

SUMMARY
The ATP-dependent chromatin assembly factor (ACF) is a dimeric motor that spaces nucleosomes to promote formation of silent chromatin. Two copies of its ATPase subunit SNF2h bind opposite sides of a nucleosome, but how these protomers avoid competition is unknown. SNF2h senses the length of DNA flanking a nucleosome via its HAND-SANT-SLIDE (HSS) domain, yet it is unclear how this interaction enhances remodeling. Using covalently connected SNF2h dimers we show that dimerization accelerates remodeling and that the HSS contributes to communication between protomers. We further identify a nucleotide-dependent conformational change in SNF2h. In one conformation the HSS binds flanking DNA, and in another conformation the HSS engages the nucleosome core. Based on these results, we propose a model in which DNA length sensing and translocation are performed by two distinct conformational states of SNF2h. Such separation of function suggests that these activities could be independently regulated to affect remodeling outcomes.
INTRODUCTION
Chromatin remodelers are molecular motors that regulate gene expression and chromatin structure by altering the position and composition of nucleosomes, the fundamental units of chromatin (Clapier and Cairns, 2009). Remodelers move nucleosomes by translocating DNA across the surface of an octamer of histone proteins, but the mechanisms by which they accomplish this remain poorly understood.
The ATP-dependent chromatin assembly factor (ACF) is an ISWI-family remodeler that generates even spacing between nucleosomes in vitro (Ito et al., 1997) and contributes to the maintenance of heterochromatin in vivo (Collins et al., 2002; Deuring et al., 2000; Fyodorov et al., 2004). ACF also moves mononucleosomes to the center of a short piece of DNA (He et al., 2006; Längst et al., 1999). These nucleosome spacing and centering activities are thought to rely on the ability of the ATPase subunit to “sense” the length of DNA flanking either side of a nucleosome and move the nucleosome preferentially toward the longer DNA (Clapier and Cairns, 2009; Kagalwala et al., 2004; Stockdale et al., 2006; Yang et al., 2006). Indeed, increasing flanking DNA length stimulates ATP hydrolysis and nucleosome remodeling by ISWI ATPases (Clapier and Cairns, 2009; He et al., 2006; Whitehouse et al., 2003; Yang et al., 2006; Zofall et al., 2004).
The conserved HAND-SANT-SLIDE (HSS) domain of the ATPase subunit (Figure 1A, top), has been identified as the region responsible for binding flanking DNA, and has thus been implicated in the characteristic DNA length sensitivity of ISWI remodelers (Clapier and Cairns, 2009; Dang and Bartholomew, 2007; Fan et al., 2004; Grüne et al., 2003; Hota et al., 2013; Narlikar et al., 2013). Recent studies demonstrated that ISWI and related Chd1 remodelers do not require rigid mechanical coupling between the flanking DNA binding domain and the ATPase domain in order to efficiently move nucleosomes (Ludwigsen et al., 2013; Nodelman and Bowman, 2013). Instead, the authors suggest that binding of the HSS to flanking DNA enhances enzymatic activity by productively anchoring the ATPase domain. In contrast, others have proposed that HSS binding to flanking DNA more directly activates the ATPase by relieving inhibition from an autoregulatory motif (Clapier and Cairns, 2012). Thus, the precise role of the HSS domain in remodeling and whether or how it communicates with the ATPase domain remain unclear.
Figure 1. SNF2h dimers remodel faster than monomers and coordinate their activities via the HSS domain.

(A) Top, domain architecture of wild type SNF2h. Bottom left, schematic of SpyCatcher method for covalent dimerization of SNF2h (Supplemental Experimental Procedures). Bottom right, schematic of dimeric SNF2h variants.
(B) Gel shift nucleosome remodeling assay comparing wild type SNF2h to [wt]-[wt] dimer. 0/60 nucleosomes (40 nM) were remodeled with the indicated concentrations of each enzyme and stopped at various time points.
(C) FRET nucleosome remodeling assay comparing wild type SNF2h to [wt]-[wt] dimer. Left, schematic of the assay setup (green donor, red acceptor) and representative kinetic traces. Right, maximum rate constants (kmax) from remodeling 7.5 nM FRET-labeled 0/60 nucleosomes with saturating amounts of each enzyme (400 nM wild type; 20 nM [wt]-[wt]). Bars are mean ± s.e.m. from three replicates.
(D) Gel shift nucleosome remodeling as in (B), but with excess, saturating nucleosomes. Left, gel of 0/60 nucleosomes (660 nM) remodeled by indicated concentrations of wild type SNF2h or [wt]-[wt]. Right, initial rate of appearance of centered product. Bars are mean ± s.e.m. from four replicates.
(E) Gel shift nucleosome remodeling as in (B), comparing SNF2h dimer variants from (A).
(F) Comparison of maximal FRET remodeling rates of SNF2h dimer variants, as in (C). [wt]-[wt] is same as (C), re-plotted for comparison. Saturating concentrations of dimers: 20 nM [wt]-[WB]; 160 nM [ΔHSS]-[wt]; 80 nM [WB/ΔHSS]-[wt]. Bars and reported values are mean ± s.e.m. from three replicates.
See also Figure S1.
Human ACF consists of the ATPase subunit, SNF2h, and an accessory subunit, Acf1. Two copies of SNF2h, can bind opposite sides of a nucleosome, and dimerization enhances the remodeling activity of both SNF2h alone and the ACF complex (Racki et al., 2009). While this dimeric architecture helps explain the ability of ACF to move nucleosomes processively and bi-directionally (Blosser et al., 2009), it raises new questions about how the two motors work together to remodel a single nucleosome instead of competing in a futile tug-of-war.
In this work we use recently described protein ligation methods to engineer constitutive SNF2h dimers, and address how each motor contributes to the remodeling reaction. We also use thermodynamic analyses and FRET-based approaches to study how the HSS domain is regulated by the nucleotide state of the ATPase active site. Our results suggest a model in which a large conformational change between DNA length-sensing and translocating forms of SNF2h enables proper nucleosome repositioning and facilitates cooperation between protomers in the SNF2h dimer.
RESULTS
Covalent linkage of SNF2h proteins reveals that dimerization enhances remodeling
Our previous work revealed positive cooperativity in the concentration dependence of SNF2h nucleosome remodeling activity (Racki et al., 2009), indicating that dimerization enables SNF2h to remodel nucleosomes more effectively. However, because SNF2h exists predominantly as a monomer in the absence of nucleosomes and only dimerizes in the presence of its substrate (Racki et al., 2009; data not shown), a quantitative comparison of the dimeric and monomeric forms of SNF2h has been technically challenging. To overcome this challenge we engineered constitutive, covalently linked SNF2h dimers using a protein ligation method termed SpyCatcher (Zakeri et al., 2012; Supplemental Experimental Procedures). The resulting covalent fusion, hereafter called SNF2h [wt]-[wt], consisted of two full-length SNF2h proteins connected at their C-termini by a 57 amino acid flexible linker and the 14kDa SpyCatcher domain (Figure 1A, left; Figures S1A–S1C).
SNF2h moves mononucleosomes containing 60 bp or less of flanking DNA toward the center (Yang et al., 2006; Figure 1B). Like wild type SNF2h, the constitutively dimeric SNF2h [wt]-[wt] also centered mononucleosomes with 60 bp of flanking DNA (0/60) in a native gel mobility shift assay (Figure 1B). Using a FRET-based assay (Yang and Narlikar, 2007; Yang et al., 2006) we compared their maximal rates of nucleosome sliding (Figure 1C), and found that SNF2h [wt]-[wt] remodeled slightly (1.5-fold) faster than wild type SNF2h. Moreover, at least 40-fold lower concentrations of SNF2h [wt]-[wt] relative to wild type SNF2h were required to achieve half maximal remodeling, consistent with SNF2h [wt]-[wt] engaging nucleosomes as a single, dimeric copy (Figures S1D–S1E). Notably, under these conditions (enzyme saturating and in excess over nucleosomes), wild type SNF2h engages nucleosomes predominantly as a dimer (Racki et al., 2009), and so the modest rate enhancement of SNF2h [wt]-[wt] likely reflects some added benefit of covalently linking the two protomers.
To obtain a quantitative estimate of the benefit of dimerization, we sought to compare the remodeling activities of SNF2h [wt]-[wt] and wild type SNF2h under conditions that would favor dissociation of the latter into monomers. We reasoned that a large excess of nucleosomes over enzyme should increase the likelihood of complexes containing a single copy of wild type SNF2h. Covalently dimerized SNF2h [wt]-[wt], by contrast, should be less susceptible to this type of disruption due to the high effective concentration of the connected proteins. We therefore measured initial rates of nucleosome remodeling with nucleosomes saturating and in excess of enzyme (>15-fold). Under these conditions, the constitutively dimeric SNF2h [wt]-[wt] moves nucleosomes 4.5-fold more rapidly than an equivalent active site concentration of wild type SNF2h (Figure 1D). This comparison provides a conservative estimate of the difference between wild type and SNF2h [wt]-[wt]. Indeed, if wild type SNF2h were entirely monomeric under these conditions, then there would be twice as much wild type SNF2h-nucleosome complex (40 nM) as covalent dimer complex (20 nM), and the difference between monomer and dimer would in fact be 9-fold.
Thus, wild type SNF2h remodels at a similar rate to SNF2h [wt]-[wt] under dimer-favoring conditions (1.5-fold more slowly; Figure 1C) but remodels significantly more slowly under monomer-favoring conditions (≥4.5-fold more slowly; Figure 1D).
The HSS domain plays a role in coordination between SNF2h protomers
The covalently connected SNF2h dimers also provided an opportunity to investigate the influence of one protomer on another. Specifically, we asked what roles the ATPase and HSS domains (Figure 1A, top) may play in enabling the two protomers in a SNF2h dimer to work together to remodel a nucleosome.
The HSS domain has been implicated in flanking DNA length sensing by ISWI enzymes, and thus in their ability to center mononucleosomes (Dang and Bartholomew, 2007; Hota et al., 2013). However, we found that deleting the HSS domain from one protomer of a SNF2h dimer ([ΔHSS]-[wt]; Figure 1A) had little or no effect on its ability to center nucleosomes (Figure 1E) or on the overall rate of remodeling (Figure 1F).
We next tested the role of the ATPase domain by introducing an active site mutation (E309A) in one protomer of the constitutive SNF2h dimer ([wt]-[WB]; Figure 1A). This mutation in the conserved Walker B ATPase motif (WB) decreases the rate of ATP hydrolysis (Racki et al., 2014) and dramatically slows remodeling in the context of wild type SNF2h (Figure S1F). In contrast to the HSS deletion, [wt]-[WB] dimers moved nucleosomes ~5-fold more slowly than [wt]-[wt] (Figure 1F). We reasoned that this result could arise from either of two distinct scenarios. One possibility is that efficient remodeling requires the action of two ATPases, and so impairing one ATPase would slow remodeling. In this case, additional mutations to the WB protomer should have little influence on remodeling, as this protomer is already dramatically impaired. Alternatively, it may be that only one ATPase is required for efficient nucleosome sliding, but the WB protomer exerts a dominant negative effect on the wild type protomer. In this scenario, it may be possible, through additional mutations to the WB protomer, to exacerbate or relieve this inhibitory effect.
To distinguish between these two possibilities, we examined the effects of combining the HSS domain deletion and the WB catalytic mutation on the same protomer in cis ([WB/ΔHSS]-[wt]; Figure 1A). Compared to the single mutant [wt]-[WB], the double mutant [WB/ΔHSS]-[wt] remodeled nucleosomes 5-fold faster, with a maximal rate comparable to [wt]-[wt] (Figure 1F). This indicates that deletion of the HSS domain in cis rescues the inhibitory effect of the WB mutation in a SNF2h dimer. Interestingly, HSS deletion has different effects on remodeling rate depending on whether it is introduced in the context of a wild type protomer ([ΔHSS]-[wt], no effect) or a catalytically impaired protomer ([WB/ΔHSS]-[wt], 5-fold increase; Figure 1F).
The above results suggest the following: 1) a single, wild type SNF2h protomer is sufficient for maximal nucleosome sliding activity. This finding disfavors a model in which both SNF2h ATPases make an obligate contribution to the remodeling reaction. 2) The role played by the HSS domain depends on the state of the ATPase domain in cis. 3) The functional interaction between the ATPase and HSS domains in cis is important for proper coordination between SNF2h protomers in trans.
Nucleotide state regulates interactions between HSS domain and the nucleosome
The ability of ISWI remodelers to generate evenly spaced chromatin is thought to rely on the ability of the HSS domain to “sense” flanking DNA length through direct binding interactions (Dang and Bartholomew, 2007; Hota et al., 2013; Yang et al., 2006). However, it remains unclear whether or how DNA binding by the HSS domain is coupled to the action of the ATPase domain (Clapier and Cairns, 2012; Hota et al., 2013; Ludwigsen et al., 2013; Mueller-Planitz et al., 2013). Our observations above suggested that the state of the ATPase active site may influence the HSS domain in cis to affect the remodeling reaction (Figure 1F, [wt]-[WB] vs. [ΔHSS/WB]-[wt]). We therefore wondered whether ATP binding and/or hydrolysis in the active site may regulate the interaction between the HSS domain and the flanking DNA. Previous studies of HSS-DNA interactions have focused primarily on the nucleotide-free (apo) state (Dang and Bartholomew, 2007; Hota et al., 2013), and it has not been directly investigated whether these interactions change during the ATPase cycle. We therefore examined the contribution of the HSS domain to nucleosome binding in the presence of different ATP analogs.
We labeled nucleosomes with a Cy3 fluorophore on a unique cysteine introduced into the N-terminal tail of histone H4 (A15C), and monitored the increase in fluorescence intensity upon SNF2h binding (Figure 2A, bottom; Supplemental Experimental Procedures). Dye attachment at this site did not interfere with nucleosome binding or remodeling (Figures S2A–S2C, data not shown). Using this method, we measured binding of SNF2h to nucleosomes in the absence (core) or presence (30/30) of symmetric, 30 bp flanking DNAs (Figure 2A, top). We observed that SNF2h binds 30/30 nucleosomes 2.1-fold more tightly than core nucleosomes in the absence of nucleotide (Figure 2B, apo; p<0.0001). The magnitude of this effect is consistent with previous reports that ISWI enzymes bind more tightly to nucleosomes with longer flanking DNA (He et al., 2006; 2008; Kagalwala et al., 2004).
Figure 2. Nucleotide state regulates binding of the HSS domain to flanking DNA or the nucleosome core.

(A) Top, schematic of core and 30/30 nucleosomes. Bottom, representative binding curves showing the increase in fluorescence intensity from various concentrations of wild type SNF2h binding to 7.5 nM H4 A15C-Cy3 nucleosomes in the presence of ADP. Points are mean ± s.e.m. from three replicates, fit to a cooperative binding model.
(B) Comparison of wild type SNF2h binding affinities for 30/30 (striped bars) and core (solid bars) nucleosomes in the presence of no nucleotide (apo), ADP or ADP-BeFx. Fold changes indicate the decrease in affinity for core relative to 30/30 nucleosomes in a given nucleotide state. Error bars are s.e.m. **** p<0.0001; n.s. not significant.
(C) Comparison of wild type SNF2h and SNF2h ΔHSS binding affinities for 30/30 (left, striped bars) or core (right, solid bars) nucleosomes in the presence of ADP. Fold changes and p-values reflect the decrease in affinity from deletion of the HSS domain. *** p<0.001; **** p<0.0001.
(D) Same as (C), but in the presence of ADP-BeFx.
(E) Simple model for the proposed nucleotide-stabilized conformational states of SNF2h. In the ADP state (top) the HSS domain is extended, binding flanking DNA. In the ADP-BeFx state (bottom) the HSS is retracted, helping bind the nucleosome core. For clarity, only one protomer is shown.
See also Figure S2.
Binding to flanking DNA was enhanced by the addition of ADP, indicated by the 4.9-fold preference for 30/30 nucleosomes over core (Figure 2B; p<0.0001). Furthermore, in the ADP state, deletion of the HSS domain greatly reduced binding to 30/30 nucleosomes (22-fold; Figure 2C, left) but had a much more modest effect on binding to core nucleosomes, which lack flanking DNA (2.1-fold; Figure 2C, right). This is consistent with a role of the HSS domain in binding flanking DNA in the ADP state (Figure 2E, top).
We next examined SNF2h binding to nucleosomes in the presence of ADP-BeFx, a nucleotide analog that can adopt configurations ranging from ATP to ADP-Pi (Del Campo and Lambowitz, 2009; Rayment et al., 1996; Thomsen and Berger, 2009). Strikingly, while the addition of ADP-BeFx slightly increased the overall affinity of SNF2h for nucleosomes, it abolished the preference for nucleosomes with flanking DNA (Figure 2B). This raised the possibility that the HSS does not make contacts with flanking DNA in the ADP-BeFx state.
If the HSS domain does not engage flanking DNA in the ADP-BeFx state, we wondered whether it might contribute to binding other parts of the nucleosome. Indeed, compared to the modest effect of HSS deletion on binding to core nucleosomes in the presence of ADP (2.1-fold), deleting this domain dramatically reduced affinity for core nucleosomes in the presence of ADP-BeFx (44-fold; Figure 2D, right). A similar effect was observed on 30/30 nucleosomes (56-fold; Figure 2D, left). Interestingly, in the presence of ADP-BeFx, deletion of the HSS domain had a similar magnitude effect on binding to a short piece of DNA (Figure S2D; 22-fold for DNA vs. 44-fold for core nucleosomes), suggesting that the contribution of this domain to core nucleosome binding may be due largely to binding of nucleosome-wrapped DNA. Importantly, the HSS may contribute to core nucleosome binding either through direct interactions between this domain and the nucleosome, or through allosteric effects on the rest of the SNF2h protein. Together, these results demonstrate that, in the ADP-BeFx state, the HSS domain does not bind flanking DNA, but rather contributes to binding the nucleosome core (Figure 2E, bottom).
The findings above suggest a nucleotide-driven switch between two distinct conformations of SNF2h: a conformation in which the HSS engages the flanking DNA (Figure 2E, top), and a conformation in which the HSS releases flanking DNA and instead stabilizes binding to the nucleosome core (Figure 2E, bottom).
Nucleotide state drives a conformational change in SNF2h
To further test the hypothesis that nucleotide state regulates the position of the HSS domain, we developed a FRET-based assay to detect the location of the HSS relative to the flanking DNA (Figure 3A). In this assay, a donor dye at the end of the flanking DNA (DNA(+20)) transfers energy to an acceptor dye attached to the HSS domain (SNF2h A930C) when the two are in close proximity.
Figure 3. A nucleotide-driven conformational change involving the HSS domain.

(A) Structural model of the SNF2h HSS domain (green) bound to a nucleosome (gray), indicating the locations of FRET probes used in (B–D). A homology model of HSS-DNA complex based on PDB:2Y9Z (Yamada et al., 2011) was generated using Phyre2 (Kelley and Sternberg, 2009) and manually docked onto the nucleosome PDB:1KX5 (Davey et al., 2002). Positions of Cy3 donor (green spheres) and Cy5 acceptor (red sphere) dyes are indicated.
(B) FRET between SNF2h A930C-Cy5 (1 μM) and 0/20 Cy3 DNA(+20) nucleosomes (200 nM). Left, schematic of label positions. Middle, representative emission spectra in the presence of ADP (blue) or ADP-BeFx (red), showing donor (~565 nm) and acceptor (~670 nm) fluorescence in response to donor excitation. Right, FRET Ratios in the presence of ADP (0.51 ± 0.004) or ADP-BeFx (0.34 ± 0.005) given as mean ± s.e.m. from three replicates. See Figures S3E–S3F for FRET determination by two alternate methods.
(C) FRET between SNF2h A930C-Cy5 (1 μM) and 0/20 H2A T11C-Cy3 nucleosomes (200 nM), in the presence of ADP (0.39 ± 0.003) or ADP-BeFx (0.56 ± 0.003), shown as in (B). See Figures S3G-S3H for FRET determination by two alternate methods.
(D) FRET between SNF2h A930C-Cy5 (1 μM) and 0/20 Cy3 DNA(−25) nucleosomes (200 nM), in the presence of ADP (0.40 ± 0.011) or ADP-BeFx (0.49 ± 0.004), shown as in (B). **** p<0.0001, *** p<0.0005
See also Figure S3 and Supplemental Experimental Procedures.
Due to the large number of endogenous cysteines in SNF2h, we used an enzymatic protein ligation strategy to label SNF2h at a single site in the HSS domain (Popp et al., 2007; 2009; Figure S3A–S3C, Supplemental Experimental Procedures). We chose a site in the SLIDE domain (A930C) predicted from molecular homology modeling to be opposite the DNA binding interface (Figure 3A; Kelley and Sternberg, 2009; Yamada et al., 2011) and labeled this site with an acceptor dye. SNF2h labeled in this way remodeled nucleosomes with rates comparable to wild type (within 2-fold; Figure S3D).
We bound acceptor-labeled SNF2h to nucleosomes labeled with donor at the end of a single 20 bp flanking DNA (Figure 3A, DNA(+20)) and measured FRET efficiency (Supplemental Experimental Procedures). The binding analysis in Figure 2 implied that the HSS domain makes stronger contacts with flanking DNA in the ADP state than in the ADP-BeFx state. Consistent with this interpretation, we found that the FRET efficiency was higher in the presence of ADP than ADP-BeFx (Figure 3B). This FRET change was robust to the method used to calculate FRET efficiency, as methods that control for intrinsic changes in donor (Figure S3E) or acceptor (Figure S3F) fluorescence also showed reduced FRET in the ADP-BeFx state. This suggests that the HSS is in closer proximity to flanking DNA in the ADP state than in the ADP-BeFx state (Figure 2E).
Our thermodynamic analysis also suggested that, in the ADP-BeFx state, rather than releasing the nucleosome altogether, the HSS domain engages the nucleosome core (Figure 2E, bottom). However, the precise location of the HSS domain in this state is unclear. We therefore introduced donor fluorophores individually at several internal locations on the nucleosome (Figure 3A; data not shown), bound each of these nucleosomes with acceptor-labeled SNF2h and measured FRET efficiency in different nucleotide states as above. Consistent with the predictions of our model, two locations within the nucleosome core exhibited increased FRET with the HSS domain in the ADP-BeFx state relative to the ADP state: one on a histone tail (H2A T11C; Figures 3A, 3C) and one on the nucleosomal DNA 25 bp from the entry/exit site (DNA (−25); Figures 3A, 3D). These probes are located on the side of the nucleosome opposite the dyad, ~120–140 Å away from the DNA (+20) probe (Figure 3A).
Together with the thermodynamic characterization above, these FRET data suggest the existence of a large conformational change in SNF2h that is driven by nucleotide state. In one conformation (stabilized by ADP) the HSS domain is extended, making contacts with flanking DNA. In the other conformation (stabilized by ADP-BeFx) the HSS retracts away from the flanking DNA and helps to engage the nucleosome core (Figure 2E).
In the Discussion section below we address the implications of these findings and propose a model for the mechanism of nucleosome remodeling by SNF2h.
Nucleosome centering relies on flanking DNA length sensitivity
It has been proposed that the characteristic mononucleosome centering activity of ISWI-family remodelers depends on their ability to sense flanking DNA length via the HSS domain (Kagalwala et al., 2004; Stockdale et al., 2006; Yang et al., 2006). We sought to test this basic premise using a recently discovered regulatory element that uncouples flanking DNA length sensing from nucleosome mobilization. This region has been examined in Drosophila ISWI in the context of a deletion series (Clapier and Cairns, 2012) and, more recently, in SNF2h where it was replaced by a short, flexible linker (Hwang et al., 2014). In the latter study, disrupting NegC selectively impaired flanking DNA length sensitivity without compromising the ability of SNF2h to move nucleosomes.
We replaced NegC with a flexible linker of equal length in the context of full length SNF2h (mNegC; Figure 4A) and found that SNF2h mNegC exhibited no defect in overall remodeling activity (Figure S4) but had dramatically reduced sensitivity to flanking DNA (Figure 4B). Whereas wild type SNF2h remodeled core nucleosomes 10-fold more slowly than 30/30 in a restriction enzyme accessibility assay (Figure 4B, left), this difference was only 1.5-fold for SNF2h mNegC (Figure 4B, right). This result is consistent with previous work, which demonstrated that the NegC element is important for DNA length sensitivity by SNF2h (Hwang et al., 2014). Interestingly, the authors observed no such effect of NegC disruption in the context of the ACF complex, suggesting that the accessory Acf1 subunit compensates for the loss of NegC. Consistent with this possibility, the authors found that the Acf1 subunit has an additional mechanism for sensing flanking DNA length via its N-terminal region (Hwang et al., 2014).
Figure 4. Impairing DNA length sensitivity abolishes nucleosome centering.
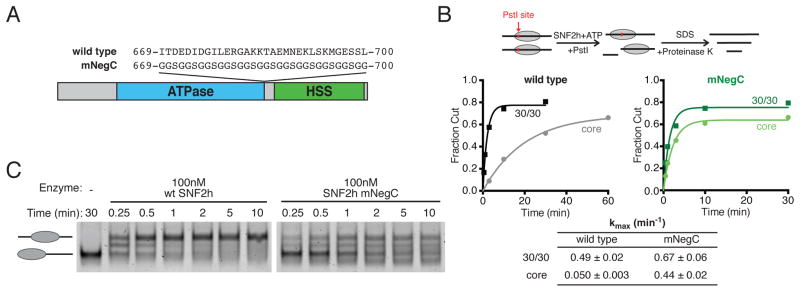
(A) Schematic showing the location of the NegC motif in the primary sequence of SNF2h, and the mutations in SNF2h mNegC.
(B) Restriction enzyme accessibility assay comparing stimulation of remodeling activity by flanking DNA for SNF2h wild type and mNegC. Top, schematic of the assay workflow. Remodeling exposes a buried PstI restriction site, DNA products are separated on a native gel and fraction of cut DNA is quantified. Middle, kinetics of nucleosomal DNA cutting over time for wild type (left) and mNegC (right) remodeling core and 30/30 nucleosomes. Saturating concentrations of enzyme were used (300 nM and 900 nM for 30/30 and core nucleosomes, respectively). Points are the mean of three or four replicates, fit to a single exponential. Bottom, maximum rate constants (kmax) obtained from fits. Values are mean ± s.e.m.
(C) Gel shift nucleosome remodeling assay comparing wild type SNF2h to mNegC. 0/60 nucleosomes (40 nM) were remodeled with the indicated concentrations of each enzyme and stopped at various time points.
See also Figure S4.
To investigate whether uncoupling DNA length sensing from nucleosome mobilization affects nucleosome centering, we next compared the remodeled products of SNF2h mNegC and wild type SNF2h on a native gel. Whereas wild type SNF2h produced primarily centered nucleosome products, SNF2h mNegC generated a steady-state distribution of products with a broad range of electrophoretic mobilities (Figure 4C). This strongly supports the hypothesis that nucleosome centering by SNF2h relies on its ability to sense flanking DNA length.
DISCUSSION
In this work we investigate the mechanism of nucleosome sliding by the human chromatin remodeling motor SNF2h. We address how the conformation of SNF2h and its engagement of the nucleosome change as a function of its ATPase cycle, as well as how two SNF2h motors work together to remodel a single nucleosome.
Two modes of nucleosome engagement suggest a two-state model for SNF2h remodeling
ISWI-family chromatin remodelers sense flanking DNA length via the conserved HSS domain (Dang and Bartholomew, 2007; Grüne et al., 2003; Hota et al., 2013). However, it is unclear how the HSS contributes to nucleosome movement, as well as whether and how it communicates with the ATPase domain (Clapier and Cairns, 2012; Ludwigsen et al., 2013; Mueller-Planitz et al., 2013; Nodelman and Bowman, 2013). Our work indicates that the nucleotide state of the active site controls binding of the HSS domain to different regions of the nucleosome (Figures 2, 3). These results suggest that the HSS domain plays an additional role in remodeling that is independent of flanking DNA binding.
One possible interpretation of this nucleotide-dependent change in HSS position (Figures 2, 3) is that SNF2h re-orients itself on the nucleosome without a conformational change in the enzyme. Such a model would require that the ATPase domain also change its position on the nucleosome as a function of nucleotide state. Alternatively, our data could reflect a conformational change in the SNF2h protein itself, in which the HSS moves with respect to the ATPase domain. We favor the latter model because our data, combined with previous observations, suggest that the ATPase domain remains bound at the same location on the nucleosome in all the nucleotide states tested here. These findings are as follows: 1) Previous work has shown that the ATPase domain of ISWI enzymes engages the histone H4 tail (Mueller-Planitz et al., 2013) and the surrounding nucleosomal DNA at super-helical location 2 (SHL2; Dang and Bartholomew, 2007) in the apo state. We have previously reported that SNF2h engages the H4 tail and SHL2 DNA in both the apo and ADP-BeFx states (Racki et al., 2009), and others have observed that the homologous yeast ISW2 protects this location in the apo and ADP states (Gangaraju et al., 2009). These previous findings suggest that the ATPase domain remains engaged at SHL2 in the apo, ADP and ADP-BeFx states. 2) In the current study, our binding analysis (Figure 2) uses a probe on the histone H4 tail. We detect fluorescence enhancement in response to SNF2h binding in all nucleotide states tested, which would suggest that the ATPase binds near this location in all these states. For these reasons, we interpret our results above to indicate a large conformational change in the SNF2h protein, rather than simply a change in binding orientation.
The conformation in which the HSS engages the nucleosome core is stabilized by ADP-BeFx, a nucleotide analogue that several lines of evidence suggest mimics an activated state of SNF2h. For example, ADP-BeFx promotes engagement of the histone H4 tail (Racki et al., 2009), a nucleosomal epitope that stimulates ATP hydrolysis, nucleosome sliding and translocation along naked DNA (Clapier et al., 2001; Clapier and Cairns, 2012; Hamiche et al., 2001). ADP-BeFx also stabilizes a restricted conformation of the nucleotide binding pocket and this change is enhanced by the histone H4 tail (Racki et al., 2014). Taken in context with these published findings, our results suggest that the HSS releases flanking DNA in an activated ATP state and instead binds the nucleosome core.
Intriguingly, recent single molecule observations indicate that SNF2h is insensitive to flanking DNA length during nucleosome translocation (Hwang et al., 2014). ISWI enzymes remodel nucleosomes in two observable phases: 1) a slow, ATP-dependent “pause” phase in which nucleosomal DNA does not move, as detected by FRET; followed by 2) a fast, ATP-dependent translocation phase in which the DNA is repositioned in several 1bp increments (Blosser et al., 2009; Deindl et al., 2013; Hwang et al., 2014). A recent study demonstrated that the duration of the pause phase is sensitive to flanking DNA length but the translocation phase is not (Hwang et al., 2014), indicating that flanking DNA length sensing and translocation occur at different times during the remodeling reaction.
Our results suggest a possible physical explanation for these single molecule observations. We propose a model in which DNA length sensing and translocation are carried out by two distinct conformational states of SNF2h (Figure 5). In this model, upon binding a nucleosome, the HSS engages flanking DNA, which helps productively orient the ATPase domain and thus stimulates ATP hydrolysis (Figure 5, top). During this phase (corresponding to the “pause” phase in single molecule experiments), ATP hydrolysis is not coupled to DNA translocation, but instead powers a slow conformational change to the translocation-competent state of SNF2h (Figure 5, vertical arrow). This process is rate limiting, making the overall reaction rate sensitive to flanking DNA. Following this switch to the translocation-competent state, in which the HSS domain engages the nucleosome core, SNF2h undergoes multiple rounds of ATP hydrolysis, leading to the translocation of several bp of nucleosomal DNA as described previously (Figure 5, bottom; Deindl et al., 2013). Finally, some process, for example the accumulation of strain on the nucleosome (Deindl et al., 2013), could lead the HSS to release the nucleosome core and re-engage the flanking DNA.
Figure 5. A model for nucleosome remodeling by SNF2h.

SNF2h initially engages nucleosomes in a non-productive ground state (top middle). Binding of the HSS domain to flanking DNA leads to productive engagement of the ATPase domain (top left and right) and ATP hydrolysis. In this DNA length-sensing state (purple box), the HSS domain binds flanking DNA, but the enzyme cannot translocate. ATP hydrolysis from the DNA length-sensing state induces a slow, rate-limiting conformational change to a translocation-competent state (vertical arrow). For clarity, this transition is only shown for one SNF2h protomer. In the translocation-competent state (orange box), ATP hydrolysis is coupled to translocation of DNA across the surface of the histone octamer. Translocation proceeds until strain on the nucleosome is released, resetting the enzyme to the length-sensing state. In this model, longer flanking DNA increases the likelihood of SNF2h undergoing a conformational change to the translocating state and, therefore, makes the overall reaction sensitive to flanking DNA length. The two SNF2h protomers take turns for binding to their respective flanking DNAs (top). This allows discrimination between long and short flanking DNA, and helps prevent the two motors from trying to move the nucleosome simultaneously in opposite directions.
See also Figure S5.
Several features of this model are consistent with previous work on the mechanism of ISWI remodelers as follows:
Two recent studies, one with Drosophila ISWI and one with yeast Chd1, have shown that flexible insertions between the ATPase domain and the DNA binding domain do not inhibit remodeling activity (Ludwigsen et al., 2013; Nodelman and Bowman, 2013). Based on these findings, it was proposed that the DNA binding domain does not communicate with the ATPase domain through transduction of mechanical force. Rather, it was suggested that binding of the HSS to flanking DNA serves to productively orient the ATPase domain on the nucleosome, consistent with the post-binding, productive engagement step we propose in Figure 5 (top). It is further possible that some level of flexibility between the ATPase and HSS domains is in fact essential to enable the transition between the DNA length-sensing state and the translocation-competent state (Figure 5, vertical arrow). Interestingly, in the case of Chd1, shortening the linker between the ATPase and DNA binding domains beyond a critical distance did compromise remodeling activity (Nodelman and Bowman, 2013). Based on this result, it was suggested that a minimal linker length between the ATPase and the DNA binding domain is needed to accommodate conformational changes during remodeling.
Our model proposes that hydrolysis of ATP from the DNA length-sensing state does not power translocation (Figure 5, vertical arrow). Consistent with this possibility, the NegC regulatory element has been shown to conditionally inhibit the coupling of ATP hydrolysis to translocation on naked DNA (Clapier and Cairns, 2012). We speculate that NegC contributes to preventing translocation in the DNA length-sensing state, thereby gating the transition to the translocation-competent state.
Published DNA footprinting results are consistent with our proposal that the HSS domain engages the nucleosome core in an activated, translocation-competent state (Figure 5, bottom). One study of the homologous yeast ISW2 complex reported the appearance of a new region of protection on nucleosomal DNA shortly following the addition of ATP or during remodeling of stalled nucleosomes (Gangaraju et al., 2009). This protected region overlaps with our two internal FRET probes (Figure 3), and we have previously observed increased protection at this location by SNF2h in the presence of ADP-BeFx (Racki et al., 2009).
Our results are thus consistent with previous work and extend current models by providing evidence for two distinct, nucleotide state-dependent conformations of SNF2h: a conformation in which the HSS binds flanking DNA, and a conformation in which the HSS engages the nucleosome core.
Cooperation and communication within SNF2h dimers
SNF2h dimerizes on nucleosomes (Racki et al., 2009), yet how two motors bound to the same nucleosome coordinate their activities to avoid competition has remained unclear. There are two general classes of models for dimer action. In one model, the two motors take turns, with only one motor moving the nucleosome at a time. In a second model, the two motors cooperate, with each motor making an obligate contribution to nucleosome sliding. Here, we address the contributions of each motor to the remodeling reaction by engineering constitutive SNF2h dimers. We find that a single wild type motor is sufficient for maximal nucleosome remodeling (Figure 1F). This is most consistent with a model in which one motor can perform the basic remodeling reaction, and the two motors in a dimer take turns moving the nucleosome back and forth. In this model, binding interactions made by the non-translocating protomer could contribute to remodeling by increasing the time the translocating motor remains bound to the nucleosome, rather than by directly increasing the rate of nucleosome sliding.
How might it be that only one SNF2h motor is activated at a time? We have previously observed that SNF2h engages nucleosomes asymmetrically in the ADP and apo states (Racki et al., 2009), states in which it also binds flanking DNA (Figure 2B). This raises the possibility that only one protomer can bind flanking DNA at a time. We therefore speculate that communication between protomers occurs at the level of binding to flanking DNA. In such a model, the two motors would have to take turns binding to their respective flanking DNAs (Figure 5, top). The longer the DNA, the greater the probability that the motor bound to that DNA would hydrolyze ATP and proceed to the translocation-competent state. The slow transition to the translocation-competent state would reduce the probability that both motors simultaneously attempt to translocate, thereby avoiding a tug-of-war.
Communication at the level of flanking DNA binding could also help explain the observed behavior of mutant SNF2h dimers (Figure 1). By this model, in SNF2h [wt]-[WB], the catalytically compromised protomer is impaired in its ability to switch to the translocation-competent state and remains bound to flanking DNA. The mutant would thus act as a dominant negative by preventing the wild type protomer from taking a turn at binding its flanking DNA (Figure S5A). However, deleting the HSS domain of the WB mutant ([ΔHSS/WB]-[wt]) would prevent it from remaining bound to the flanking DNA, allowing the wild type motor to bind flanking DNA and proceed to translocation unimpeded (Figure S5B).
If flanking DNA length sensing and nucleosome translocation are carried out by two different conformations of SNF2h, then it may be possible to regulate these two processes independently of one another. The model proposed here (Figure 5) provides a framework for interrogating how protein binding partners or contextual cues (i.e. histone modifications) may selectively alter one process or the other, in order to promote defined remodeling outcomes.
EXPERIMENTAL PROCEDURES
For greater detail on the methods used in this work, see Supplemental Experimental Procedures.
Protein expression and purification
All SNF2h variants were expressed as N-terminal 6xHis fusions in E. coli and affinity purified. Tags were removed by TEV protease and proteins were further purified by ion exchange and size exclusion chromatography.
Nucleosome labeling and reconstitution
Mononucleosomes were reconstituted from purified, recombinant histones and PCR products containing the 601 positioning sequence as previously described (Lowary and Widom, 1998; Shahian and Narlikar, 2012). Fluorescent DNA labels were introduced by PCR with labeled primers (Integrated DNA Technologies, IBA Life Sciences) and histone labels were introduced prior to octamer refolding using dye maleimides, similar to previously described (Shahian and Narlikar, 2012).
SpyCatcher covalent dimerization
SNF2h proteins containing a C-terminal (GGS)9 linker and SpyTag or SpyCatcher (Zakeri et al., 2012) were purified by affinity and ion exchange chromatography, then mixed in equimolar amounts overnight at 4°C during TEV cleavage. Dimers were separated from monomers by size exclusion. The pDEST14-SpyCatcher plasmid was obtained from AddGene (Plasmid #35044).
Nucleosome remodeling assays
Native gel shift remodeling assays were performed similar to previously reported (Yang et al., 2006), at 25–30°C in Reaction Buffer (12–15 mM HEPES pH 7.5, 70 mM KCl, 0.02% (v/v) Igepal (NP-40), 2–4% (v/v) glycerol, 5 mM MgCl2, 2 mM ATP), quenched at various time points with excess ADP and plasmid DNA, and separated by native PAGE (5% polyacrylamide, 0.5X TBE). FRET remodeling was performed similar to previously reported (Racki et al., 2009; Shahian and Narlikar, 2012) at 25°C in Reaction Buffer with 11% (v/v) glycerol. Experiments were performed in a K2 fluorometer (ISS) with a 550 nm short-pass excitation filter, 535 nm long-pass emission filter, and excitation and emission monochromators set to 515nm and 670nm, respectively. Restriction enzyme accessibility (REA) assays were performed similar to previously reported (Narlikar et al., 2001) at 25°C in Reaction Buffer with 8% (v/v) glycerol, 7 mM MgCl2 and 3 U/μL PstI (NEB). At time points, aliquots were quenched with 1% (w/v) SDS and 35 mM EDTA, digested with 3.3 mg/mL Proteinase K, and separated by native PAGE (10% polyacrylamide, 1X TBE). All reactions were started with the addition of nucleosomes. Gels were visualized with SYBR Gold (Life Technologies).
Nucleosome binding assays
7.5 nM nucleosomes labeled with Cy3 on H4 A15C were incubated with varying concentrations of SNF2h for 35–45 minutes at room temperature in 15 mM HEPES pH 7.5, 70 mM KCl, 0.02% (v/v) Igepal (NP-40), 3mM MgCl2 and 4% (v/v) glycerol. Reactions with nucleotide contained an additional 3 mM MgCl2, 3 mM ADP and (for ADP-BeFx) 3 mM BeCl2 and 15 mM NaF. Reactions were assembled in opaque 384-well plates (Corning) and imaged on an Analyst AD platereader (Molecular Devices) with a 520 nm bandpass excitation filter, 580 nm bandpass emission filter and 561 nm dichroic mirror.
Labeling and sortase-mediated transpeptidation of SNF2h
SNF2h was expressed in two fragments: one containing the HSS domain (aa730-1052) and one containing the ATPase (aa1-729) with its C-terminus mutated to contain the cognate site for the S. aureus sortase A transpeptidase (Popp et al., 2009). All native Cys residues in the HSS were mutated to Ser, and a unique Cys was introduced (A930C). HSS-A930C was labeled with Cy5-maleimide (Lumiprobe) and ligated to the ATPase fragment by S. aureus sortase A to reconstitute full-length, labeled SNF2h (Figures S1A–S1C).
FRET between HSS and Nucleosomes
1 μM Cy5-labeled SNF2h was incubated with 200 nM 20/0 nucleosomes (labeled at various positions with Cy3) in: 7 mM HEPES pH 7.5, 70 mM KCl, 0.02% (v/v) Igepal (NP-40), 7% (v/v) glycerol, 5 or 6 mM MgCl2, 2 or 3 mM ADP. ADP-BeFx reactions also contained 2 or 3 mM BeCl2 and 10 or 15 mM NaF. Samples were incubated for 5–10 min at 25°C and measured in a fluorometer as above. See also Figures S1E–S1H.
Data fitting and FRET calculations are described in Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Olaf Schneewind (University of Chicago) for the Sortase expression plasmid. We thank John Gross, Hiten Madhani, Daniele Canzio, Richard Isaac and Coral Zhou for helpful comments on the manuscript, Julia Tretyakova for histone purification and members of the Narlikar laboratory for helpful discussions. This work was funded by a National Science Foundation pre-doctoral fellowship to J.D.L and a grant from the National Institutes of Health (#GM073767) to G.J.N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blosser TR, Yang JG, Stone MD, Narlikar GJ, Zhuang X. Dynamics of nucleosome remodelling by individual ACF complexes. Nature. 2009;462:1022–1027. doi: 10.1038/nature08627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Längst G, Corona DF, Becker PB, Nightingale KP. Critical role for the histone H4 N terminus in nucleosome remodeling by ISWI. Mol Cell Biol. 2001;21:875–883. doi: 10.1128/MCB.21.3.875-883.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. Regulation of ISWI involves inhibitory modules antagonized by nucleosomal epitopes. Nature. 2012:1–6. doi: 10.1038/nature11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins N, Poot RA, Kukimoto I, García-Jiménez C, Dellaire G, Varga-Weisz PD. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet. 2002;32:627–632. doi: 10.1038/ng1046. [DOI] [PubMed] [Google Scholar]
- Dang W, Bartholomew B. Domain architecture of the catalytic subunit in the ISW2-nucleosome complex. Mol Cell Biol. 2007;27:8306–8317. doi: 10.1128/MCB.01351-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- Deindl S, Hwang WL, Hota SK, Blosser TR, Prasad P, Bartholomew B, Zhuang X. ISWI Remodelers Slide Nucleosomes with Coordinated Multi-Base-Pair Entry Steps and Single-Base-Pair Exit Steps. Cell. 2013;152:442–452. doi: 10.1016/j.cell.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo M, Lambowitz AM. Structure of the Yeast DEAD box protein Mss116p reveals two wedges that crimp RNA. Mol Cell. 2009;35:598–609. doi: 10.1016/j.molcel.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuring R, Fanti L, Armstrong JA, Sarte M, Papoulas O, Prestel M, Daubresse G, Verardo M, Moseley SL, Berloco M, et al. The ISWI chromatin-remodeling protein is required for gene expression and the maintenance of higher order chromatin structure in vivo. Mol Cell. 2000;5:355–365. doi: 10.1016/s1097-2765(00)80430-x. [DOI] [PubMed] [Google Scholar]
- Fan HY, Narlikar GJ, Kingston RE. Noncovalent modification of chromatin: different remodeled products with different ATPase domains. Cold Spring Harb Symp Quant Biol. 2004;69:183–192. doi: 10.1101/sqb.2004.69.183. [DOI] [PubMed] [Google Scholar]
- Fyodorov DV, Blower MD, Karpen GH, Kadonaga JT. Acf1 confers unique activities to ACF/CHRAC and promotes the formation rather than disruption of chromatin in vivo. Genes Dev. 2004;18:170–183. doi: 10.1101/gad.1139604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangaraju VK, Prasad P, Srour A, Kagalwala MN, Bartholomew B. Conformational changes associated with template commitment in ATP-dependent chromatin remodeling by ISW2. Mol Cell. 2009;35:58–69. doi: 10.1016/j.molcel.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grüne T, Brzeski J, Eberharter A, Clapier CR, Corona DFV, Becker PB, Müller CW. Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol Cell. 2003;12:449–460. doi: 10.1016/s1097-2765(03)00273-9. [DOI] [PubMed] [Google Scholar]
- Hamiche A, Kang JG, Dennis C, Xiao H, Wu C. Histone tails modulate nucleosome mobility and regulate ATP-dependent nucleosome sliding by NURF. Proc Natl Acad Sci USA. 2001;98:14316–14321. doi: 10.1073/pnas.251421398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Fan HY, Garlick JD, Kingston RE. Diverse regulation of SNF2h chromatin remodeling by noncatalytic subunits. Biochemistry. 2008;47:7025–7033. doi: 10.1021/bi702304p. [DOI] [PubMed] [Google Scholar]
- He X, Fan HY, Narlikar GJ, Kingston RE. Human ACF1 alters the remodeling strategy of SNF2h. J Biol Chem. 2006;281:28636–28647. doi: 10.1074/jbc.M603008200. [DOI] [PubMed] [Google Scholar]
- Hota SK, Bhardwaj SK, Deindl S, Lin Y-C, Zhuang X, Bartholomew B. Nucleosome mobilization by ISW2 requires the concerted action of the ATPase and SLIDE domains. Nat Struct Mol Biol. 2013 doi: 10.1038/nsmb.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WL, Deindl S, Harada BT, Zhuang X. Histone H4 tail mediates allosteric regulation of nucleosome remodelling by linker DNA. Nature. 2014;512:213–217. doi: 10.1038/nature13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell. 1997;90:145–155. doi: 10.1016/s0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Kagalwala MN, Glaus BJ, Dang W, Zofall M, Bartholomew B. Topography of the ISW2-nucleosome complex: insights into nucleosome spacing and chromatin remodeling. Embo J. 2004;23:2092–2104. doi: 10.1038/sj.emboj.7600220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJE. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Längst G, Bonte EJ, Corona DF, Becker PB. Nucleosome movement by CHRAC and ISWI without disruption or trans-displacement of the histone octamer. Cell. 1999;97:843–852. doi: 10.1016/s0092-8674(00)80797-7. [DOI] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Ludwigsen J, Klinker H, Mueller-Planitz F. No need for a power stroke in ISWI-mediated nucleosome sliding. EMBO Rep. 2013 doi: 10.1038/embor.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller-Planitz F, Klinker H, Ludwigsen J, Becker PB. The ATPase domain of ISWI is an autonomous nucleosome remodeling machine. Nat Struct Mol Biol. 2013;20:82–89. doi: 10.1038/nsmb.2457. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Phelan ML, Kingston RE. Generation and interconversion of multiple distinct nucleosomal states as a mechanism for catalyzing chromatin fluidity. Mol Cell. 2001;8:1219–1230. doi: 10.1016/s1097-2765(01)00412-9. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and Functions of ATP-Dependent Chromatin-Remodeling Enzymes. Cell. 2013;154:490–503. doi: 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodelman IM, Bowman GD. Nucleosome sliding by Chd1 does not require rigid coupling between DNA-binding and ATPase domains. EMBO Rep. 2013;14:1098–1103. doi: 10.1038/embor.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nat Chem Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- Popp MW-L, Antos JM, Ploegh HL. Site-specific protein labeling via sortase-mediated transpeptidation. Curr Protoc Protein Sci. 2009;Chapter 15(Unit15.3) doi: 10.1002/0471140864.ps1503s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racki LR, Naber N, Pate E, Leonard JD, Cooke R, Narlikar GJ. The Histone H4 Tail Regulates the Conformation of the ATP-Binding Pocket in the SNF2h Chromatin Remodeling Enzyme. J Mol Biol. 2014;426:2034–2044. doi: 10.1016/j.jmb.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racki LR, Yang JG, Naber N, Partensky PD, Acevedo A, Purcell TJ, Cooke R, Cheng Y, Narlikar GJ. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature. 2009;462:1016–1021. doi: 10.1038/nature08621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I, Smith C, Yount RG. The active site of myosin. Annu Rev Physiol. 1996;58:671–702. doi: 10.1146/annurev.ph.58.030196.003323. [DOI] [PubMed] [Google Scholar]
- Shahian T, Narlikar GJ. Analysis of changes in nucleosome conformation using fluorescence resonance energy transfer. Methods Mol Biol. 2012;833:337–349. doi: 10.1007/978-1-61779-477-3_20. [DOI] [PubMed] [Google Scholar]
- Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem. 2006;281:16279–16288. doi: 10.1074/jbc.M600682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ND, Berger JM. Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell. 2009;139:523–534. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse I, Stockdale C, Flaus A, Szczelkun MD, Owen-Hughes T. Evidence for DNA translocation by the ISWI chromatin-remodeling enzyme. Mol Cell Biol. 2003;23:1935–1945. doi: 10.1128/MCB.23.6.1935-1945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Frouws TD, Angst B, Fitzgerald DJ, DeLuca C, Schimmele K, Sargent DF, Richmond TJ. Structure and mechanism of the chromatin remodelling factor ISW1a. Nature. 2011;472:448–453. doi: 10.1038/nature09947. [DOI] [PubMed] [Google Scholar]
- Yang JG, Narlikar GJ. FRET-based methods to study ATP-dependent changes in chromatin structure. Methods. 2007;41:291–295. doi: 10.1016/j.ymeth.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ. The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol. 2006;13:1078–1083. doi: 10.1038/nsmb1170. [DOI] [PubMed] [Google Scholar]
- Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci USA. 2012;109:E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Persinger J, Bartholomew B. Functional role of extranucleosomal DNA and the entry site of the nucleosome in chromatin remodeling by ISW2. Mol Cell Biol. 2004;24:10047–10057. doi: 10.1128/MCB.24.22.10047-10057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.