal of RANKL, M-CSF or both. Experimental results were pooled from 5 independent experiments. All values were expressed as mean ±SEM. *p<0.05, *** p<0.001 WT versus MIF-/-.
Supplementary Figure 3. WT bone marrow cells were treated with M-CSF (30 ng/ml) and RANKL (50 ng/ml) for 7 days, and with the MEK inhibitor PD98059 (PD; 10 μM) or DMSO vehicle (Veh).