

Abstract
Localized surface plasmon resonance (LSPR) spectroscopy is a label-free chemical and biological molecular sensing technique whose sensitivity depends upon development of nanostructured transducers. Herein, we report an electrodeposition method for fabricating nanostructured gold films (NGFs) that can be used as transducers in LSPR spectroscopy. The NGF was prepared by electrodepositing gold from potassium dicyanoaurate solution onto a flat gold surface using two sequential controlled potential steps. Imaging by scanning electron microscopy reveals a morphology consisting of randomly configured block-like nanostructures. The bulk refractive index sensitivity of the prepared NGF is 100 ± 2 nm RIU−1 and the initial peak in the reflectance spectrum is at 518 ± 1 nm under N2(g). The figure of merit is 1.7. In addition, we have studied the interaction between carbohydrate (mannose) and lectin (Concanavalin A) on the NGF surface using LSPR spectroscopy by measuring the interaction of 8-mercaptooctyl-α-D-mannopyranoside (αMan-C8-SH) with Concanavalin A by first immobilizing αMan-C8-SH in mixed SAMs with 3,6-dioxa-8-mercaptooctanol (TEG-SH) on the NGF surface. The interaction of Con A with the mixed SAMs is confirmed using electrochemical impedance spectroscopy. Finally, the NGF surface was regenerated to its original sensitivity by removing the SAM and the bound biomolecules. The results from these experiments contribute toward the development of inexpensive LSPR based sensors that could be useful for studying glycan–protein interactions and other bioanalytical purposes.
Keywords: Lectin, Mannose, Monolayer, Plasmon, Gold
1. Introduction
Localized surface plasmon resonance (LSPR) spectroscopy based on the development of noble metal nanostructures with tunable and responsive plasmonic behavior has become of broad interest. 1–5 LSPR spectroscopy can provide a label-free and sensitive technique for biosensing or assays that has great potential to be miniaturized or developed into array formats. The sensitivity of LSPR spectroscopy depends on the properties of the nanostructure used as a transducer. Nanostructures of the coinage metals such as copper,6 silver,7 and gold8 are being actively studied as LSPR-based transducers. LSPR can be observed for nanostructures having features much smaller than the wavelength of the incident light. The LSPR response to change in refractive index in the medium surrounding the nanostructure depends on the composition, shape, size, and local dielectric properties. Although silver shows a stronger LSPR response compared to gold or copper, gold is preferred due to its chemical stability. A recent effort has been reported to electrodeposit gold around gold-silver core-shell nanoparticles on indium tin oxide coated glass to preserve the stronger response of silver.9 Nanostructures having different shapes such as triangles, spheres, cubes, and rods produce different peak wavelengths, full widths at half maxima and hence different LSPR bulk sensitivity.10 In general, nanostructures having sharper features yield higher refractive index sensitivity.11 It has also been found that increasing the size of nanoparticles red shifts the resonance peak position and increases the bulk refractive index sensitivity; however, the peak becomes broader decreasing the figure of merit (FOM) due to radiation damping.12,13
Common techniques for fabricating nanostructured transducers include immobilization of nanoparticles on chemically modified substrates,14,15 nanolithography (including nanosphere lithography, 16,17 and electron-beam lithography18,19), and evaporation of a thin layer of metal on a glass surface followed by thermal annealing. 20 Although immobilized nanoparticles (e.g., nanorods, nanostars, nanoprisms, nanorice) show good LSPR responses, there can be some disadvantages with regard to stability and reproducibility. 21 In addition, aggregation of free nanoparticles in solution is a potential challenge and nanoparticles may not be completely free from stabilizers used to avoid aggregation, which will affect the sensitivity measurements and binding experiments.22 To avoid these limitations, nanolithography techniques have been developed using templates to fabricate different nanostructures. One of the popular nanolithography techniques is nanosphere lithography. 23 In this method, polystyrene nanospheres of various diameters are used as deposition masks on glass substrates. These nanospheres self-assemble in hexagonally close-packed pattern on substrate, such that metals can be deposited in gaps between the nanospheres. The nanospheres can then be removed by sonication of the substrate in organic solvents leaving behind the triangular or spherical nanostructures in a periodic array.24,25 This method is popular because it is cheaper, simpler, and does not require sophisticated instrumentation.26 However, there are possibilities for the formation of various types of defects in this method as a result of nanosphere polydispersity, site randomness, point defects, line defects, and polycrystalline domains.24 The concentration of nanospheres directly plays a role in the arrangement of nanospheres on the substrate,24 which means a variety of structures may be formed on the same substrate. An alternate strategy involves depositing gold caps on SiO2 nanospheres randomly arranged on a gold surface, for which a good LSPR response was found.27 Electron beam lithography can make nanostructures precisely without any defects;28 however, this technique is expensive and requires more time and expertise.24 Evaporating a thin layer of metal on glass surface followed by annealing is also a cheaper and simpler technique;29 however, the nanostructures produced are polydisperse. Annealing of evaporated thin Au films at high temperatures can help to control the morphology and improve the LSPR response.30 All of these examples show that more research remains to be done in this field for producing sensitive nanostructures so that LSPR spectroscopy can become a method of choice for biochemical sensing. Besides LSPR spectroscopy, these nanostructured transducers are also used in surface enhanced Raman spectroscopy (SERS),24 a very sensitive analytical technique whose detection limit approaches the single molecular level,31,32 which once again emphasizes the importance of research in nanostructure fabrication.
LSPR has been compared to traditional SPR33 and is found to be quite competitive on the basis of a number of features, especially cost. SPR experiments are based upon propagating surface plasmons, often at the surface of a flat gold film, whose thickness should be near 50 nm, and supported on a prism or waveguide. Many of the SPR experiments reported use commercial Biacore instruments along with supplied sensor chips. SPR can be done in a variety of modes, the most popular being measurement of the shift of the resonance angle with analyte binding to the gold surface modified with some sort of recognition layer. Both SPR and LSPR can be conducted in imaging mode; for SPR the element size must be approximately 10 microns, while for LSPR single supported nanoparticles and changes occurring on them can be imaged.34 For a basic LSPR measurement on an ensemble of nanostructures, either by transmission or reflection, the cost of instrumentation is a small fraction (as little as 1/60th) of the cost of a commercial Biacore instrument, thus far adopted as a standard by much of the life science community. Real-time detection is possible with LSPR as it is with SPR. For LSPR done in transmission mode, extinction at a specific wavelength or resonant wavelength versus time can be followed, while in reflection mode reflectivity at a chosen wavelength or resonant wavelength versus time can be followed. As noted by Van Duyne, the refractive index sensitivity of LSPR is much lower than that of SPR; however, the plasmon decay length is much shorter for LSPR (typically 5–15 nm) than for SPR (200–300 nm), and hence a high level of sensitivity to molecular binding at the surface can still be achieved. The lower bulk refractive index sensitivity of LSPR does provide an advantage of simplicity in that close temperature control is less essential. Recent reviews have covered the variety of nanostructures developed for use with LSPR.35,36
SPR has played a major role in probing many types of biomolecular interactions,37 including protein–carbohydrate and lectin–glycoprotein binding. The applications of SPR to study carbohydrate binding interactions have been reviewed,38 and compared with other analytical methods. The use of imaging SPR to study binding to carbohydrate arrays is especially promising for screening carbohydrate–protein interactions.39,40 Approaches based on coupling derivatized carbohydrates to activated SAMs, often in the presence of a diluting species terminated in oligoethylene glycol units known to minimize non-specific protein adsorption, have been pursued using Diels–Alder reactions,41 disulfide-thiol exchange,42 and click chemistry.43 Use of Biacore sensor chips pre-modified with a carboxymethylated dextran gel to which amine derivative glycans can be bound after NHS activation has been reported.44 This widely used type of sensor chip has the potential complication that the lectin Con A, for example, has an affinity for the dextran component.45 Mixed SAMs of a carbohydrate component and diluting species have also been prepared directly and studied using SPR.46 Recently, a method for directly attaching underivatized glycans by photochemically activated C–H bond insertion onto SAMs terminated in a perfluorophenylazide group was reported.39 Efforts have been reported to precisely control the spacing between sugars using cyclic peptides presenting a specified number of mannose units and to examine the influence of this on the multivalency and clustering effects that can occur during lectin binding.47,48
The studies reported in which LSPR has been applied to studying protein binding to a carbohydrate modified nanostructure have primarily been carried out in transmission mode. In an early study, the results for studying a protein–carbohydrate interaction using LSPR and SPR were directly compared.49 Mixed SAMs of a triethylene glycol terminated disulfide and a maleimide terminated analog were formed on silver triangular nanoprisms formed by nanosphere lithography on glass slides. Reaction of maleimide with a mannose thiol derivative gave about 5% mannose coverage available for interaction with Con A. Experiments were conducted in transmission mode, and both the peak wavelength and the magnitude of its shift due to Con A binding were found to depend on the aspect ratio of the nanoprisms. The modified Ag triangular nanoprisms were resistant to non-specific protein binding and were suitable for following Con A binding in real-time by monitoring the peak wavelength as a function of time, with comparable results for SPR found by monitoring the resonance angle versus time using a Biacore instrument. The response during the dissociation phase was markedly different for LSPR than for SPR, and also dependent on the aspect ratio of the triangular nanoprisms which was found to influence the plasmon decay length. Au nanoparticles supported on glass have been modified by polymer brushes with many pendant glucose residues and LSPR was used to determine a binding constant from real-time analysis of 5.0 ± 0.2 × 105 M−1 noted as larger than that for Con A binding to methyl α-D-glucopyranoside of 2.4 ± 0.1 × 103 M−1 in solution and attributed to multipoint binding effects.50 The use of supported gold nanoparticles modified with a polymer brush having pendant mannose units was applied to follow Con A binding,51 resulting in an apparent association constant determined from analysis of real-time association kinetics data of 7.4 ± 0.1 × 106 M−1, noted as much greater than that for Con A tomethyl α-D-mannopyranoside in solution of 7.6 ± 0.2 × 103 M−1, with the difference attributed to multipoint binding effects. Au nanoparticles bound to glass modified by 3-aminopropyltrimethoxysilane were modified by dodecanethiol SAMs into which a N-acetylglucosamine glycolipid was inserted, and shift in the LSPR peak wavelength due to Con A binding was observed in transmission mode in real-time for both association and dissociation.52 A recent study aimed at optimizing supported Au nanoislands formed on glass by evaporation. 53 Mixed SAMs were formed of alkanethiol derivatives with a penta(ethylene glycol) segment terminated in –OH, mannose, or galactose. The response to Con A binding in terms of peak wavelength shift or shift in extinction at a fixed optimal wavelength was found to be greatest for Au islands of average height 2.5 nm. The optimal combination of refractive index sensitivity and plasmon decay length was required to obtain the maximum response to Con A binding. Analysis of real-time binding kinetics data gave a value for Ka of 7.7 × 106 M−1. Glyconanoparticles presenting different sugars have been shown to differentiate between a set of 4 lectins using a pattern recognition approach.54
In this paper, we demonstrate an electrodeposition method for preparing a nanostructured gold film (NGF) which can be used as a LSPR-based transducer. While much work has been reported on the preparation of LSPR transducers by evaporation of gold to form thin nanostructured metal films, electrodeposition can provide an additional convenient route for fabrication of LSPR transducers. Electrodeposition onto indium tin oxide coated glass was previously shown to produce rough flower-like deposits with a peak wavelength of 675 nm.55 The NGF prepared by this method compares well to nanostructured gold films prepared by other methods. We show how the LSPR peak wavelength and bulk refractive index sensitivity vary with changes in nanostructure’s shape and size which can be controlled by changing the electrodeposition parameters. We also demonstrate how LSPR spectroscopy can be performed on the NGF surface to follow carbohydrate–lectin interactions. The interaction between carbohydrate and lectin is chosen because this type of interaction is predominant in many fundamental biological processes such as cellular recognition, inflammation, signal transduction, cell adhesion, and cancer cell metastasis.56,57 Concanavalin A (Con A), 104 kDa, originally obtained from Jack bean, Canavalia ensiformis, is used as a model lectin.58 It is a mannose specific lectin59 and exists as tetramer above pH 7.0 and dimer below 6.0.60 Here, we show that the interaction of Con A with mixed self-assembled monolayers (SAMs) of a thiolated α-mannoside (αMan-C8-SH) and 3,6-dioxa-8-mercaptooctanol can be followed using LSPR on the NGF prepared by electrodeposition.
2. Results
2.1. General procedure
Figure 1 shows a general outline of the preparation and usage of the nanostructured gold films (NGFs) for use in LSPR spectroscopy. Figure 1A outlines the preparation of NGF by first forming flat gold film electrodes by stripping of epoxy coated glass slides off of Au sputtered on silicon wafers, and then electrodepositing gold onto these from a solution of potassium dicyanoaurate to produce the nanostructures. Figure 1B shows a sketch of the electrochemical cell used to carry out the electrodeposition. In Figure 1C, the LSPR apparatus, reflection probe, and flow cell are depicted. Figure 2 shows a depiction of binding of lectin Concanavalin A to a mixed SAM containing the thiolated mannoside (αMan-C8-SH) and 3,6-dioxa-8-mercaptooctanol (TEG-SH) molecule.
Figure 1.
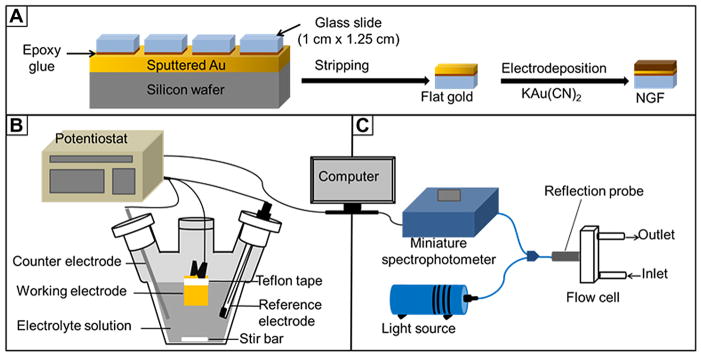
Schematic depiction of: (A) nanostructured gold film (NGF) preparation steps. (B) Electrochemical setup for nanostructured gold film preparation. (C) Optical set up for localized surface plasmon resonance spectroscopy in reflection mode.
Figure 2.
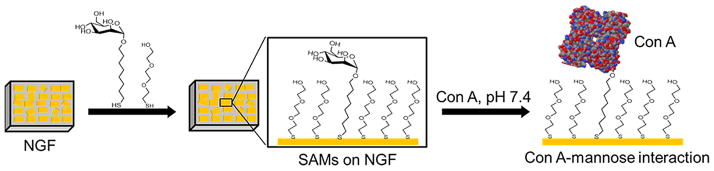
Schematic diagram showing binding of Concanavalin A to thiolated mannoside in mixed self-assembled monolayers on a nanostructured gold film (NGF) surface.
2.2. Nanostructured gold film (NGF) preparation
NGFs were prepared using one-step or two-step chronoamperometry (CA) to electrodeposit gold from basic aqueous solutions of potassium dicyanoaurate (50 mM in 0.25 M Na2CO3) onto already prepared flat gold films on glass slides. Electrodeposited Au films were formed by one-step CA by applying potentials of −0.8 V, −1.0 V, −1.2 V, or −1.4 V (vs Ag|AgCl (KCl satd)) for 60 s or 90 s. The films formed at −1.4 V were not very stable and peeled off of the glass slides. However, films formed at −1.2 V or above were stable. Those formed at −1.0 V or above showed lower bulk refractive index sensitivity than those formed at −1.2 V. The films formed in one-step at −1.2 V for 90 s show a distinct LSPR peak, while those formed in one-step at −1.2 V for 60 s do not. Application of a two-step CA with −1.2 V as the initial potential for 60 s followed by a second potential of −1.0 V, −1.4 V, or −1.6 V for an additional 30 s was then explored. The time for the second potential was fixed at 30 s as the observed peak became broader when 60 s was used as the second deposition time.
The current versus time curves obtained during preparation of NGF at the different potentials are shown in Figure 3A. The curves show a sharp jump in current when the potential is stepped, followed by a decay to a plateau. The jump and decay is attributed to the sudden reorganization of the electric double layer which discharges during the current decay,61 while the plateau represents the Faradaic current associated with reduction of Au(I) in Au(CN)2− to Au(0) at the interface between the growing gold nanostructures and the electrolyte solution. These curves show a general trend of increasing current with more negative applied potential in the second step. The corresponding LSPR spectra of the different NGFs under N2(g) are shown in Figure 3B. The data are presented as R−1 versus wavelength, where R is the percent reflectance. As noted by Van Duyne62, LSPR spectra show a minimum in R in reflection mode and a maximum in extinction in transmission mode. We have found that R−1 at a fixed wavelength plotted versus bulk refractive index varies linearly. The peak wavelength for NGF prepared by depositing Au at −1.2 V for a total duration of 90 s is 516.9 nm whereas the structure that was formed by depositing at this potential for 60 s shows a shoulder with an indistinct peak just near 500 nm. The reflectance spectrum of bare gold has a shoulder near 500 nm63,64 similar to that observed in Figure 3B for gold electrodeposited at −1.2 V for 60 s. This suggested that these films are behaving similar to bulk gold and that a longer deposition time and a second step to more negative potentials is required to red shift the peak wavelength. All of the other preparations display peak wavelengths near 517 nm (see Table 1). These values are close to but lower than that reported for Au nanoparticles on glass in air for which peak wavelengths from 521.0 nm to 525.5 nm were reported dependent on size.65 It has been reported that the plasmon peak wavelength due to scattering can be blue shifted relative to that due to extinction for gold nanodots in an array on the end of an optical fiber, by as much as 7 nm for nanodots 180–200 nm in diameter and 55 nm in height.66 The morphology of the NGF changed when we changed the potential applied in the second step, as can be seen in the SEM images in Figure 4. The NGFs prepared at −1.2 V for 60 s and 90 s look similar and each has randomly distributed nanostructured features with those for the 90 s deposition being larger with smoother boundaries (Fig. 4A and B). The nanostructures formed upon application of a second potential of −1.0 V for 30 s are diverse but relatively smaller (Fig. 4C). Figure 4D and E represents images of NGFs prepared at −1.2 V for 60 s followed by −1.4 V for 30 s and −1.6 V for 30 s, respectively. Rectangular brick-shaped nanostructured features are now evident all over the surface having lengths of around 200 nm and widths of around 100 nm.
Figure 3.
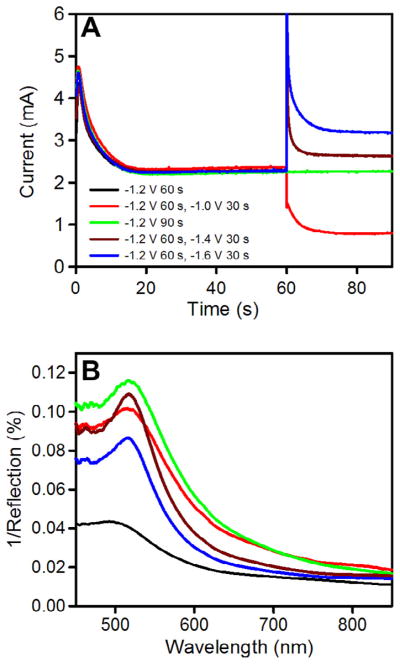
(A) Chronoamperometric curves obtained during the preparation of nanostructured gold films (NGFs) by applying different potentials (vs Ag|AgCl (KCl, satd) for the times indicated in a 50 mM aqueous solution of potassium dicyanoaurate (KAu(CN)2) in 0.25 Msodium carbonate. (B) LSPR spectra of the corresponding NGFs recorded under nitrogen gas, plotted as inverse of percent reflectance versus wavelength.
Table 1.
Dependence of the refractive index sensitivity, figure of merit, and peak wavelength on the deposition time(s) and potential(s) (vs Ag|AgCl) for the electrodeposited nanostructured gold films reported in this study
| Conditions | Refractive index sensitivity (nm/RIU) | Figure of merit (FOM) | Peak wavelength (nm) |
|---|---|---|---|
| −1.2 V 60 s | 52 ± 4 | 0.9 | 499.4 |
| −1.2 V 60 s, −1.0 V 30 s | 58 ± 2 | 0.8 | 517.3 |
| −1.2 V 90 s | 58 ± 2 | 0.8 | 516.9 |
| −1.2 V 60 s, −1.4 V 30 s | 84 ± 2 | 1.4 | 517.5 |
| −1.2 V 60 s, −1.6 V 30 s | 100 ± 2 | 1.7 | 517.5 |
Conditions for which two potentials and times are listed represent two sequential electrodeposition steps.
Figure 4.
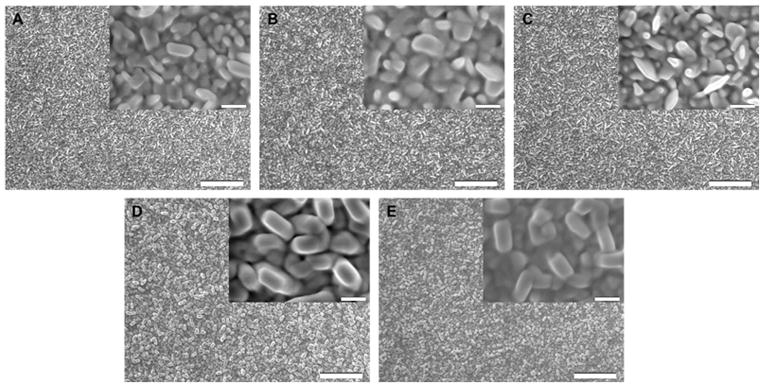
SEM images of nanostructured gold films prepared by electrodeposition from 50 mM potassium dicyanoaurate (KAu(CN)2) in 0.25 M Na2CO3 solution with all potentials versus Ag|AgCl (KCl, satd): (A) at −1.2 V for 60 s, (B) at −1.2 V for 90 s, (C) −1.2 V for 60 s followed by −1.0 V for 30 s, (D) at −1.2 V for 60 s followed by −1.4 V for 30 s, and (E) −1.2 V for 60 s followed by −1.6 V for 30 s. Scale bars: 2 μm. Insets are the corresponding higher magnification SEM images (scale bars: 0.2 μm).
2.3. Bulk refractive index sensitivity test
To check the bulk refractive index sensitivity (RIS) of the NGFs we injected liquids of different refractive indices, water (n = 1.33), 15% glycerol (n = 1.35), 30% glycerol (n = 1.37), 45% glycerol (n = 1.39), 60% glycerol (n = 1.41), and 75% glycerol (n = 1.43), over the NGF surface inside a flow cell and recorded the LSPR spectra. The RIS value provides the shift in the peak LSPR wavelength with increase in the bulk refractive index of the surrounding medium. More recent approaches to assessing nanostructure sensitivity to adsorbed proteins or other molecular layers involve jointly optimizing both RIS and the effective surface plasmon decay length.67–69 We found that the NGF prepared by the two-step electrodeposition using a potential −1.2 V for 60 s followed by −1.4 V or −1.6 V for 30 s exhibits a greater red shift in the peak wavelength with increasing refractive index than the other preparations.
Figure 5A and B shows the representative bulk RIS test performed on NGF prepared by applying −1.2 V for 60 s followed by −1.6 V for an additional 30 s. For this preparation of NGF, the peak wavelength under N2(g) was found to be 518 ± 1 nm. A linear dependence of peak wavelength on bulk refractive index can be seen in Figure 5B with a slope of 99 nm RIU−1. Table 1 shows the sensitivity comparison of NGFs prepared under the different reported conditions. It has been found that the structures prepared by applying a second potential more negative than −1.2 V show greater bulk refractive index sensitivity and improved figure of merit (see Table 1, the standard deviations are for 4 or 5 trials on each NGF preparation). The figure of merit (FOM) is defined as the RIS/fwhm where fwhm is the full width at half maximum of the LSPR peak in nm. Higher values of FOM are associated with improved detection limits for LSPR based sensors.31 Formation of more distinctly nanostructured brick-like features having aspect ratios of approximately 2 with evident inter-nanostructure gaps may be the reason that this film structure shows a better plasmonic response compared to other structures formed.70,71 When we increased the second potential to −1.8 V, it resulted in peeling of the gold film, and thus −1.6 V was a practical lower limit. A linear dependence of wavelength on refractive index is shown in Figure 5D, based upon the LSPR spectra shown in Figure 5C for NGF prepared by applying only one potential step of −1.2 V for 90 s. The slope gives an RIS value of 60 nm RIU−1, lower than for the other preparation.
Figure 5.

(A) Bulk refractive index response of nanostructured gold films prepared using electrodeposition conditions of −1.2 V for 60 s, and then −1.6 V for 30 s. LSPR spectra obtained at different refractive indices (n) = 1, 1.33, 1.35, 1.37,1.39, 1.41, and 1.43 using nitrogen, water, 15% glycerol, 30% glycerol, 45% glycerol, 60% glycerol, and 75% glycerol, respectively. The nitrogen peak is the lowest and the peaks move upward in the graph with increasing refractive index. (B) Plot of peak wavelength versus refractive index for the spectra shown in (A). (C) LSPR spectra showing the bulk RIS response at the same series of refractive index values for nanostructured gold film prepared using electrodeposition at −1.2 V for 90 s. (D) Plot of peak wavelength versus refractive index for the spectra shown in (C).
Higher bulk RIS and FOM have been reported previously for different shapes and sizes of nanostructures.11 Even if shape and size are similar, nanostructures made from silver show higher sensitivity compared to nanostructures made from gold.11 However, it has also been found that nanostructures can show better bulk refractive index sensitivity when their initial LSPR peak occurs at a higher wavelength.72 For gold nanostructures displaying a LSPR peak wavelength slightly above 500 nm, values of 44 nm RIU−1 (nanospheres, λmax = 527 nm, FOM = 0.6) and 83 nm RIU−1 (nanocubes, λmax = 538 nm, FOM = 1.5) have been reported.73 Free nanoparticles have higher sensitivity compared to films but are more difficult to handle as stabilizing agents are needed to avoid aggregation. 22 Therefore, the NGF prepared here with a RIS of 100 ± 2 nm RIU−1 (based on 4 trials) and a FOM = 1.7 could help contribute to overcoming some of the shortcomings of nanoparticle based biosensors and perform with good sensitivity within the visible region.
Recent studies have indicated that in addition to the bulk refractive index sensitivity, the surface plasmon decay length (lsp) should also be consider in optimization of the LSPR response for analyte binding to a recognition layer on the metal surface.67–69 For a given thickness of the recognition layer, such as a carbohydrate terminated SAM, and analyte such as the protein Con A (dimensions 6.3 nm × 8.7 nm × 8.9 nm, tetrameric form, Protein Data Bank ID 3CNA), it is proposed to optimize the combined effect of the RIS and the effective plasmon decay length by matching the dimensions of the analyte layer to the decay length and thereby achieve optimal response to analyte binding to the transducer surface. It is not yet known which parameters for the electrodeposited NGF structures control the value of the effective plasmon decay length. A method exists for estimating the plasmon decay length for the nanostructure prepared under different conditions, such as deposition potential and time in the NGF studied here, but also other variables such as concentration and temperature of the electrodeposition solution could be considered as well as post-preparation annealing steps. In the reported method for estimating surface plasmon decay length, successively greater numbers of bilayers of the alternating cationic and anionic polyelectrolyte pairing of poly(allylamine) and poly(styrene sulfonate) are formed on the surface of the transducer by an alternate dipping procedure between two solutions. The shift in LSPR peak wavelength is plotted versus the number of deposited polyelectrolyte bilayers and reported values of thickness per bilayer of d = 2.09 ± 0.03 nm and of n = 1.56 for these bilayers are used in fitting the wavelength shift to the equation Δλpeak = m Δn [1 − exp(−2d/lsp)], wherein m is the refractive index sensitivity (RIS) and Δn is the difference in refractive index between the adsorbate and the surrounding medium. For transmission measurements, change in extinction at a selected wavelength may also be fit to layer thickness. An extension of this equation for wavelength shift for the case of analyte binding to a recognition layer has been given as Δλpeak = m Δn exp(−2d1/lsp) [1 − exp(−2d2/lsp)] where d1 is the thickness of the recognition layer and d2 is the thickness of the bound layer of analyte.49 If we do not consider variation in the value of m, and consider only the exponential terms dependent on lsp, and use an approximate value of d1 = 1.5 nm for the SAM, and assume that Con A adopts a laying down orientation and can completely cover the surface when binding is saturated so that d2 = 6.3 nm can be taken as an approximation, calculation suggests that the optimal value of lsp is near 7.6 nm which would approximately ‘match’ the thickness of the recognition + analyte layers. If the coverage of the Con A layer was not complete, then the effective value of d2 is reduced and a smaller optimal value of lsp will be obtained. The value of m, the bulk refractive index response, must also be considered so both parameters would be needed for optimization of the response. A lsp value of 5–6 nm was reported for silver triangular nanoprisms.49
2.4. Assay of lectin–carbohydrate interaction
Since the LSPR peak position depends on the change in refractive index due to SAM formation and then protein adsorption around the nanostructures, this effect can be applied to studying biomolecular interactions. When binding occurs on the surface of nanostructures, the refractive index around the surface of the nanostructures increases compared to the initial refractive index, which red shifts the LSPR peak. We have studied the interaction between mannose and Con A on the surface of NGF to determine its sensitivity and application to studying carbohydrate–protein interactions. NGFs prepared by applying −1.2 V for 60 s, followed by −1.6 V for 30 s were studied as these showed a comparatively higher bulk refractive index sensitivity compared to the other NGF preparations as well as the best value for figure of merit of 1.7 (see Table 1). Mixed SAMs (prepared from a 1:3 molar ratio in solution) of thiolated α-mannoside αMan-C8-SH and TEG-SH were formed on the NGF. Mixed SAMs with TEG-SH are formed instead of single component SAMs to control nonspecific interactions, decrease steric interference effects between ligand head groups and to control the surface density of bound Con A.74–76 The LSPR peak wavelength of NGF modified with these mixed SAMs is found to be 521.3 nm which is 2.3 nm red shifted from the unmodified NGF surface which was initially at 519.0 nm under a nitrogen gas environment, as seen in Figure 6A. After Con A was bound onto the SAM modified NGF, by injecting 0.5 μM Con A inside the flow cell for 1 h followed by washing thoroughly with buffer and water, the peak wavelength was then found at 526.1 nm when obtained under a nitrogen gas environment, a 4.8 nm shift from the SAM modified NGF. Figure 6B shows the result for Con A binding to these SAMs on the surface of the NGF prepared in one-step electrodeposition at −1.2 V for 90 s. The peaks are broader, as expected since the figure of merit is lower for this structure. A red shift in peak wavelength for Con A binding of 4–5 nm is observed but the location is harder to discern due to the broader nature of the peaks. The wavelength shift due to SAM formation is also harder to discern due to the broader peaks. Given the sharper nature of the peaks, the NGF prepared by applying −1.2 V for 60 s, followed by −1.6 V for 30 s appears preferable as a LSPR substrate. We also injected BSA through the flow cell over the same SAM-modified NGF prepared by applying −1.2 V for 60 s for one hour but saw only a very small shift in peak magnitude and peak wavelength as seen in Figure 6C, presumably due to some non-specific interaction of BSA with the surface. After the interaction with BSA, buffer was flushed through the cell, prior to studying interaction of the SAM with Con A. In buffer, the peak wavelength for the bare NGF prepared by applying −1.2 V for 60 s, followed by −1.6 V for 30 s was found to be 546 ± 2 nm, and after Con A exposure for 60 min shifted by 3 nm.
Figure 6.

LSPR spectra on nanostructured gold films (NGFs) prepared at −1.2 V for 60 s followed by −1.6 V for 30 s: unmodified (black), modified with mixed SAM (red) of αMan-C8-SH and TEG-SH prepared from 1:3 solution molar ratio (1 mM total concentration in ethanol, 2 h), and after binding of protein (green): (A) Concanavalin A (Con A, 0.5 μM in 10 mM Tris buffer, pH 7.4, 0.1 M NaCl, 1 mM CaCl2, 1 mM MnCl2) for 1 h. (B) LSPR spectra on NGF prepared at −1.2 V for 90 s and then modified with SAM and exposed to Con A as was done in (A). (C) LSPR spectra on nanostructured gold films (NGFs) prepared at −1.2 V for 60 s followed by −1.6 V for 30 s (black), modified with SAM (red) and exposed to bovine serum albumin (BSA, 0.5 μM in 10 mM PBS (pH 7.4)) for 1 h (green). These spectra were measured under nitrogen gas. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To further confirm lectin binding to the mannose presenting SAMs, we performed a mannose-Con A interaction study using electrochemical impedance spectroscopy (EIS). The EIS method has been applied to study the formation of supported bilayers and streptavidin binding to incorporated biotinylated phospholipid77 and to study ion binding to biomimetic SAMs.78 EIS has also been applied to study carbohydrate–lectin interactions in SAMs,43,79,80 and the use of the technique as well as other electrochemical methods to probe glycan–protein interactions has recently been reviewed.81,82 We previously applied square-wave voltammetry to study binding of a Con A–alkaline phosphatase conjugate to immobilized glycoproteins.83 We also have studied the application of EIS to Con A binding to a mannose presenting SAM on nanoporous gold.84 The Nyquist plot obtained from EIS for the SAM modified NGF shows a characteristic semicircle in the higher frequency region near the origin whose diameter represents charge transfer resistance (Rct) and a straight line in the lower frequency region further away from the origin representing diffusion processes. The Nyquist plot of unmodified NGF, SAM modified NGF, and the SAM modified NGF after Con A binding is shown in Figure 7. The Nyquist plot for the unmodified NGF gives a very small charge transfer resistance value (Rct = 10 Ω), as shown in Figure 7. A small semicircle in the Nyquist plot at high frequencies has been reported previously for bare gold surfaces.85–87 Values of Rct can only be directly compared if reported in units of Ω cm2 to account for electrode surface area. The geometric surface area of our NGF used as an electrode that is exposed to the solution is 1.0 cm2, and the roughness factor is estimated to be near 1.4 on the basis of gold oxide stripping. Thus value of Rct for the bare NGF surface is thus estimated to be 14 Ω cm2 accounting for electrode area. This value can be compared with those of 24.4 Ω cm2 on bare Au(111)87, and of 34 Ω cm2 (reported on a polished gold electrode of 1.6 mm diameter whose electroactive surface area was determined by application of the Randles-Sevcik equation)85, and 35.2 Ω cm2 (reported on a bare gold electrode of 2 mm diameter) using the electrode areas reported.86 Upon analyzing the mixed SAMs on NGF we found a subsequent increase in the interfacial charge transfer resistance (Rct = 30 Ω) which increases considerably after incubation with 0.5 μM Con A in buffer (Rct = 225 Ω) indicating specific binding of Con A to mannose in the SAM.
Figure 7.
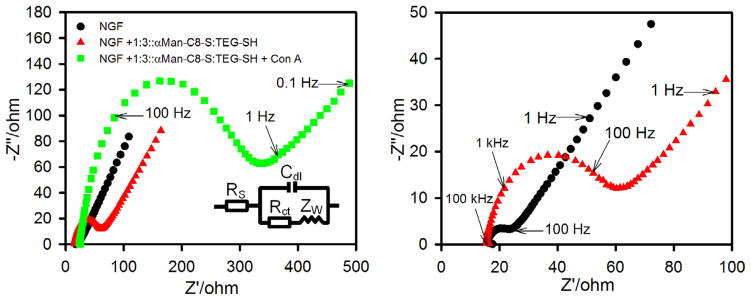
Nyquist plots for Faradic impedance spectra of nanostructured gold films (NGFs) in 10 mM PBS (pH 7.4) containing 5 mM K3[Fe(CN)6] and 5 mM K4[Fe(CN)6]: unmodified NGF (black circles), NGF modified by mixed SAM of αMan-C8-SH and TEG-SH (red triangles), and after Con A binding from a 0.5 μM (50 μgmL−1) solution (green squares). The impedance spectra were recorded in the frequency range of 100 kHz to 0.1 Hz at 0.2 V bias potential (vs Ag|AgCl (KCl, satd) and with AC potential amplitude of 10 mV. Inset is equivalent circuit used to model impedance data. The panel on the right shows an expanded view of the higher frequency data for the bare NGF and the SAM-modified NGF. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Both LSPR and EIS show a clear response for mannose-Con A binding; however, an advantage of LSPR over EIS is that it can be used to study binding directly in situ in real time. Therefore, we performed a real time interaction study of mannose and Con A on the surface of NGF, shown in Figure 8. We have found that R−1 at a fixed wavelength varies linearly with bulk refractive index, as shown in Figure 8A (slope = 2.28 RIU−1), making this variable useful for representing real-time studies. The slope of R−1 at fixed wavelength versus refractive index was found to vary with the exact positioning of the reflection probe, but the highest values were obtained for the NGF prepared by electrodeposition at −1.2 V for 60 s followed by −1.6 V for 30 s. Figure 8B shows the real-time response over a series of Con A concentrations from 5 μgmL−1 (0.05 μM) to 100 μgmL−1 (1.0 μM). The LSPR response is plotted as change in R−1 at 557 nm versus time. This wavelength was chosen above the LSPR peak for the SAM modified NGF in buffer (546 ± 2 nm) and such that R−1 would increase during binding to produce readily followed binding kinetics plots. Real-time interaction studies done in transmission mode are usually performed by following the change in extinction at a chosen fixed wavelength above the peak.53 For this purpose, we placed the mixed SAM modified NGF in the flow cell and tris buffer was flowed through. The reflectance was recorded at a constant wavelength of 557 nm. After recording the reflectance versus time in circulating buffer for 300 s, Con A was injected into the flow cell over a period of 20–30 s until the cell volume was completely filled by the protein solution. We can clearly see the increase in R−1 with time arising from the specific binding between mannose and Con A on the modified NGF surface, and some contribution of nonspecific binding which may be to patches of bare Au exposed by defects or incompletely covered Au areas. There can also be a minor contribution due to a small increase in the refractive index of the bulk solution. Injection of the tris buffer to fill the cell after 15 min of interaction with the Con A solution results in a decrease in the R−1, attributed to washing away of some non-specifically bound Con A from the surface and some desorption of Con A that was bound to mannose.
Figure 8.
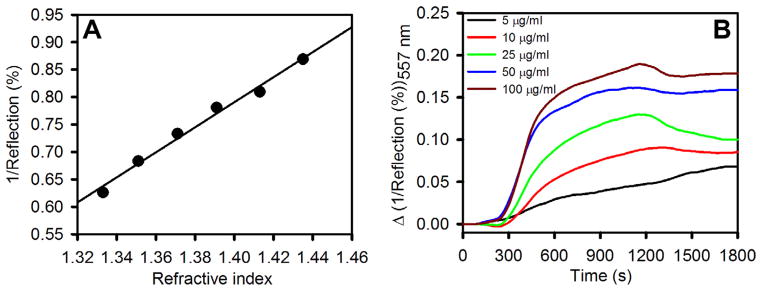
(A) Plot of the inverse of the percent reflectance at the peak wavelength versus bulk refractive index for the nanostructured gold film prepared at −1.2 V for 60 s followed by −1.6 V for 30 s. (B) Real-time LSPR response of SAM modified nanostructured gold films (NGFs) to Con A at a series of concentrations (in 10 mM Tris buffer, pH 7.4, 0.1 MNaCl, 1 mMCaCl2, 1 mMMnCl2): 5 μg mL−1, 10 μg mL−1, 25 μg mL−1, 50 μgmL−1, and 100 μg mL−1. NGF was surface modified with a SAM of αMan-C8-SH and TEG-SH (1:3 solution molar ratio in ethanol, 1 mM total concentration).
Real-time LSPR data have been used to determine values of association rate constants (kon) and of dissociation rate constants (koff) and hence determine Kd values (Kd = koff/kon).50,51,53 Values of kon = 2.0 × 104 M−1 s−1 and koff = 2.6 × 10−3 s−1 corresponding to Kd = 130 nM were reported for Con A interacting with SAMs of a thiolated PEG linker terminated in a mannose unit on 5 nm Au islands.53 As noted by Bellapadrona et al., a range of values of koff between 2.48 × 10−4 s−1 and 1.2 × 10−2 s−1, and of kon ranging from 5.2 × 103 M−1 s−1 to 1.4 × 105 M−1 s−1, corresponding to Kd values ranging from 42 nM to 423 nM have been reported for the Con A–mannose interaction depending upon measurement method, details of surface modification, and experimental conditions.53 Future more detailed kinetic studies using the NGF transducers will be used to determine Kd values for carbohydrate–protein interactions.
2.5. Regeneration of NGF
Finally, we regenerated the NGF surface as this can reduce the cost of potential use as a transducer for biosensor development. NGF modified with same SAMs as described above was immersed in 0.50 μM Con A (in tris buffer containing 0.1 M NaCl, 1 mM CaCl2, and 1 mM MnCl2) for 60 minutes and then rinsed with buffer and Milli-Q water. We then dipped the biomolecule functionalized (SAM + bound Con A) NGF plate in freshly prepared piranha solution for 45 s and rinsed thoroughly with Milli-Q water. The NGF regenerated after this step was tested using different methods. Figure 9A compares LSPR spectra of unmodified NGF and NGF obtained after regeneration. The peak wavelength is the same for both spectra; however, there is a small shift in the magnitude. We believe that the change in magnitude is partly due to a small change in position or orientation of the NGF relative to the reflection probe while loading it back into the flow cell after regeneration.
Figure 9.
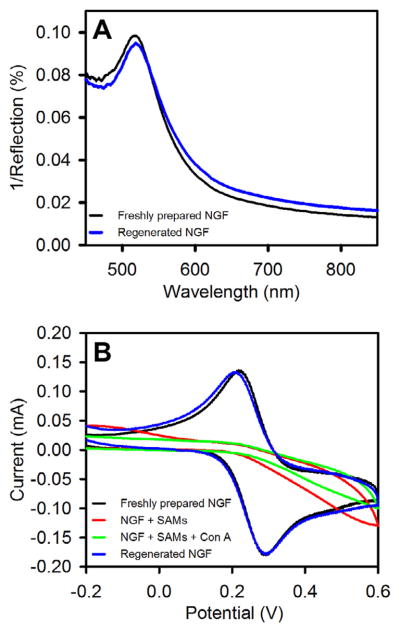
Regeneration experiments on nanostructured gold films (NGFs). NGF was modified with mixed SAMs of αMan-C8-SH and TEG-SH prepared from 1:3 solution molar ratio (1 mM total concentration in ethanol, 2 h). SAM modified NGF was exposed to Concanavalin A (0.5 μM in 10 mM tris buffer, pH 7.4, 0.1 M NaCl, 1 mM CaCl2, 1 mM MnCl2) for 1 h and then rinsed prior to regeneration by immersion in piranha solution for 45 s. (A) LSPR spectra of unmodified NGF (black) and regenerated (blue) nanostructured gold films measured under N2(g) environment. (B) Cyclic voltammograms of unmodified (black) NGF, SAM-modified NGF (red), SAM-modified NGF after Con A exposure (green), and NGF regenerated by brief exposure to piranha solution (blue). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We have also performed cyclic voltammetry experiment to see if some biomolecules are still attached on the surface of NGF and capable of blocking a redox probe. Figure 9B shows the cyclic voltammograms of unmodified NGF, NGF after SAM formation, NGF after Con A binding to the SAM, and NGF after regeneration. It can be seen that unmodified NGF and NGF after regeneration have very similar anodic and cathodic peaks, whereas NGF modified by SAM and also then with bound Con A do not show distinct anodic or cathodic peaks due to blocking of charge transfer at the electrode surface. Exposure to Con A further reduces the current somewhat relative to that seen in the CV of the SAM modified NGF. Finally, we performed the refractive index sensitivity test on regenerated NGF and found no discernable difference compared to sensitivity of unmodified NGF.
3. Discussion
Here we have prepared nanostructured gold films (NGFs), suitable for use in LSPR based spectroscopy, using electrochemical deposition techniques onto flat gold electrodes. We have found that the structure prepared by a two-step electrodeposition technique using a first potential of −1.2 V for 60 s and second potential more negative than −1.2 V for 30 s can produce sensitive nanostructured films with reproducible bulk RIS of up to 100 ± 2 nm RIU−1 and with a good figure of merit of 1.7. The NGFs formed have randomly arranged brick-like nanostructures having lengths of about 200 nm and widths of about 100 nm. Comparison of Figure 4A with 4D and E shows that gold deposition during the second potential step favors enlargement and dominance of the roughly rectangular features of a larger aspect ratio. The prepared NGFs are easy to handle and chemically stable, showing LSPR peak within the visible region, suitable for use as LSPR related transducers. In order to optimize the response, other variables such as electrolyte solution concentrations, temperature, and annealing steps combined with measurement of plasmon decay lengths remain to be investigated.
We used the NGF prepared at −1.2 V for 30 s followed by −1.6 V for 30 s for label-free detection of carbohydrate–protein interactions showing how LSPR peak shifts due to specific interaction of mannose and Con A while no significant shift in the peak was observed for the mannose–BSA interaction. We are also able to demonstrate that LSPR based technique can be used to perform a real time bioassay of carbohydrate–lectin interactions. Finally, NGF modified with a SAM and used to bind Con A was easily regenerated to its original structure and sensitivity, supporting the possible use of NGF as a biosensor/bioassay transducer. Improved plasmonic substrates not only support LSPR based biosensor development but can also be used along with SERS technique for detecting small molecules.
4. Experimental
4.1. Materials
Silicon wafers (3″ N〈100〉, 1–10 Ω-cm, 356–406 μm thick) of prime grade were purchased from Nova Electronic Materials, LLC (Flower Mound, TX). Fisherbrand plain microscope slides (25 × 75 × 1 mm) were purchased from Fisher Scientific (Fairlawn, NJ) and cut to 12.5 × 10 × 1 mm. Sulfuric acid and 30% hydrogen peroxide required to prepare piranha solution were purchased from Fisher Scientific. Methanol (HPLC grade), ethanol (HPLC grade), glycerol, potassium dicyanoaurate(I), 11-mercaptoundecanoic acid (MUA), and N-hydroxysuccinimide (NHS) were purchased from Sigma–Aldrich, Inc. (Milwaukee, WI). Epoxy (EPO-TEK 377) was purchased from Epoxy Technology (Billerica, MA). Sodium carbonate anhydrous, sodium acetate, calcium chloride dihydrate, and manganese(II) chloride tetrahydrate were obtained from Sigma–Aldrich, Inc. (Allentown, PA). Bovine serum albumin (BSA), Concanavalin A (Con A), tris buffer, and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride were purchased from Sigma-Aldrich, Inc. (Saint Louis, MO). Milli-Q water (18.3 MΩ cm) was used for making all solutions. 8-Mercaptooctyl-α-D-mannopyranoside (αMan-C8-SH) and thiolated triethylene glycol, 3,6-dioxa-8-mercaptooctanol (TEG-SH) were synthesized using published methods.84
4.2. Nanostructured gold film preparation details
4.2.1. Cleaning
Cut glass slides were cleaned using piranha solution (3:1, H2SO4/30% H2O2) for 45 min, (Warning: Because of its oxidizing nature, piranha reacts violently with organic compounds, so care must be taken while handling), rinsed with copious amount of Milli-Q water, and stored in methanol. They were dried in oven at 125 °C for 30 min and cooled prior to use.
4.2.2. Flat gold preparation
Flat gold was prepared using published method with some modifications. 88 In brief, a Hummer VI sputter coater (Anatech Ltd) was used to sputter gold onto the silicon wafer for 20 min, adjusting the current to 10 mA so as to obtain a thickness of about 200 nm. Then, one drop of the two components of epoxy, thoroughly mixed, was used to attach a cleaned glass slide to the gold sputtered silicon wafer. Multilayers of silicon wafer-gold-epoxy-coverslip were cured at 150 °C for 2 h. After cooling, glass slides were stripped off of the silicon wafer using a slight force with the help of tweezers to expose a flat gold surface. The gold films were found to be stable in solution on the glass surface; whereas on the silicon surfaces, detachment of the gold film in solution was often observed.
4.2.3. Nanostructured gold film preparation
Potentiostat/Galvanostat model 273A (EG&G Princeton Applied Research) operated by PowerSuite software and using a three-electrode glass cell having Ag|AgCl (KCl satd) (0.22 V vs SHE) as the reference electrode (CH Instruments Inc., Austin, TX) and platinum wire (99.997% purity, 0.5 mm diameter) as the counter-electrode (Alfa Aesar, Ward Hill, MA) was used. A thin film of nanostructured gold was prepared onto the flat gold surface (working electrode) from a 50 mM aqueous solution of potassium dicyanoaurate(I) electrolyte consisting of 0.25 M sodium carbonate by applying different potentials for different times at room temperature. Argon gas was bubbled through the solution for 10 min prior to measurement.
4.3. LSPR peak (λmax) stabilization
For stabilizing the λmax of NGF, it was treated with water and ethanol for 1 h each while stirring followed by drying under N2 gas. This step is necessary to perform in order to avoid solvent induced changes to NGF structure which in turn will change the peak position.
4.4. Localized surface plasmon resonance spectroscopy
The LSPR spectra of NGF were obtained in reflection geometry (Fig. 1C). The setup consists of a white light source (HL-2000 Tungsten Halogen Light, Ocean Optics), spectrophotometer (Jaz, Ocean Optics) connected to a computer having SpectraSuite software (Ocean Optics), a reflection probe (Ocean Optics, QR400-7-SR), and home-built flow Teflon cell of internal volume 500 μL. The reflection probe consists of a bundle of 7 optical fibers with 6 illumination fibers around 1 read fiber. The probe was mounted on a linear translation stage (Newport Oriel). The distance between probe tip surface and the Au surface was kept constant at close to 4mm. The reflection spectra obtained were plotted as R−1 versus wavelength in nanometers using SigmaPlot 12.0 (Systat Software Inc.) and Origin-Pro 8.5.0 SR1 (OriginLab Corporation) was used to determine the full-width at half maximum (fwhm) of peaks. The spectrometer was calibrated using an Ocean Optics HG-1 mercury argon calibration source and fitting of the set of expected wavelengths versus detector array pixel number to a cubic equation as prescribed in the operator’s manual. The resulting calibration gave differences between measured and expected at the 21 standard wavelengths used between −0.97 nm and +0.44 nm.
4.5. Determination of refractive index sensitivity
The refractive index sensitivity of the gold surfaces was determined by measuring the reflectivity spectra with the Teflon flow cell filled with glycerol solutions of 15%, 30%, 45%, 60%, and 75% by weight. The refractive index values of the solutions were taken from the website of the Dow Chemical Company (http://www.dow.com/optim/optim-advantage/physical-properties/refractive.htm). Measurement was also done under water and in air. The peak wavelength and value of reflectance R (in percent) were determined for each peak and then peak wavelength and R−1 were plotted versus refractive index.
4.6. Mannose–Con A binding assay
Mixed self-assembled monolayers (SAMs) of αMan-C8-SH and TEG-SH were prepared on the surface of NGF by co-adsorption from ethanol solution having total concentration 1 mM (molar ratio 1:3 of αMan-C8-SH/TEG-SH) for 2 h at room temperature. After coadsorption, the NGF plate was washed with ethanol, dried under nitrogen, and placed inside the flow cell. Tris-buffer (10 mM, pH 7.4) containing NaCl (0.1 M), CaCl2 (1 mM), and MnCl2 (1 mM) was passed through the flow cell followed by Con A and allowed to interact for 1 h, washed with the buffer followed by water, and dried using nitrogen gas before collecting LSPR reflection spectra. However for the real-time interaction study all the spectra were collected in buffer. For the real-time study, Con A concentrations of 5, 10, 25, 50, and 100 μgmL−1 were used. The Con A was allowed to bind to the SAM for 15 min, and then buffer was re-introduced into the flow cell. During the real-time study, the reflectivity at 557 nm was recorded. A wavelength sufficiently above the peak was chosen so that R−1 would increase with Con A binding during the real-time LSPR experiment.
4.7. Electrochemical impedance spectroscopy
A PARSTAT2273 potentiostat along with PowerSine software was used to acquire impedance data. Experiment was carried out using a conventional three electrode system, 5 mM each of K3[Fe(CN)6] and K4[Fe(CN)6] (1:1 molar ratio) redox probe prepared in 10 mM phosphate buffer at pH 7.4, a frequency range of 100 kHz to 0.1 Hz and a bias potential of 0.2 V (vs Ag|AgCl), and an AC amplitude of 10 mV. Zsimpwin 3.21 software (Princeton Applied Research, Oak Ridge, TN) was used for data analysis. The data are presented as Nyquist plots showing the real and imaginary parts of the impedance at each measurement frequency. Argon gas was bubbled through the solution for 10 min prior to measurement.
4.8. Cyclic voltammetry (CV)
CV was performed for checking surface purity of regenerated NGF using 1 mMK4[Fe(CN)6] in 0.1 M KCl by scanning the potential from −0.2 V to 0.6 V versus Ag|AgCl (KCl satd) at a scan rate of 100 mV s−1. Argon gas was bubbled through the solution for 10min prior to measurement. In order to estimate surface area of the NGF, gold oxide formation and stripping were performed in 0.5 M H2SO4 between the potential limits of 0.0 V and 1.5 V versus Ag|AgCl (KCl satd) at 100mV s−1. A reported and widely used conversion factor of 450 μC cm−2 was used to estimate surface area.89–91
4.9. Scanning electron microscopy (SEM)
SEM imaging and energy dispersive spectroscopy (EDS) were performed using field emission scanning electron microscope (6320F, JEOL USA, Inc.). Imaging was carried out at a working distance of 8 mm at an accelerating voltage of 8 kV.
Acknowledgments
This work was supported by NIGMS (R01-GM090254). The authors are also grateful to Dr. David Osborn (MIST laboratory, Center for Nanoscience, UMSL) for valuable discussions and guidance during SEM imaging.
References
- 1.Mock JJ, Hill RT, Tsai YJ, Chilkoti A, Smith DR. Nano Lett. 2012;12:1757. doi: 10.1021/nl204596h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jia K, Bijeon JL, Adam PM, Ionescu RE. Anal Chem. 2012;84:8020. doi: 10.1021/ac301825a. [DOI] [PubMed] [Google Scholar]
- 3.Dalfovo MC, Salvarezza RC, Ibañez F. J Anal Chem. 2012;84:4886. doi: 10.1021/ac300491h. [DOI] [PubMed] [Google Scholar]
- 4.Blaber MG, Henry A-I, Bingham JM, Schatz GC, Van Duyne RP. J Phys Chem C. 2011;116:393. [Google Scholar]
- 5.Endo T, Kerman K, Nagatani N, Takamura Y, Tamiya E. Anal Chem. 2005;77:6976. doi: 10.1021/ac0513459. [DOI] [PubMed] [Google Scholar]
- 6.Chan GH, Zhao J, Hicks EM, Schatz GC, Van Duyne RP. Nano Lett. 2007;7:1947. [Google Scholar]
- 7.Jensen TR, Malinsky MD, Haynes CL, Van Duyne RP. J Phys Chem B. 2000;104:10549. [Google Scholar]
- 8.Mayer KM, Lee S, Liao H, Rostro BC, Fuentes A, Scully PT, Nehl CL, Hafner JH. ACS Nano. 2008;2:687. doi: 10.1021/nn7003734. [DOI] [PubMed] [Google Scholar]
- 9.Dong P, Wu Y, Guo W, Di J. Plasmonics. 2013;8:1577. [Google Scholar]
- 10.Wiley BJ, Im SH, Li Z-Y, McLellan J, Siekkinen A, Xia Y. J Phys Chem B. 2006;110:15666. doi: 10.1021/jp0608628. [DOI] [PubMed] [Google Scholar]
- 11.Mayer KM, Hafner JH. Chem Rev. 2011;111:3828. doi: 10.1021/cr100313v. [DOI] [PubMed] [Google Scholar]
- 12.Sönnichsen C, Franzl T, Wilk T, Plessen Gv, Feldmann J. New J Phys. 2002;4:93. doi: 10.1103/PhysRevLett.88.077402. [DOI] [PubMed] [Google Scholar]
- 13.Zhao J, Pinchuk AO, McMahon JM, Li S, Ausman LK, Atkinson AL, Schatz GC. Acc Chem Res. 2008;41:1710. doi: 10.1021/ar800028j. [DOI] [PubMed] [Google Scholar]
- 14.Joshi GK, McClory PJ, Muhoberac BB, Kumbhar A, Smith KA, Sardar R. J Phys Chem C. 2012;116:20990. [Google Scholar]
- 15.Huang H, Tang C, Zeng Y, Yu X, Liao B, Xia X, Yi P, Chu PK. Colloids Surf, B. 2009;71:96. doi: 10.1016/j.colsurfb.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 16.Haes AJ, Zhao J, Zou S, Own CS, Marks LD, Schatz GC, Van Duyne RP. J Phys Chem B. 2005;109:11158. doi: 10.1021/jp051178g. [DOI] [PubMed] [Google Scholar]
- 17.Ormonde AD, Hicks ECM, Castillo J, Van Duyne RP. Langmuir. 2004;20:6927. doi: 10.1021/la0494674. [DOI] [PubMed] [Google Scholar]
- 18.Barbillon G, Bijeon JL, Plain J, de la Chapelle ML, Adam PM, Royer P. Surf Sci. 2007;601:5057. [Google Scholar]
- 19.Cao W, Elsayed-Ali HE. Mater Lett. 2009;63:2263. [Google Scholar]
- 20.Karakouz T, Tesler AB, Bendikov TA, Vaskevich A, Rubinstein I. Adv Mater. 2008;20:3893. [Google Scholar]
- 21.Park DK, Kim HI, Kim JP, Park JS, Lee SY, Yang S-M, Lee J, Chung C-H, Sim SJ, Yoo PJ. Langmuir. 2010;26:6119. doi: 10.1021/la100598v. [DOI] [PubMed] [Google Scholar]
- 22.Nusz GJ, Marinakos SM, Curry AC, Dahlin A, Hook F, Wax A, Chilkoti A. Anal Chem. 2008;80:984. doi: 10.1021/ac7017348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones MR, Osberg KD, Macfarlane RJ, Langille MR, Mirkin CA. Chem Rev. 2011;111:3736. doi: 10.1021/cr1004452. [DOI] [PubMed] [Google Scholar]
- 24.Haynes CL, Van Duyne RP. J Phys Chem B. 2001;105:5599. [Google Scholar]
- 25.Jensen TR, Duval ML, Kelly KL, Lazarides AA, Schatz GC, Van Duyne RP. J Phys Chem B. 1999;103:9846. [Google Scholar]
- 26.Haes A, Duyne R. Anal Bioanal Chem. 2004;379:920. doi: 10.1007/s00216-004-2708-9. [DOI] [PubMed] [Google Scholar]
- 27.Takei H, Bessho N, Ishii A, Okamato T, Beyer A, Henning V, Golzhauser A. Langmuir. 2014;30:2297. doi: 10.1021/la403407g. [DOI] [PubMed] [Google Scholar]
- 28.Raphael MP, Christodoulides JA, Mulvaney SP, Miller MM, Long JP, Byers JM. Anal Chem. 2011;84:1367. doi: 10.1021/ac2023266. [DOI] [PubMed] [Google Scholar]
- 29.Tesler AB, Chuntonov L, Karakouz T, Bendikov TA, Haran G, Vaskevich A, Rubinstein I. J Phys Chem C. 2011;115:24642. [Google Scholar]
- 30.Jia K, Bijeon J-L, Adam P-M, Ionescu RE. Plasmonics. 2013;8:143. [Google Scholar]
- 31.Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Nat Mater. 2008;7:442. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- 32.Stiles PL, Dieringer JA, Shah NC, Van Duyne RP. Annu Rev Anal Chem. 2008;1:601. doi: 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- 33.Stuart DA, Haes AJ, Yonzon CR, Hicks ECM, Van Duyne RP. IEEE Proc Nanobiotechnol. 2005;152:13. doi: 10.1049/ip-nbt:20045012. [DOI] [PubMed] [Google Scholar]
- 34.McFarland AD, Van Duyne RP. Nano Lett. 2003;3:1057. [Google Scholar]
- 35.Szunerits S, Boukherrroub R. Chem Commun (Cambridge) 2012:8999. doi: 10.1039/c2cc33266c. [DOI] [PubMed] [Google Scholar]
- 36.Petryayeva E, Krull UJ. Anal Chim Acta. 2011;706:8. doi: 10.1016/j.aca.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 37.de Mol NJ, Fischer MJE, editors. Surface Plasmon Resonance. Methods and Protocols. Vol. 627 Springer; New York: 2010. [Google Scholar]
- 38.Safina G. Anal Chim Acta. 2012;712:9. doi: 10.1016/j.aca.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 39.Tyagi A, Wang X, Deng L, Yan M. Biosens Bioelectron. 2010;26:344. doi: 10.1016/j.bios.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fais M, Karamanska R, Allman S, Fairhurst SA, Innocenti P, Fairbanks AJ, Donohoe TJ, Davis BG, Russell DA, Field RA. Chem Sci. 2011;2:1952. [Google Scholar]
- 41.Houseman BT, Mrksich M. Chem Biol. 2002;9:443. doi: 10.1016/s1074-5521(02)00124-2. [DOI] [PubMed] [Google Scholar]
- 42.Smith EA, Thomas WD, Kiessling LL, Corn RM. J Am Chem Soc. 2003;125:6140. doi: 10.1021/ja034165u. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Luo S, Tang Y, Yu L, Hou K-Y, Cheng J-P, Zeng X, Wang PG. Anal Chem. 2006;78:2001. doi: 10.1021/ac051919+. [DOI] [PubMed] [Google Scholar]
- 44.Nahálková J, Švitel J, Gemeiner P, Danielsson B, Pribulová B, Petruš L. J Biochem Biophys Methods. 2002;52:11. doi: 10.1016/s0165-022x(02)00016-7. [DOI] [PubMed] [Google Scholar]
- 45.Ballerstadt R, Schultz J. Sens Actuators, B. 1998;46:50. [Google Scholar]
- 46.Dhayal M, Ratner D. Langmuir. 2009;25:2181. doi: 10.1021/la8031122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilczewski M, Van der Heyden A, Renaudet O, Dumy P, Coche-Guerente L, Labbe P. Org Biomol Chem. 2008;6:1114. doi: 10.1039/b716214f. [DOI] [PubMed] [Google Scholar]
- 48.Chen Y-X, Zhao L, Huang Z-P, Zhao Y-F, Li Y-M. Bioorg Med Chem Lett. 2009;19:3775. doi: 10.1016/j.bmcl.2009.04.090. [DOI] [PubMed] [Google Scholar]
- 49.Yonzon CR, Jeoung E, Zou S, Schatz GC, Mrksich M, Van Duyne RP. J Am Chem Soc. 2004;126:12669. doi: 10.1021/ja047118q. [DOI] [PubMed] [Google Scholar]
- 50.Morokoshi S, Ohhori K, Mizukami K, Kitano H. Langmuir. 2004;20:8897. doi: 10.1021/la049201x. [DOI] [PubMed] [Google Scholar]
- 51.Kitano H, Takahashi Y, Mizukami K, Matsuura K. Colloids Surf, B. 2009;70:91. doi: 10.1016/j.colsurfb.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 52.Guo C, Boullanger P, Jiang L, Liu T. Biosens Bioelectron. 2007;22:1830. doi: 10.1016/j.bios.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 53.Bellapadrona G, Tesler AB, Grünstein D, Hossain LH, Kikkeri R, Seeberger PH, Vaskevich A, Rubinstein I. Anal Chem. 2011;84:232. doi: 10.1021/ac202363t. [DOI] [PubMed] [Google Scholar]
- 54.Surangi H, Jayawardena N, Wang X, Yan M. Anal Chem. 2013;85:10277. doi: 10.1021/ac402069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Praig VG, Piret G, Manesse M, Castel X, Boukherrroub R, Szunerits S. Electrochim Acta. 2008;53:7838. [Google Scholar]
- 56.Jelinek R, Kolusheva S. Chem Rev. 2004;104:5987. doi: 10.1021/cr0300284. [DOI] [PubMed] [Google Scholar]
- 57.Linman MJ, Taylor JD, Yu H, Chen X, Cheng Q. Anal Chem. 2008;80:4007. doi: 10.1021/ac702566e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edelman GM, Cunningham BA, Reeke GN, Jr, Becker JW, Waxdal MJ, Wang JL. Proc Natl Acad Sci USA. 1972;69:2580. doi: 10.1073/pnas.69.9.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perçin I, Yavuz H, Aksöz E, Denizli A. Biotechnol Prog. 2012;28:756. doi: 10.1002/btpr.1552. [DOI] [PubMed] [Google Scholar]
- 60.Fraser AR, Hemperly JJ, Wang JL, Edelman GM. Proc Natl Acad Sci USA. 1976;73:790. doi: 10.1073/pnas.73.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li YG, Lasia A. J Appl Electrochem. 1997;27:643. [Google Scholar]
- 62.Willets KA, Van Duyne RP. Annu Rev Phys Chem. 2007;58:267. doi: 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
- 63.Loebich O. Gold Bull. 1972;5:2. [Google Scholar]
- 64.Campos-Fernandez C, Azofeifa DE, Hernandez-Jimenez M, Ruiz-Ruiz A, Vargas WE. Opt Mater Exp. 2011;1:85. [Google Scholar]
- 65.Yuan J, Hajebifard A, George C, Berini P, Zou S. J Colloid Interface Sci. 2013;410:1. doi: 10.1016/j.jcis.2013.07.070. [DOI] [PubMed] [Google Scholar]
- 66.Lin Y, Zou Y, Lindquist RG. Biomed Opt Exp. 2011;2:478. doi: 10.1364/BOE.2.000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nath N, Chilkoti A. Anal Chem. 2004;76:5370. doi: 10.1021/ac049741z. [DOI] [PubMed] [Google Scholar]
- 68.Nusz GJ, Curry AC, Marinakos SM, Wax A, Chilkoti A. ACS Nano. 2009;3:795. doi: 10.1021/nn8006465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kedem O, Tesler AB, Vaskevich A, Rubinstein I. ACS Nano. 2011;5:748. doi: 10.1021/nn102617d. [DOI] [PubMed] [Google Scholar]
- 70.Becker J, Trügler A, Jakab A, Hohenester U, Sönnichsen C. Plasmonics. 2010;5:161. [Google Scholar]
- 71.Duan H, Hu H, Kumar K, Shen Z, Yang JKW. ACS Nano. 2011;5:7593. doi: 10.1021/nn2025868. [DOI] [PubMed] [Google Scholar]
- 72.Zhao J, Jensen L, Sung J, Zou S, Schatz GC, Van Duyne RP. J Am Chem Soc. 2007;129:7647. doi: 10.1021/ja0707106. [DOI] [PubMed] [Google Scholar]
- 73.Chen H, Kou X, Yang Z, Ni W, Wang J. Langmuir. 2008;24:5233. doi: 10.1021/la800305j. [DOI] [PubMed] [Google Scholar]
- 74.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chem Rev. 2005;105:1103. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 75.Lahiri J, Isaacs L, Grzybowski B, Carbeck JD, Whitesides GM. Langmuir. 1999;15:7186. [Google Scholar]
- 76.Patel N, Davies MC, Hartshorne M, Heaton RJ, Roberts CJ, Tendler SJB, Williams PM. Langmuir. 1997;13:6485. [Google Scholar]
- 77.Stelzle M, Weismueller G, Sackmann E. J Phys Chem. 1993;97:2974. [Google Scholar]
- 78.Gafni Y, Weizman H, Libman J, Shanzer A, Rubinstein I. Chem Eur J. 1996;2:759. [Google Scholar]
- 79.La Belle JT, Gerlach JQ, Svarovsky S, Joshi L. Anal Chem. 2007;79:6959. doi: 10.1021/ac070651e. [DOI] [PubMed] [Google Scholar]
- 80.Loaiza OA, Lamas-Ardisana PJ, Jubete E, Ochoteco E, Loinaz I, Cabanero G, Garcia I, Penades S. Anal Chem. 2011;83:2987. doi: 10.1021/ac103108m. [DOI] [PubMed] [Google Scholar]
- 81.Cunningham S, Gerlach JQ, Kane M, Joshi L. Analyst. 2010;135:2471. doi: 10.1039/c0an00276c. [DOI] [PubMed] [Google Scholar]
- 82.Sanchez-Pomales G, Zangmeister R. Int J Electrochem. 2011;5:56. [Google Scholar]
- 83.Pandey B, Bhattarai JK, Pornsuriyasak P, Fujikawa K, Catania R, Demchenko AV, Stine KJ. J Electroanal Chem. 2014;717–718:47. doi: 10.1016/j.jelechem.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pandey B, Tan YH, Fujikawa K, Demchenko AV, Stine KJ. J Carbohydr Chem. 2012;31:466. doi: 10.1080/07328303.2012.683909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ding S-J, Chang B-W, Wu C-C, Lai M-F, Chang H-C. Anal Chim Acta. 2005;554:43. [Google Scholar]
- 86.McEwen GD, Chen F, Zhou A. Anal Chim Acta. 2009;643:26. doi: 10.1016/j.aca.2009.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Janek RP, Fawcett WR, Ulman A. Langmuir. 1998;14:3011. [Google Scholar]
- 88.Stamou D, Gourdon D, Liley M, Burnham NA, Kulik A, Vogel H, Duschl C. Langmuir. 1997;13:2425. [Google Scholar]
- 89.Finklea HO, Snider DA, Fedyk J. Langmuir. 1990;6:371. [Google Scholar]
- 90.Guo Y-G, Zhang H-M, Hu J-S, Wan L-J, Bai C-L. Thin Solid Films. 2005;484:341. [Google Scholar]
- 91.Cao L, Yan P, Sun K, Kirk DW. Electroanalysis. 2009;21:1183. [Google Scholar]