

Abstract
Epidemiologic studies have shown that diabetics receiving the biguanide metformin, as compared to sulfonylureas or insulin, have a lower incidence of breast cancer. Metformin increases levels of activated AMPK and decreases circulating IGF-1; encouraging its potential use in both cancer prevention and therapeutic settings. In anticipation of clinical trials in non-diabetic women, the efficacy of metformin in non-diabetic rat and mouse mammary cancer models was evaluated.
Metformin was administered by gavage or in the diet, at a human equivalent dose, in standard mammary cancer models: (1) methylnitrosourea (MNU)-induced ER+ mammary cancers in rats, and (2) MMTV-Neu/P53KO ER- mammary cancers in mice.
In the MNU rat model, metformin dosing (150 or 50 mg/Kg BW/day, by gavage) was ineffective in decreasing mammary cancer multiplicity, latency, or weight. Pharmacokinetic studies of metformin (150 mg/kg BW/day, by gavage) yielded plasma levels (Cmax and AUC) higher than humans taking 1.5 g/day. In rats bearing small palpable mammary cancers, short-term metformin (150 mg/kg BW/day) treatment increased levels of phospho-AMPK and phospho-p53 (Ser20) but failed to reduce Ki67 labeling or expression of proliferation-related genes. In the mouse model, dietary metformin (1500 mg/kg diet) did not alter final cancer incidence, multiplicity, or weight.
Metformin did not prevent mammary carcinogenesis in two mammary cancer models; raising questions about metformin efficacy in breast cancer in non-diabetic populations.
Keywords: Metformin, Prevention, Mammary Cancer
Introduction
Population studies of Metformin
Metformin (1,1-dimethylbiguanide) is an anti-hyperglycemic drug prescribed for the management of type 2 diabetes (1), and is used worldwide by approximately 120 million people (2).Recently, there has been great interest in the use of metformin as a potential chemopreventive/therapeutic agent based on epidemiologic studies showing a lower incidence of cancer and lower toxicity in diabetics taking this drug compared to those taking sulfonylurea or insulin (3,4). In 1995, Evans, et al (3) reported an association of metformin use with reduced cancer risk in diabetic populations. Among the cancers shown to be lower was breast cancer (5,6). These findings helped support a large ongoing clinical trial of metformin in a therapeutic adjuvant setting for breast cancer (7,8).
Molecular, cellular, and physiological studies with Metformin
One area of current research is focused on how metformin alters energy metabolism in cancers. Metformin clearly leads to decreased liver gluconeogenesis and reduced circulating glucose and insulin in diabetics, although the mechanism is unclear. Metformin is thought to inhibit complex I in the electron transport chain in the inner mitochondrial membrane (9); increasing the AMP/ATP ratio in the cell and stimulating AMP-activated protein kinase (AMPK). Dependent on liver kinase B1 (LKB1) activity (10), AMPK is a primary intracellular energy sensor (11). Activated AMPK inhibits the mammalian target of rapamycin (mTOR) signaling network to decrease cell proliferation and increase apoptosis. However, understanding mTOR is complicated by the fact that it modulates multiple intracellular and extracellular pathways, therefore defining an exact mechanism may be complex (12). Furthermore, Metformin may act indirectly at cancer sites by altering liver metabolism and reducing systemic risk factors (e.g. glucose, insulin, IGF-1 signaling), or metformin may act directly to influence cancer cell metabolism at the target site. Though the role of LKB1/AMPK in the activity of metformin is convincing, published studies have led to multiple proposed alternative mechanisms of action such as altered glucagon signaling or altered growth factor signaling via receptor tyrosine kinases (13,14). For breast cancer, the two major pathways that have been routinely discussed are: (a) activation of AMP kinase secondary to the primary effect on LKB1 (11-13) altering the expression of various genes involved in gluconeogenesis; and (b) metformin reducing levels of IGF1 a mechanism which may be relevant for a variety of cancers (15).
Animal studies with Metformin
Although numerous publications on metformin have focused on cell culture studies (1,16), in vivo animal data employing in situ arising mammary cancer models are more limited (17,18). We examined the effects of metformin in three animal models of breast cancer routinely used to screen for chemopreventive agents. In all three studies we employed standard rodent chow. The specific models were: (a) the MNU-induced model of ER+ mammary cancer in rats. These tumors respond to the same hormonal agents observed in ER+ human breast cancer (19,20), and are similar by array analysis to well differentiated human ER+ tumors (21); (b) the MMTV-Neu/P53KO model of ER- mammary cancer in mice. This is a model of ER- Neu over-expressing cancers in humans which overwhelmingly have P53 alterations (22,23); and (c) a therapeutic xenograft assay with cells derived from the C31 T antigen model of breast cancer, which by array analysis appears similar to the human basal/triple negative subtype of breast cancer (24). The data for this therapeutic model which employed a different mode of agent delivery are presented as supplementary material in this manuscript. In all three assays, metformin failed to inhibit cancer formation or growth. Pharmacokinetic data in the MNU rat model showed that metformin achieved plasma levels slightly higher than those achieved at a standard dose in humans, and did modulate biochemical endpoints such as AMP kinase levels and phosphorylation of P53. Finally, short-term treatment with metformin failed to decrease Ki67 levels by IHC, and failed to decrease the expression of multiple proliferation related genes (Ki67, ccnb1, Top2A) determined by RT-PCR in MNU mammary cancers of rats, whereas similar treatment with tamoxifen strikingly decreased Ki67 levels. These latter studies showed that metformin was ineffective as a chemopreventive agent despite achieving substantial plasma levels and altering known pharmacodynamic endpoints e.g. phosphorylated P53.
Materials and Methods
MNU-induced rat mammary cancer model
The MNU-induced rat mammary cancer model was performed as previously described (20,23). In brief, female Sprague-Dawley rats (Harlan Sprague-Dawley, Inc., Indianapolis, IN), were placed on standard Teklad diet (4% fat by weight; 8% fat by caloric intake) and administered the carcinogen MNU (75 mg/kg BW) by IV injection at 50 days of age. Five days after MNU, the animals were dosed by gavage with metformin; 150 or 50 mg/kg BW/day in saline. The animals received the treatment until termination of the study at 126 days after MNU injection. The rats were weighed 1×/week, palpated for mammary tumors 2×/week, and observed daily for signs of toxicity. Mammary tumors were excised, weighed, and processed for histological classification at the termination of the study. Differences in latency and cancer weights were determined by log rank analysis and Mann-Whitney, respectively.
Short-term biomarker determination
A pre-surgical animal model was used to examine the effect of metformin on pharmacodynamic biomarkers. This model most closely approximates the pre-surgical intervention that has often been employed in clinical breast cancer trials (25). For determination of biomarkers (IHC or RT-PCR), animals bearing small MNU-induced ER+ mammary cancers were treated for 7 days with metformin (150 mg/Kg BW/day), or tamoxifen (100 mg/kg of diet) as a positive control. Animals were sacrificed and tumors were either snap frozen (RT-PCR), placed in Zamboni's fixative (IHC), or fixed in formalin for 24 hours.
Immunohistochemistry and Dapi staining
Mammary cancers were harvested and fixed in Zamboni's fixative and embedded in Optimal Cutting Temperature Compound (Tissue Tek, Sakura Finetex USA, Inc. Torrance, CA). Samples were washed with TPBS (1xPBS-0.3% Triton X-100) and blocked overnight with 5% normal donkey serum TPBS. Primary antibodies were prepared in 1% normal donkey serum TPBS and incubated for 7 h with samples at room temperature and then washed with TPBS overnight. Samples were washed with additional fresh TPBS for 1 h. Secondary antibodies were prepared using 1% normal donkey serum TPBS and added to each well. Secondary antibodies were left on the samples 5 h at room temperature, washed in TPBS overnight, and washed again in PBS a second overnight. After secondary antibody staining/washing, Dapi was diluted in PBS at 1:50,000. The samples were incubated for 30 minutes followed by several washes in PBS with gentle gyro-rotatory shaking for one hour. After washing was complete, samples were mounted on coverslips using 1.35%-1.5% Noble agar, then dehydrating using progressively greater percentages of alcohol, cleared with xylene and mounted on coverslips using cytoseal. All samples were attached to coverslips and allowed to set at room temperature until dry (1-5 min), and mounted onto slides using Cytoseal XYL (Richard-Allan Scientific, 8312-4). Slides were examined with a Nikon Confocal Microscope. Antibodies Employed: Primaries: p-Akt (Ser473) Rb - Cell Signaling: 4060S, Lot #14 p53 Total Mo – Cell Signaling: 2524, Lot #4 p-AMPK (Tyr172) Rb – Santa Cruz: sc-33524, Lot #C1011 p-p53 (Ser392) Gt – Santa Cruz: sc-7997, Lot #L1809 p-mTOR (Ser2448) Rb – Cell Signaling: 5536S, Lot #1 p-p53 (Ser20) Gt – Santa Cruz: sc-18078, Lot #C0510; Secondaries: Dylight488 Gt – Jackson Immuno Research: 705-485-147, Lot #91400 Cy3 Rb- Jackson-Immuno Research: 711-165-152, Lot #97894 Cy5 Mo – Jackson Immuno Research: 715-176-151, Lot #76886.
Proliferation by RT-qPCR
Sample preparation and first-strand cDNA synthesis
MNU-induced mammary tumors from non-treated, tamoxifen-treated, and metformin-treated (n=10) rats were processed as formalin-fixed, paraffin embedded tissue blocks. Two 5 μscrolls were taken from each tumor block and total RNA was extracted using the Roche High Pure miRNA Isolation Kit (Roche Applied Science, Indianapolis, IN). The quantity of RNA was assessed using the NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Inc., Rockland, DE). First-strand cDNA was synthesized from 600 ng RNA using a 2 pmol mixture of reverse primers, 100 ng of random hexamers, and Superscript III reverse transcriptase (1st Strand Kit; Invitrogen, Carlsbad, CA). The reaction was held at 55°C for 60 minutes followed by a 15-minute step at 70°C. The cDNA was washed on a QIAquick PCR purification column (Qiagen Inc., Valencia, CA) and was stored at -80°C in TE (10mM Tris-HCl, pH 8.0, 0.1 mM EDTA) until qPCR analysis.
PCR and relative quantification
Primer sets were selected to have similar GC contents and Tms using the LightTyper Probe Design software (version: 2.0.B.22) (Roche Applied Science, Indianapolis, IN). Each primer set was validated using a pool of cDNA from different mammary cancers processed as formalin fixed, paraffin embedded tissue blocks. Five-μl PCR reactions were prepared in 384-well plates using a robotic Evolution P3 Precision Pipetting Platform (PerkinElmer Ltd, Shelton, CT). Each reaction contained 2.5 μL Roche LC 480 SYBR Green I Master Mix 2×, 0.4 μM of each primer, and 1.25 ng cDNA. A pool of cDNA from rat mammary cancers was provided in each run as a calibrator reference and assigned a value of 10 ng for each gene. Samples and calibrator were run in duplicate. PCR amplification was performed using the LC480 (Roche Applied Science, Indianapolis, IN) with an initial denaturation step (95°C, 8 minutes) followed by 45 cycles of denaturation (95°C, 4 seconds), annealing (56°C, 6 seconds with 2.5°C/s transition), and extension (72°C, 6 seconds with 2°C/sec transition). Fluorescence (530 nm) from the SYBR® Green I dsDNA dye was acquired for each cycle after the extension step. The specificity of the PCR was determined by post-amplification melting curve analysis. Reactions were automatically cooled to 65°C and slowly heated at 2°C/s to 99°C while continuously monitoring fluorescence (10 acquisitions/1°C). Relative copy numbers were calculated from an external standard curve (efficiency 1.8) and correcting to the Cp of the 10ng calibrator. To control for variation in RNA quality, the copy number for each gene was further normalized to actb, which served as a housekeeper gene. Values from replicate samples were averaged and data were log2 transformed. The primers employed for the RT-PCR studies were ccnb1 R3F gaggtggaactggatgag ccnb1 R3R ggcttggagagggagtatcaac; Mki67 R3F gtctcttggcactcacag Mki67 R3R tggtggagttactccaggagac; Top 2A R1F cctccggcagagactagagaTop 2A R1R ttgcttctttgccctggta.
Pharmacokinetic Studies
Female Sprague-Dawley rats were administered metformin (150 mg/kg BW/day) by gavage for 14 days. Plasma samples were collected on Days 1 and 14. Metformin concentrations in plasma were determined using an LC/MS/MS method as modified from Tucker et al (26) (See Supplementary Materials). Plasma concentration-time data were analyzed by non-compartmental methods using the program WinNonlin Version 4.1 (Pharsight Corporation, Mountainview, CA). Additional materials can be found in the supplemental information (e.g. Supplementary Fig 1).
MMTV-Neu/P53KO ER- mouse mammary cancer model
MMTV-Neu+/-/p53 KO+/- mice were generated and maintained in IACUC approved laboratories at the University of Alabama at Birmingham as previously described (23).MMTV-Neu transgenic mice (strain FVB/N-Tg (MMTV-Neu) 202 Mul/J) were purchased from the Jackson Laboratory. A p53 deficient line (p53 NS-T) was purchased on a C57BL background (Taconic) and back-crossed at least five times onto a FBV/N background. For generation of MMTV-Neu+/-/p53 KO+/- females, p53-/- males were crossed to MMTV-Neu+/+ females. MMTV-Neu females were generated by crossing MMTV-Neu+/+ males with FVB/NJ females. The animal rooms were maintained at 22 ± 2°C and lighted 12h/day in a facility specially designed for administering chemical carcinogens to animals. Animals were allowed free access to the diet (Teklad 4% mash) and water throughout the duration of the experiments. Mice were placed on control diet or diet containing 1500 mg metformin/kg of diet beginning at 60 days of age. Metformin was obtained from the National Cancer Institute (NCI) Prevention Repository and incorporated into the mash diet by mixing with a liquid-solid blender (Patterson-Kelly Co). Mice were examined weekly for the development of palpable mammary tumors beginning at 4 months of age. Mice were kept on the control or metformin-containing diets until approximately 11 months of age.
ER negative mouse mammary tumor orthotopic model (Supplementary Figure 2)
Mammary tumor cells derived from female C3(1)Tag tumor bearing mice (24) were implanted into the mammary fat pads of female SCID mice. Once tumors reached 125 mg mice were treated with metformin (100 or 150 mg/KgBW) by i.p. injection in saline (0.1 ml/10 grams of body weight) daily for a period of 14 days. Neither dose significantly inhibited the growth of the C3(1)/Tag tumor xenografts.
Results
Effects of Metformin on ER+ Mammary Cancers
Two different doses of metformin (150 or 50 mg/kg BW/day) were administered by gavage to rats beginning 5 days after MNU administration; with the higher dose being equivalent to a human dose of 1.5 g/day based on the FDA human equivalent dosing calculation. Neither dose significantly altered tumor latency (Figure 1A) or multiplicity (Figure 1B). However, metformin was associated, with a slight increase [0.1>P>.05] in the final tumor weights at the end of the study (Fig 1C).
Figure 1. Effects of Metformin on the Development of MNU-Induced ER+Mammary Cancers.
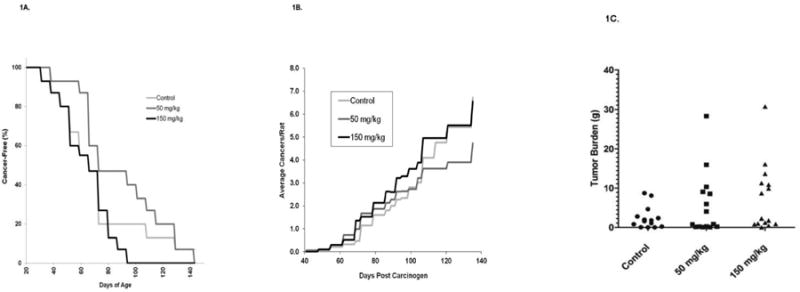
Female Sprague-Dawley rats were administered MNU at 50 days of age. Rats were administered daily metformin (150 or 50 mg/kg BW/day, by gavage) beginning 5 days after MNU. (A) Development of Palpable Cancers. Rats were examined 2×/week for palpable cancer development. Differences in cancer latency were not significantly different (P>.05). (B) Effects of metformin on cancer multiplicity. (C) Effects of metformin on final cancer weights. Tumors from individual rats were weighed at the termination of the study (140 days post MNU). The cancer weights were not significantly higher in the groups treated with metformin when compared to control rats.
Effects of Metformin in the ER- mouse transgenic model
Metformin administered in the diet (1500 mg/kg) to MMTV-Neu/heterozygous P53/KO mice failed to alter survival of the mice (Figure 2A), tumor multiplicity (Fig 2B) or decrease tumor weights of treated mice (Figure 2C). Metforminin an ER- mouse mammary tumor xenograft model (Supplementary Figure 2) was administered at 100 or 150 mg/KgBW i.p., twice daily. Neither dose significantly inhibited the growth of the C3(1)/Tag tumor xenografts.
Figure 2. Effects of Metformin on the Development of Mammary Carcinomas in MMTV-Neu/P53KO mice.
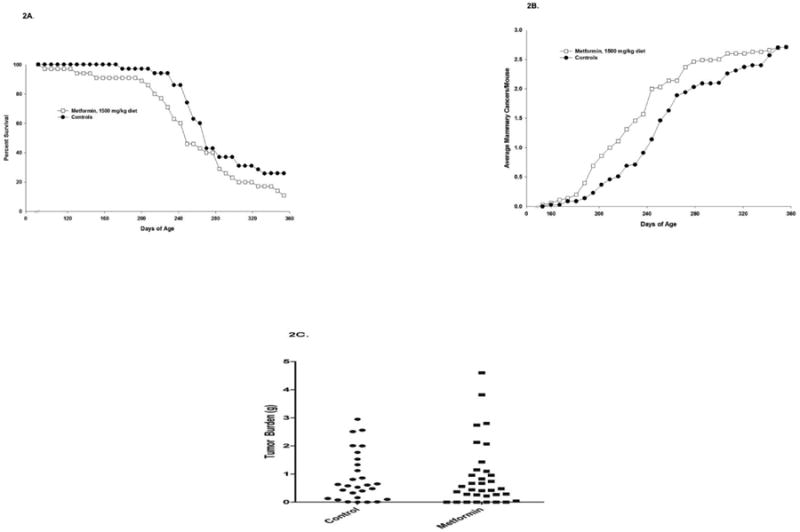
Weanling mice were characterized for the presence both the MMTV-Neu transgene and the KO of one copy of the tumor suppressor gene p53. Mice were placed on control diet or diet containing 1500 mg/kg metformin at 60 days of age. Metformin did not significantly increase survival (P<.05) (A), and also did not impact incidence, multiplicity, (B), or cancer weight (C).
Biomarker studies on Metformin in MNU rat model
As can be seen in Figure 3A and B, a limited (albeit statistically significant) increase in phospho-AMPK was observed in the tumors of rats treated for 7 days with metformin. Interestingly, a substantial increase in the levels of phosphorylated p53 at serine 20 and a smaller increase at position 392 were observed, along with greatly increased levels of phosphorylated p70S6 kinase at threonine 389. Upon further examination of levels of p53 phosphorylation in MCF cells treated in cultures with metformin, p53 was similarly phosphorylated at these two sites confirming the in vivo results (Supplementary Figure 3). We also examined the effects of metformin on the proliferation of metformin-treated tumors employing Ki67. Treatment with metformin did not decrease the proliferation index (∼30% increase, 0.1<P < 0.05) (Figure 4A). In contrast, tamoxifen, a highly effective preventive agent, decreased the Ki67 index by roughly 75% (P < 0.05). Finally, the effect of metformin on proliferation-associated genes (Figure 4B) was examined. Gene expression of the proliferation related genes mki67, ccnb1 and top2a was decreased by tamoxifen but not metformin, in agreement with the effects on Ki67 observed with IHC.
Figure 3. Effects of Metformin on Expression of Multiple Biomarkers Employing IHC.

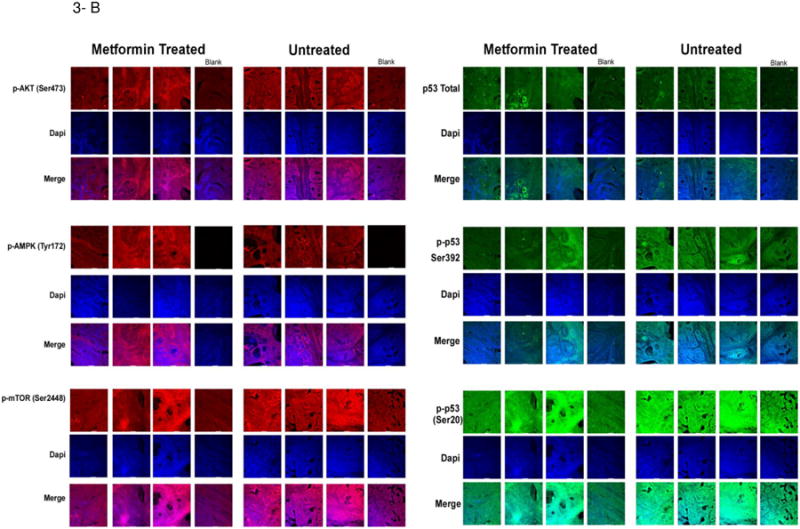
Rats bearing MNU-Induced cancers were treated with vehicle or metformin (150 mg/kg BW/day) for a period of 7 days. At that time, rats were sacrificed and tumors removed. Tissue slices were prepared and stained as described in Material and Methods. Representative staining is shown. (A) Shows alterations of expression in the various phosphorylated proteins in control mammary tumors following short term metformin exposure; (B) Shows IHC slides staining for the various phosphoproteins, DAPI and a merged image.
Figure 4. Effects of Metformin on Expression of Proliferation Related Biomarkers.
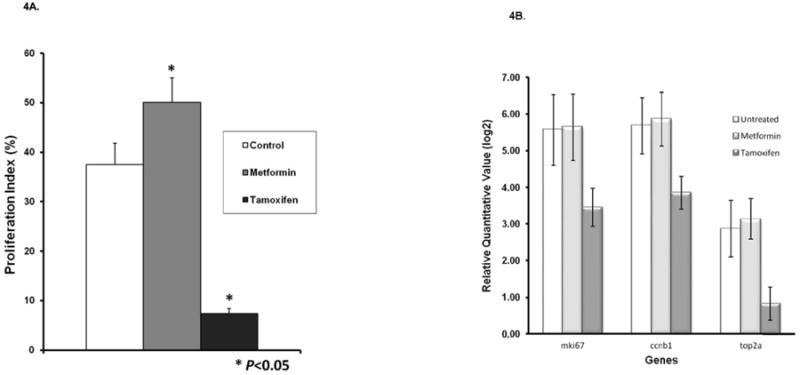
Rats were treated as described in Materials and Methods. Both expression of proliferation related genes and Ki67 values were determined in formalin-fixed samples. (A) Effects of metformin and a positive control (tamoxifen) on proliferation index in mammary cancers. Results are based on counting at least 2,000 cells for each of the tumors. The Ki67 labeling index was significantly increased in cancers treated with metformin and decreased in cancers treated with tamoxifen (P<.05) when compared with controls. (B) Effects of metformin and tamoxifen on expression of proliferation related genes.
Pharmacokinetic studies with Metformin in female Sprague-Dawley rats
Plasma concentrations were measured in female Sprague-Dawley rats on day 1 and day 14 after treatment with 150 mg/kgBW/day by gavage. Metformin plasma profiles and pharmacokinetic data are presented in Supplementary Figure 1 and Supplementary Table 1 and 2. Mean plasma concentrations of 7.47 ± 0.79 μg/ml and 6.72 ± 0.39 μg/ml were achieved 1 hour after the oral dose on day 1 and day 14, respectively (Table 1). The steady-state (AUC0-24h) value of 44.6 μg/ml×hr on day 14 was similar to the AUC0-24h value of 47.7 μg/ml×hr on day 1, indicating the drug did not accumulate in plasma during the daily administration schedule.
Table 1. Mean Plasma Concentration-Time Data and Pharmacokinetic Estimates.
| Time (h) | Group 1 (Day 1) | Group 2 (Day 14) |
|---|---|---|
| Mean Plasma Concentration (ng/ml) (± SD) | Mean Plasma Concentration (ng/ml) (± SD) | |
| 0 | 0 | 97 ± 42 |
| 1 | 7470 ± 790 | 6720 ± 390 |
| 2 | 7080 ± 1490 | 4610 ± 1090 |
| 4 | 4920 ± 1410 | 5030 ± 1240 |
| 8 | 1430 ± 570 | 1470 ± 480 |
| 24 | 70 ± 14 | 129 ± 44 |
| Pharmacokinetic Estimates | ||
|
| ||
| Tmax (h) | 1 | 1 |
| Cmax (ng/ml) | 7470 | 6720 |
| Half-life (h) | 3.7 | 4.6 |
| AUC0-24h (ng/ml × h) | 47670 | 44560 |
Discussion
Metformin, an anti-diabetic, has generated a great deal of interest in recent years as a potential preventive and therapeutic agent (1,2). Part of the initial interest was based on epidemiologic studies showing that individuals taking metformin (as contrasted with other anti-diabetic agents) had a lower incidence of cancer; although certain recent epidemiologic studies have not confirmed this observation. Among the cancers shown to be lower was breast cancer (5,6). Although certain of the more recent epidemiologic data has shown more limited efficacy. There has been great enthusiasm for the use of metformin because of its relatively low toxicity, and the fact that it alters energy metabolism which is felt to be altered in most cancers. Furthermore, the main mechanistic alterations induced by metformin are: (a) effects on LKB1, which secondarily effects activity of cyclic AMP kinase and the mTOR pathway and/or (b) effects on the IGF1 pathway (15). Both of these pathways would appear relevant for a wide variety of cancers. In vitro studies have confirmed the expected mechanistic results and show metformin is preferentially effective in tumor cells (1,16). There have also been a variety of xenograft studies (27). However, there have been relatively few studies employing in situ arising mammary cancers that are typically employed in prevention studies. Given this limited number of studies in prevention models, we examined the use of metformin in two standard mammary cancer prevention models, and in a graft model more closely approximating the basal subtype of breast cancer. The MNU model of mammary cancer which develops ER+ cancers appears by gene array analysis to be similar to highly differentiated ER+ mammary cancers in humans (21). The model has been shown to be sensitive to hormonal manipulations that decrease ER+ tumors in women including SERMs, aromatase inhibitors, and ovariectomy (19,20). We failed to observe any preventive activity of metformin at a dose comparable to standard human doses. The higher dose was chosen to be the equivalent of a human dose of approximately 1.5 g/day based on standard FDA scaling factors. Some what surprisingly, we found that the Cmax and AUC values (Table 1) of this metformin dose were roughly 2-3 folds higher than the Cmax (3.1 ± 0.93 μg/ml) and AUC (18.4 ± 6.5 μg/ml×hr) values for a dose in humans of 1.5 g/day (26). Despite the fact the PK data were higher than that achieved in humans; no cancer preventive activity in the rat model was observed. Of note, the peak concentration of metformin achieved in plasma of approximately 7.5 μg/mL, equivalent to a concentration of 60μM, is markedly less than a media concentration of ≥1mM (and more typically 2.5-10 mM) employed in cell culture to achieve efficacy [16] (Supplementary Figure 2). We observed qualitative increases in final tumor weights at both doses of metformin, which was marginally significant at the 150mg/Kg BW/day dose (.05<P<.075). A prior study by Zhu, et al (17) in MNU treated rats with a modified method similarly failed to significantly inhibit tumor formation until doses > 3× higher were employed.
We employed two mouse models of breast cancer to test the efficacy of metformin. The first model was the MMTV-Neu P53/KO model which develops ER- tumors with an altered P53 and over expression of Neu (22,23). This is similar to ER- Neu over expressing tumors in humans which similarly show over expression of Neu and mutations in P53. The Neu model has been shown to be sensitive to the preventive effects of both EGFr inhibitors and RXR agonists (28,29); which can profoundly increase latency and decrease tumor incidence. In the MMTV-Neu model, metformin was administered in the diet at a dose similar to the human equivalent dose. The reason we employed diet for the mouse study was due to the practical limitations of administering metformin by gavage for an extended time period. The dose employed (1500 mg/kg diet) is in line with the human equivalent dose of 1.5 g/day employing the standard scaling factors. The results show that metformin failed to demonstrate any preventive activity (Figures 2A, B). There was a prior report showing a significant, albeit not very striking, effect of metformin at a similar dose when given in water (30). The pharmacokinetics of the two is likely to be somewhat different. However, even the metformin data in the prior study, although administered early in life, was only marginally effective; increasing tumor latency by only 7%. This must be viewed in the context of highly effective agents which can increase latency more than 60% and strongly decrease tumor incidence (28,29). We also performed a more limited therapeutic study of metformin employing a tumor graft derived from a transgenic C31 T antigen mouse (31) and administered to immunosuppressed SCID mice. These tumors appear by array analysis to be quite similar to basal type breast cancer in humans (24). This study (Supplementary Figure 2) examined the therapeutic effects of metformin following i.p. administration; no efficacy was observed. The i.p. dose was employed because a lung cancer prevention model had found it to be highly effective (32); although administration by this route is unlikely for aprevention study in humans. We have examined the efficacy of metformin in additional in situ models in other organs including colon, head and neck and urinary bladder and failed to achieve positive results in animals on a standard chow diet (data not shown; Lubet and Grubbs). There is, however, a preclinical study in a pancreatic model which achieved strong efficacy after dietary administration of metformin at doses similar to those we employed in the MMTV-Neu/P53KO model which was intended to parallel the human equivalent dose (33).
We determined whether the treatments employed altered potential biomarkers in the MNU model (25). The model parallels a pre-surgical model in humans. Specifically, a small palpable ER+ tumor is allowed to develop and the rat was treated short-term with metformin (or tamoxifen as a positive control) (Figure 4A). We first looked at Ki67 labeling in cancers treated short-term with metformin, and found that metformin increased proliferation in the treated tumors. In contrast, tamoxifen (which greatly decreases tumor incidence) decreased Ki67 roughly 75%. These studies must be seen in the context of our prior studies which showed that a wide variety of agents which inhibit mammary cancer formation (SERMS, aromatase inhibitors, EGFR inhibitors, RXR agonists etc.) inhibit proliferation, while ineffective agents do not (25). The effects of metformin and tamoxifen were then examined for their effects on expression of Ki67, ccnb1 and Top2A by RT-PCR (Figure 4B). The specific genes employed were initially defined as part of a set of genes associated with proliferation in human breast cancer (34,35). Genes involved in cell cycle regulation (i.e., proliferation) are prognostic for determining risk of recurrence in women with ER+ breast cancer receiving endocrine therapy alone (34-36). We selected 3 proliferation genes (MKI67, CCNB1, and TOP2A) that are important for prognosis in ER+ human breast tumors, and determined their change in expression in ER+ cancers in the rat after treatment with tamoxifen or metformin. The proliferation markers MKI67 and CCNB1 are both included in the risk algorithms for OncotypeDx and Prosigna (i.e PAM50) (34-36). Topoisomerase 2α protein expression is highly correlated with Ki-67 protein expression in breast cancer and may provide additional predictive information in response to anthracycline regimens (37-39). The effects of metformin on AMP kinase and a number of phosphorylated proteins involved in cell cycling (Figure 3) were also evaluated. A limited, but significant, increase in AMP kinase was observed. We also saw an increased phosphorylation of P53 at serines 20 and 392 sites. Unexpectedly, an increased level of P30S6 kinase, a ribosomally associated kinase, was also seen. Increased levels of the latter might be expected with an agent that causes increased proliferation of tumors (as shown in Figure 4A). It was observed almost 10 years ago that P53 is a substrate for AMPK. In fact, P53 (via downstream proteins) significantly contributes to the overall response to gluconeogenesis and may induce autophagy (38-40). We subsequently determined whether phosphorylation was increased at these sites in MCF-7 cells exposed in cell culture to metformin (Supplementary Figure 3). Similar increases in these P53 phosphorylation sites in MCF-7 treated cells occurred. However, the doses employed in vitro (>1mM)are much higher than the Cmax achieved in plasma either in our studies or in human studies. These changes are obviously not efficacy biomarkers, since metformin was ineffective as a preventive agent in this model. Nevertheless, they may be pharmacodynamic biomarkers indicating that metformin has reached the target organ and has a physiologic effect. One of the more interesting aspects of metformin efficacy was an early paper by Thompson and colleagues (38,40) showing metformin was preferentially active in cells with a knockout of P53. Although our data does not include animal cancers with a clear P53 mutation or knockout P53, the MMTV-Neu ER- mammary cancer model is heterozygous for P53 deletion and is likely to lose the second copy of P53 (16).
Our lack of efficacy in commonly employed mammary cancer models is disconcerting. These results argue for testing agents employing standard prevention models at doses close to the human equivalent dose employing the same route of administration. Our PK data show that reasonable doses were administered. These results encourage clinical trials at the Phase IIA levels using biomarkers more directly related to preventive efficacy (41,42) and not purely biochemical parameters (activated AMP kinase) as the primary endpoint. In fact, there were two recent biomarker studies in breast cancer employing metformin, with proliferation as a potential biomarker. While the smaller trial that lacked a placebo control yielded positive results (41), the larger trial was negative and, in fact, observed an increase in proliferation index in women with a BMI <25 (42); similarly to our present data (Figure 4A). It is hoped that this will temper enthusiasm for large Phase II or III trials in the absence of clear Phase IIA data; particularly in the more general population. Perhaps the greatest clinical question deals with the ongoing Phase III adjuvant trial employing metformin in an adjuvant setting at a dose of 1.7 g/day; which is close to the dose we employed (7,8). The majority of these participants are likely either to have ER+ and/or Neu over expressing tumors; where our present data looks clearly ineffective. This trial specifically precludes the inclusion of diabetics where the epidemiologic data looks somewhat promising. It is emphasized that the present data is in animals that were on a standard diet; i.e., animals were neither diabetic nor pre-diabetic. It certainly is possible that in a model with the altered physiology associated with diabetes or pre-diabetes one may observe preventive activity.
Supplementary Material
Acknowledgments
Funding Source: NCI Contract Number HHSN261201200021I awarded to Dr. Clinton J. Grubbs, University of Alabama at Birmingham
Footnotes
There is no Conflict of Interest with any Authors.
References
- 1.Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2:778–90. doi: 10.1158/2159-8290.CD-12-0263. [DOI] [PubMed] [Google Scholar]
- 2.Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011;9:33. doi: 10.1186/1741-7015-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PloS One. 2012;7:e33411. doi: 10.1371/journal.pone.0033411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chlebowski RT, McTiernan A, Wactawski-Wende J, Manson JE, Aragaki AK, Rohan T, et al. Diabetes, metformin, and breast cancer in postmenopausal women. J Clin Oncol. 2012;30:2844–52. doi: 10.1200/JCO.2011.39.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosco JL, Antonsen S, Sorensen HT, Pedersen L, Lash TL. Metformin and incident breast cancer among diabetic women: a population-based case-control study in Denmark. Cancer Epidemiol Biomarkers Prev. 2011;20:101–11. doi: 10.1158/1055-9965.EPI-10-0817. [DOI] [PubMed] [Google Scholar]
- 7.Goodwin PJ, Stambolic V, Lemieux J, Chen BE, Parulekar WR, Gelmon KA, et al. Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat. 2011;126:215–20. doi: 10.1007/s10549-010-1224-1. [DOI] [PubMed] [Google Scholar]
- 8.Niraula S, Dowling RJ, Ennis M, Chang MC, Done SJ, Hood N, et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat. 2012;135:821–30. doi: 10.1007/s10549-012-2223-1. [DOI] [PubMed] [Google Scholar]
- 9.Falcone AB, Mao RL, Shrago E. A study of the action of hypoglycemia-producing biguanide and sulfonylurea compounds on oxidative phosphorylation. J Biol Chem. 1962;237:904–9. [PubMed] [Google Scholar]
- 10.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–46. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homoestasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signaling by decreasing production of cyclic AMP. Nature. 2013;494:256–60. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–69. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia; an update. Nat Rev Cancer. 2012;12:159–69. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 16.Gallagher EJ, LeRoith D. Diabetes, cancer, and metformin: connections of metabolism and cell proliferation. Ann N Y Acad Sci. 2011;1243:54–68. doi: 10.1111/j.1749-6632.2011.06285.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Z, Jiang W, Thompson MD, McGinley JN, Thompson HJ. Metformin as an energy restriction mimetic agent for breast cancer prevention. J Carcinog. 2011;10:17. doi: 10.4103/1477-3163.83043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bojkova B, Orendas P, Garajova M, Kassayova M, Kutna V, Ahlersova E, et al. Metformin in chemically-induced mammary carcinogenesis in rats. Neoplasma. 2009;56:269–74. doi: 10.4149/neo_2009_03_269. [DOI] [PubMed] [Google Scholar]
- 19.Gottardis MM, Jordan VC. Antitumor actions of keoxifene and tamoxifen in the N-nitrosomethylurea induced rat mammary carcinoma model. Cancer Res. 1987;47:4020–4. [PubMed] [Google Scholar]
- 20.Lubet RA, Steele VE, DeCoster R, Bowden C, You M, Juliana MM, et al. Chemopreventive effects of the aromatase inhibitor vorozole (R83842) in the methylnitrosourea-induced mammary cancer model. Carcinogenesis. 1998;19:1345–51. doi: 10.1093/carcin/19.8.1345. [DOI] [PubMed] [Google Scholar]
- 21.Chan MM, Lu X, Merchant F, Iglehart JD, Miron PL. Gene expression profiling of NMU-induced rat mammary tumors: cross species comparison with human breast cancer. Carcinogenesis. 2005;26:1343–53. doi: 10.1093/carcin/bgi100. [DOI] [PubMed] [Google Scholar]
- 22.Zelazny E, Li B, Anagnostopoulos AM, Coleman A, Perkins AS. Cooperating oncogenic events in murine mammary tumorigenesis: assessment of ErbB2, mutant p53, and mouse mammary tumor virus. Exp Mol Pathol. 2001;70:183–93. doi: 10.1006/exmp.2001.2357. [DOI] [PubMed] [Google Scholar]
- 23.Lubet RA, Boring D, Steele VE, Ruppert JM, Juliana MM, Grubbs CJ. Lack of efficacy of the statins atorvastatin and lovastatin in rodent mammary carcinogenesis. Cancer Prev Res. 2009;2:161–7. doi: 10.1158/1940-6207.CAPR-08-0134. [DOI] [PubMed] [Google Scholar]
- 24.Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013;14(11):R125. doi: 10.1186/gb-2013-14-11-r125. E Published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christov K, Grubbs CJ, Shilkaitis A, Juliana MM, Lubet RA. Short term modulation of cell proliferation and apoptosis and preventive/therapeutic efficacy of various agents in a mammary cancer model. Clin Cancer Res. 2007;13:5488–96. doi: 10.1158/1078-0432.CCR-07-0404. [DOI] [PubMed] [Google Scholar]
- 26.Tucker GT, Casey C, Phillips PJ, Connor H, Ward JD, Woods HF. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol. 1981;12:235–46. doi: 10.1111/j.1365-2125.1981.tb01206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alguire C, Amrein L, Bazile M, David S, Zakikhani M, Pollak M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene. 2011;30:1174–82. doi: 10.1038/onc.2010.483. [DOI] [PubMed] [Google Scholar]
- 28.Wu K, Zhang Y, Xu XC, Hill J, Celestino J, Kim HT, et al. The retinoid X receptor-selective retinoid, LGD1069, prevents the development of estrogen receptor-negative mammary tumors in transgenic mice. Cancer Res. 2002;62:6376–80. [PubMed] [Google Scholar]
- 29.Lu C, Speers C, Zhang Y, Xu X, Hill J, Stenbis E, et al. Effect of epidermal growth factor receptor inhibitor on development of estrogen receptor-negative mammary tumors. J Natl Cancer Inst. 2003;95:1825–33. doi: 10.1093/jnci/djg117. [DOI] [PubMed] [Google Scholar]
- 30.Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. ExpGerontol. 2005;40:685–93. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Yoshidome K, Shibata MA, Maroulakou IG, Liu ML, Jorcyk CL, Gold LG, et al. Genetic alterations in the development of mammary and prostate cancer in the C3(1)/Tag transgenic mouse model. Int J Oncol. 1998;12:449–53. [PubMed] [Google Scholar]
- 32.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen-induced lung tumorigenesis. Cancer Prev Res. 2010;3:1066–76. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohammed A, Janakiram NB, Brewer M, Ritchie RL, Marya A, Lightfoot S, et al. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl Oncol. 2013;6:649–59. doi: 10.1593/tlo.13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 35.Nielsen TO, Parker JS, Leung S, Voduc D, Ebbert M, Vickery T, et al. A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin Cancer Res. 2010;16:5222–32. doi: 10.1158/1078-0432.CCR-10-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Filipits M, Nielsen TO, Rudas M, Greil R, Stöger H, Jakesz R, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res. 2014;20:1298–305. doi: 10.1158/1078-0432.CCR-13-1845. [DOI] [PubMed] [Google Scholar]
- 37.Lynch BJ, Guinee DG, Jr, Holden JA. Human DNA topoisomerase II-alpha: a new marker of cell proliferation in invasive breast cancer. Hum Pathol. 1997;28:1180–8. doi: 10.1016/s0046-8177(97)90256-2. [DOI] [PubMed] [Google Scholar]
- 38.Tokiniwa H, Horiguchi J, Takata D, Kikuchi M, Rokutanda N, Nagaoka R, et al. Topoisomerase II alpha expression and the Ki-67 labeling index correlate with prognostic factors in estrogen receptor-positive and human epidermal growth factor type-2-negative breast cancer. Breast Cancer. 2012;19:309–14. doi: 10.1007/s12282-011-0291-4. [DOI] [PubMed] [Google Scholar]
- 39.Chen S, Huang L, Liu Y, Chen CM, Wu J, Shao ZM. The predictive and prognostic significance of pre- and post-treatment topoisomerase IIα in anthracycline-based neoadjuvant chemotherapy for local advanced breast cancer. Eur J Surg Oncol. 2013;39:619–26. doi: 10.1016/j.ejso.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 40.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–52. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 41.Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L, et al. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res Treat. 2011;128:783–94. doi: 10.1007/s10549-011-1612-1. [DOI] [PubMed] [Google Scholar]
- 42.Bonanni B, Putoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol. 2012;30:2593–600. doi: 10.1200/JCO.2011.39.3769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.